
Abstract
We investigated the role of carbon monoxide as a neural modulator of extracellular glutamate concentration in rat hippocampus CA1 in transient forebrain ischemia by using metalloporphyrins, which block the production of carbon monoxide through the inhibition of heme oxygenase (HO) activity. Infusion of 10 and 100 μM zinc protoporphyrin IX, which inhibits nitric oxide synthase activity as well as HO activity, significantly increased glutamate concentration compared with that on the vehicle-treated side. However, infusion of 100 μM tin mesoporphyrin IX, which inhibits only HO activity, did not affect glutamate concentration in ischemia. Our results therefore do not support the hypothesis that carbon monoxide acts as a neural messenger through the modulation of extracellular glutamate concentration in ischemia.
Like nitric oxide (NO), carbon monoxide (CO), which is also a noxious gas that activates guanylyl cyclase, is a candidate neural messenger (Verma et al., 1993; Dawson and Snyder, 1994). Heme ring cleavage by heme oxygenase (HO) generates CO and biliverdin, which is rapidly reduced to bilirubin (Sun et al., 1990). Blocking of CO production with zinc protoporphyrin IX (Zn PP-9), an HO inhibitor, prevents the induction of long-term potentiation (LTP) in hippocampal slices (Stevens and Wang, 1993; Zhuo et al., 1993), blocks the effect of metabotropic glutamate receptor activation in rat brain slices (Glaum and Miller, 1993), inhibits release of corticotropin-releasing hormone in cell culture (Parkes et al., 1994; Pozzoli et al., 1994), and reduces depolarization-induced glutamate release in synaptoneurosomes (Shinomura et al., 1994). Although these in vitro studies indicate that CO may act as a neural messenger, no in vivo studies have examined the role of CO in cerebral ischemia. Furthermore, Meffert et al. (1994) reported that Zn PP-9 inhibits not only HO, but also NO synthase (NOS) activity, while tin mesoporphyrin IX (Sn MP-9) inhibits HO activity with little effect on NOS activity. Therefore, previous data concerning the role of CO, obtained by using Zn PP-9, may reflect the inhibitory effects on both HO and NOS activities. To test the hypothesis that CO acts as a neural messenger modulating extracellular glutamate concentration in ischemia, we evaluated the effects of infusing Zn PP-9 and Sn MP-9 in the hippocampus CA1 on glutamate concentration in rats with transient forebrain ischemia, using an in vivo brain microdialysis technique. We also evaluated the effect of infusing bilirubin, an end product of heme catabolism that acts as an antioxidant (Stocker et al., 1987a,b).
MATERIALS AND METHODS
Male Wistar rats weighing ∼350 g were used. Transient forebrain ischemia was achieved by using a modification of the four-vessel occlusion method (Pulsinelli and Brierly, 1979; Schmidt-Kastner et al., 1989). On day 1, following anesthesia with 2.5% halothane, both vertebral arteries were permanently occluded by electrocautery. Two microdialysis probe/platinum electrode assemblies (EPT-BDP-I-2-01; Eicom, Kyoto, Japan) were placed stereotaxically at the CA, region of the hippocampus bilaterally (bregma, 3.6 mm; lateral, 2.2 mm; depth, 2.8 mm). Simultaneously, a thermocouple probe (52K/J thermometer; John Fluke Mfg., WA, U.S.A.) was placed in the subdural space through a burr hole. On day 2, rats were reanesthetized with 2.5% halothane for insertion of a femoral arterial catheter and a venous catheter. Each rat was then tracheotomized, paralyzed with pancuronium bromide (0.6 mg/kg i.v.), and ventilated with a mixture of 30% oxygen, 70% nitrous oxide, and 1.0% halothane. Both carotid arteries were transiently occluded with microaneurysm clips for 30 min. Animals that did not show flattened EEG at the ischemic insult were excluded from the study. During the experiment, brain and rectal temperatures were controlled at ∼37°C with an external heating lamp and a heating pad, respectively. Arterial gases, blood glucose, and hematocrit were examined during the experiment. At the end of the experiment, animals were killed with an overdose of pentobarbital sodium.
In all rats, both microdialysis probes were perfused with Ringer's solution (155 mM NaCl, 5.5 mM KCl, 2.3 mM CaCl2) starting 2.5 h prior to carotid occlusion at a rate of 2.0 μl/min. Animals were divided into four groups as follows: Group 1 (n = 7), group 2 (n = 8), and group 4 (n = 7) received 10 and 100 μM Zn PP-9 (Paesel, Frankfurt, Germany) and 100 μM Sn MP-9 (Porphyrin Products, UT, U.S.A.), respectively, dissolved in Ringer's solution with 1.0% polyoxyethylene sorbitan monooleate (pH 7.0) (Wako, Tokyo, Japan) in the left hippocampus and Ringer's solution with 1.0% polyoxyethylene sorbitan monooleate as the control in the right hippocampus. Group 3 (n = 6) received 100 μM Zn PP-9 dissolved in the same solution as for group 1 in the left hippocampus and 100 μM Zn PP-9 with 100 μM bilirubin (Wako) in the opposite hippocampus. The infusions were started 0.5 h prior to the occlusion and stopped at 1 h postocclusion.
Glutamate concentration in the dialysates was measured by HPLC with fluorescence detection after precolumn derivatization with o-phthaldialdehyde (Lindroth and Mopper, 1979). The glutamate response was calibrated with internal and external standards. CBF was measured by the hydrogen clearance method (Aukland et al., 1964) using a digital blood flow meter (NMG-D1; Unique Medical, Tokyo, Japan).
The statistical significance of differences in glutamate concentration and CBF between with and without treatment before ischemia and in glutamate concentration and CBF between both hippocampi in each group was analyzed by the one-sample Wilcoxon test.
RESULTS
Although statistically significant changes in physiological parameters in each group were observed (the paired t test), these changes were within the ranges of normal physiologic responses in forebrain ischemia and were thought unlikely to have materially affected the conclusions reached about the drugs used.
The changes in CBF in hippocampal CA1 are given in Table 1. Preischemia CBF in the four groups ranged from 76.2 to 87.5 ml 100 g−1 min−1, and a statistically significant difference was observed between the two hippocampi in group 1 only. Before the onset of ischemia, no treatment in any of the four groups changed the CBF value significantly. During ischemia and recirculation, no significant difference in CBF values between the two hippocampi in any group was observed. Thus, there was no significant difference in CBF values at preischemia, during ischemia, or during recirculation among the four groups.
Hippocampal CBF (ml 100 g−1 min−1) on both sides in the four groups
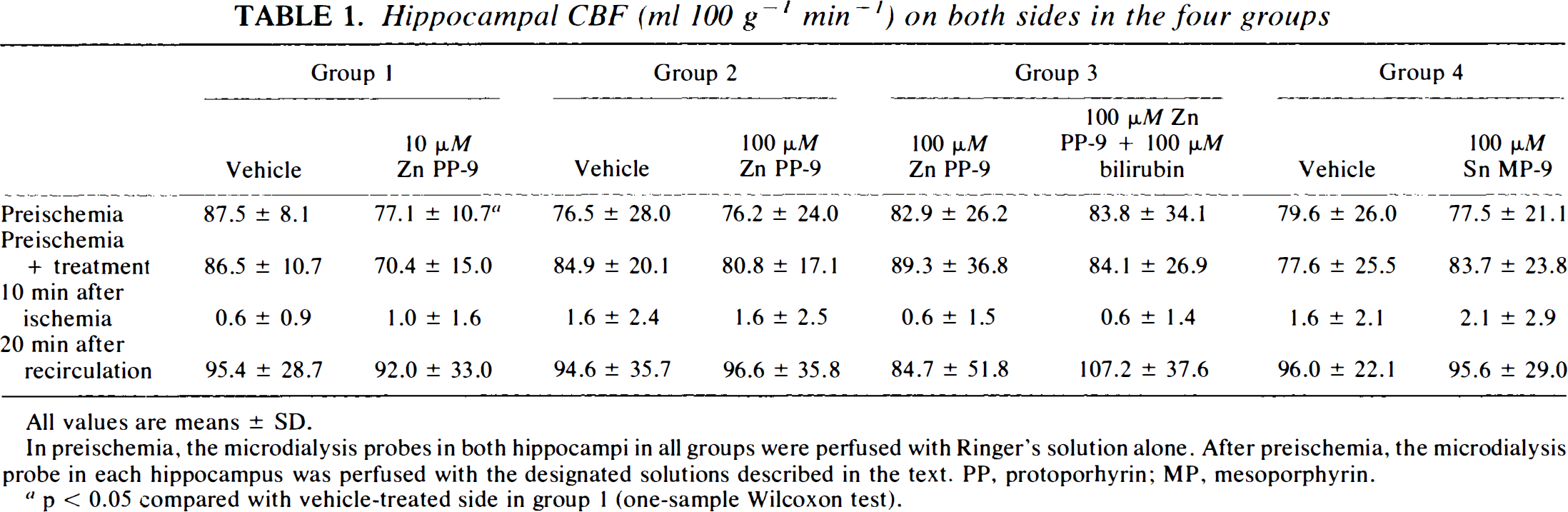
All values are means ± SD.
In preischemia, the microdialysis probes in both hippocampi in all groups were perfused with Ringer's solution alone. After preischemia, the microdialysis probe in each hippocampus was perfused with the designated solutions described in the text. PP, protoporhyrin; MP, mesoporphyrin.
p < 0.05 compared with vehicle-treated side in group 1 (one-sample Wilcoxon test).
Before onset of ischemia, there was no significant difference in extracellular glutamate levels between the two hippocampi in any group. None of the treatments changed the extracellular glutamate level before ischemia compared with that in Ringer's solution in each group. In group 1, topical infusion of 10 μM Zn PP-9 significantly increased glutamate concentration, which reached 2,497 ± 1,649 and 2,738 ± 2,444% of baseline compared with 1,255 ± 1,123 and 1,296 ± 1,142% of baseline at the 30-min ischemia and 10-min recirculation time points in the vehicle-treated side, respectively (p < 0.05, one-sample Wilcoxon test). In group 2 (Fig. 1a), topical infusion of 100 μM Zn PP-9 also significantly increased glutamate concentration at the 30-min ischemia and 10-min recirculation time points compared with the vehicle-treated side (p < 0.05, one-sample Wilcoxon test). In group 3, there was no significant difference in glutamate concentration between the side treated with 100 μM Zn PP-9 and the other treated with 100 μM Zn PP-9 and 100 μM bilirubin. In group 4 (Fig. 1b), glutamate concentrations in the vehicle-treated side and the 100 μM Sn MP-9-treated side reached 1,226 ± 838 and 1,050 ± 493% of baseline at the 10-min recirculation time point, respectively, and no significant difference in glutamate concentration between the two sides was observed.
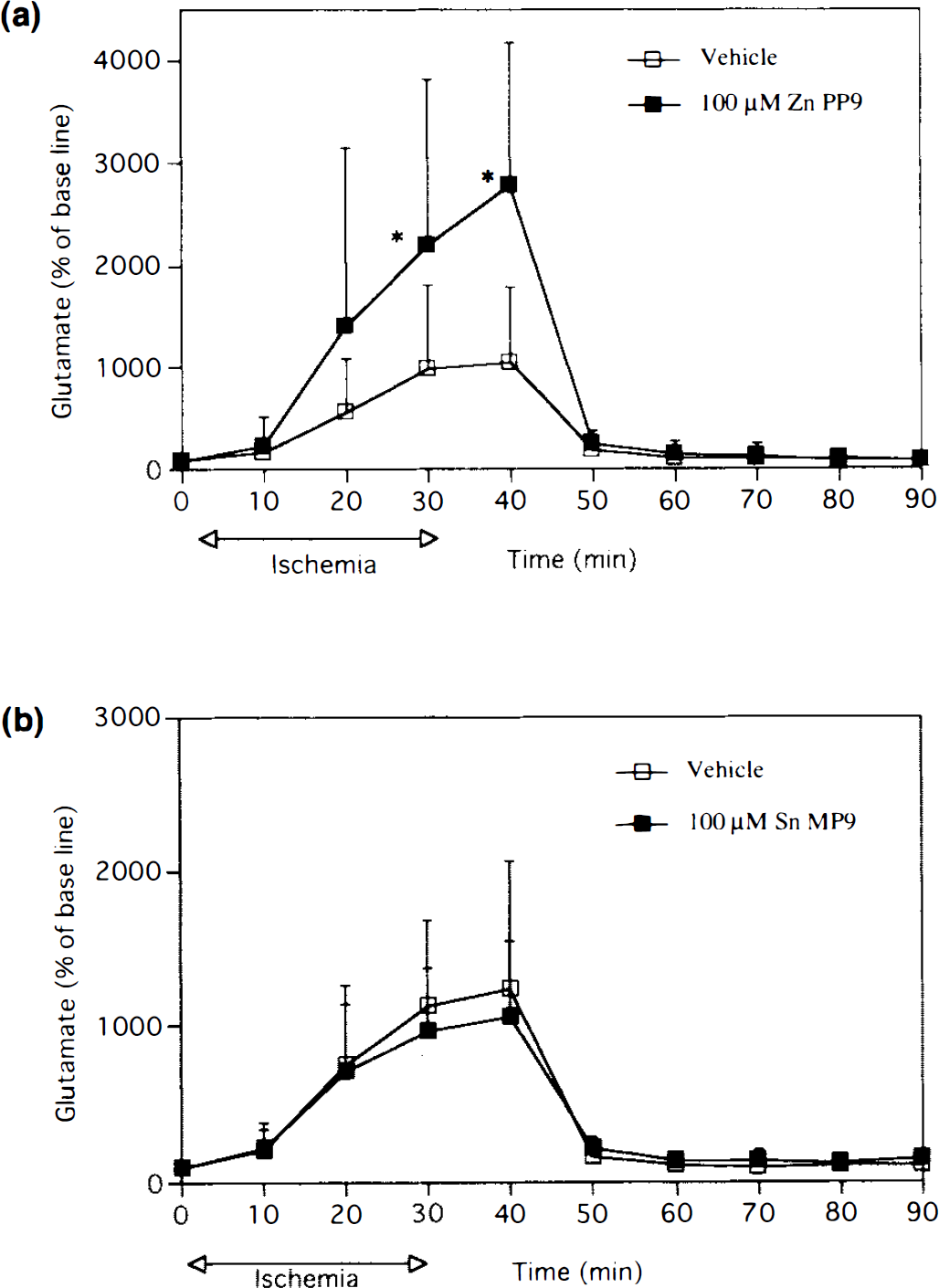
Effects of 100 μM zinc protoporphyrin IX (Zn PP-9) and 100 μM in mesoporphyrin IX (Sn MP-9) on the concentration of extracellular glutamate in transient forebrain ischemia,
DISCUSSION
A role of CO as a neural messenger has been proposed (Verma et al., 1993; Dawson and Snyder, 1994), but its influence in cerebral ischemia has not been studied. Our main finding is that the administration of Sn MP-9 does not affect extracellular glutamate concentration during forebrain ischemia, while the administration of Zn PP-9 significantly increases glutamate concentration during ischemia and following recirculation. Since Zn PP-9 reduces NOS activity in a dose-dependent fashion, while Sn MP-9 has no significant effect on NOS activity (Meffertet al., 1994), we assume that our data using Zn PP-9 may reflect inhibition of NOS activity in addition to inhibition of HO activity. Thus, our finding that Sn MP-9 had no effect on glutamate concentration is not consistent with the hypothesis that CO acts as a neural messenger modulating extracellular glutamate concentration in ischemia. This confirms the work of Meffert et al. (1994), who have concluded that CO does not act as a mediator in either LTP induction or expression/maintenance, because the ability of metalloporphyrins to block LTP in rat hippocampal slices was better correlated with their inhibition of NOS than with their inhibition of HO, which basically coincides with our conclusion.
Although it is generally accepted that neuronal NO production contributes to the development of ischemic brain injury (Huang et al., 1994), the effect of NOS inhibitors on glutamate concentration in ischemia is still controversial. In focal ischemia, it was found that an NOS inhibitor suppressed the increase in extracellular glutamate (Buisson et al., 1993). In forebrain ischemia, however, an NOS inhibitor increased extracellular glutamate concentration during ischemia (Nanri et al., 1994) or on reperfusion (Zhang et al., 1993, 1995), although the mechanism involved is unclear. Therefore, our finding of an increase in glutamate concentration by Zn PP-9 in forebrain ischemia may reflect the inhibitory effect on NOS activity. To examine other possible explanations for this result, we evaluated the effect of bilirubin on extracellular glutamate in ischemia, because bilirubin, which is also suppressed by Zn PP-9, can efficiently scavenge peroxyl radicals at micromolar concentrations in vitro (Stocker et al., 1987a,b). Our results suggested that the glutamate increase caused by Zn PP-9 was not related to suppression of bilirubin production. Further, Zn PP-9 had no influence on CBF. Thus, the effect of Zn PP-9 seems to arise predominantly through its modification of NOS levels. We present a schema showing the proposed relation between extracellular glutamate and CO in cerebral ischemia in Fig. 2.
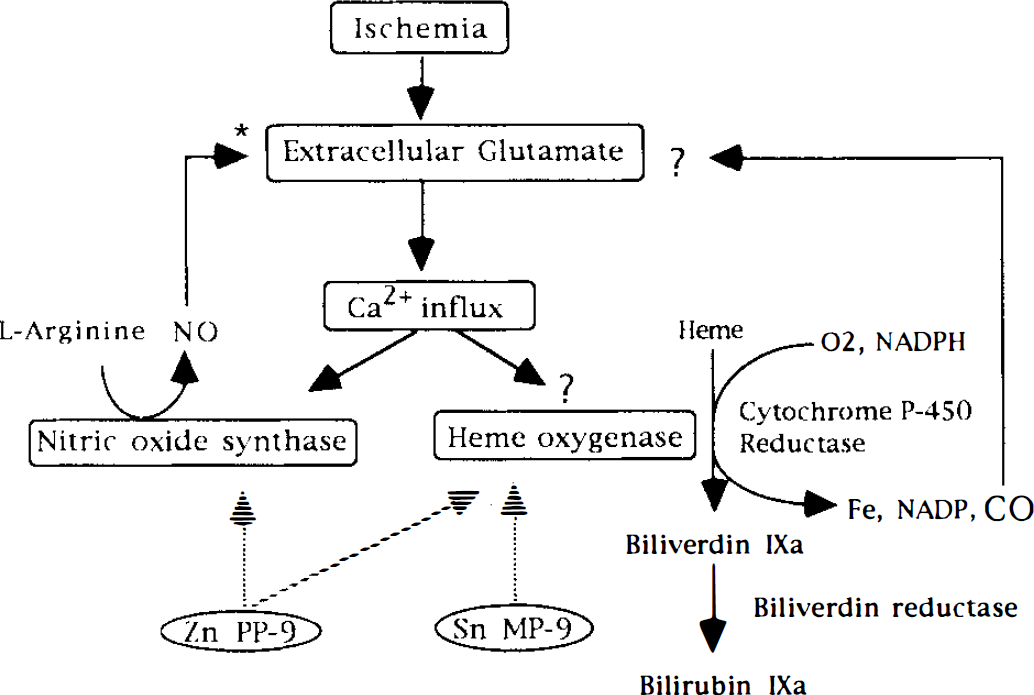
Schema showing the proposed relation between extracellular glutamate and CO in cerebral ischemia. Excessive accumulation of extracellular glutamate in cerebral ischemia may increase intracellular Ca2+ through the activation of glutamate receptors. NO is involved as a neural messenger by modulating extracellular glutamate concentration in cerebral ischemia (*). Although the mechanism of the activation of heme oxygenase (HO) in ischemia is unclear, HO cleaves the heme ring into CO and biliverdin. Biliverdin is rapidly reduced to bilirubin. Zinc protoporphyrin IX (Zn PP-9) inhibits both HO and NO synthase activities, while tin mesoporphyrin IX (Sn MP-9) inhibits only HO activity. Sn MP-9 did not affect glutamate concentration in ischemia in this study. A dotted arrow indicates the inhibitory effect by the drug.
In conclusion, our data do not support a role of CO in the modulation of extracellular glutamate concentration, although the possibility remains that CO may have other functions as a neural messenger in the brain.
Footnotes
Acknowledgment:
We thank Drs. A. M. Hakim and K. Ichimori for useful suggestions. The skillful technical assistance of Y. Takahari, S. Ueno, and Y. Shinozaki in the laboratories for experimental animals and physiologic research is gratefully acknowledged. This investigation was supported by a research grant from Tokai University School of Medicine (1994).