
Abstract
The Tat-NR2B9c peptide has shown clinical efficacy as a neuroprotective agent in acute stroke. Tat-NR2B9c is designed to prevent nitric oxide (NO) production by preventing postsynaptic density protein 95 (PSD-95) binding to
INTRODUCTION
Brain ischemia leads to acute cell death, in part through sustained activation of
Postsynaptic density protein 95 (PSD-95) serves as a scaffolding protein for the clustering of synaptic receptors, ion channels, and associated signaling proteins. 3 It binds both to neuronal NO synthase (nNOS) and to the NR2B subunits of NMDAr. 4 Targeted disruption of these binding interactions with a Tat-conjugated peptide comprising the nine C-terminal residues of NR2B blocks NMDA-induced NO production. This peptide, termed ‘Tat-NR2B9c’ or ‘NA-1’ also blocks excitotoxic neuronal death, reduces acute ischemic cell death in animal stroke models, and has shown efficacy as a neuroprotective agent in clinical stroke.2,5,6
Given the crucial role of NO in excitotoxic injury, the disruption of NMDAr/nNOS interaction is a likely mechanism for the neuroprotective effects observed with Tat-NR2B9c. However, superoxide production by NADPH oxidase (NOX2) is equally required for peroxynitrite formation and for excitotoxic and ischemic cell death.1,2,7–9 These factors raise the possibility that disruption of NMDAr/NOX2 interactions might also (or alternatively) contribute to the mechanism of this promising stroke therapeutic. The present studies confirm that Tat-NR2B9c blocks NMDA-induced superoxide formation by a mechanism that prevents phosphorylation of the NOX2 p47phox subunit.
MATERIALS AND METHODS
MATERIALS
Studies were approved by the SFVAMC Animal Studies subcommittee and conducted according to the current Guide for the Care and Use of Laboratory Animals (Public Health Service Policy on Humane Care and Use of Laboratory Animals). The Tat-NR2B9c was a kind gift from M Tymianski, Toronto, Canada. The gp91ds-Tat and scrambled sequence (control) peptides were purchased from CPC Scientific (Sunnyvale, CA, USA). The control peptide sequence was CSFNSYELGSLCYGRKKRRQRRR, with underlining denoting the Tat component. All other reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA) except where noted.
Cell Cultures
Neuronal monocultures were prepared from the cortices of embryonic mice as described.
10
Studies were initiated in cultures at days 10 to 12
Superoxide Detection and Immunostaining
Oxidation of dihydroethidium to fluorescent ethidium species (Eth) was used to monitor intracellular superoxide levels as described. 10 The Eth signal was quantified as fluorescence area × mean intensity and normalized to cell number. Intracellular calcium imaging was performed with Fura FF and quantified as previously described. 10 For immunostaining, cultures were fixed in 4% formaldehyde and incubated overnight with primary antibodies: anti-4-hydroxynonenal (Abcam, Cambridge, MA, USA); anti-Ser139-Histone H2A.X (Cell Signaling Technology, Danvers, MA, USA), or anti-Ser328 phospho-p47phox,11 together with mouse anti-microtubule-associated protein 2 (Chemicon, Temecula, CA, USA). Antibody binding was visualized with fluorescently-tagged secondary antibodies and quantified as the mean fluorescence intensity within the area defined by MAP-2 fluorescence (neurons). Most studies were independently replicated by at least two different observers, and in all cases the individual performing data analysis was blinded to the experimental conditions.
Cell Death
Peptides and drugs were added from concentrated stocks 15 minutes before the addition of NMDA, and the NMDA was removed by medium exchanges after 30 minutes. Twenty-four hours later live neurons were identified by calcein green retention, and dead neurons were identified by Hoeschst 33258. 10 Live and dead neurons were counted in three randomly chosen fields in four wells per plate of each experiment, and results were expressed as the percent of neurons that were dead.
Statistics
The
RESULTS
To determine whether NR2B binding to PSD-95 is required for NMDAr-triggered superoxide production, we treated neuronal cultures with 100 μmol/L NMDA for 30 minutes together with Tat-NR2B9c over a range of concentrations and evaluated superoxide formation by the dihydroethidium method. At the highest concentration (0.5 μmol/L), Tat-NR2B9c suppressed NMDA-induced superoxide production by about 75% (Figures 1A and 1B). This effect was comparable to that achieved with the gp91ds-Tat peptide (Figures 1B and 1C), which prevents assembly of the active neuronal NOX2 complex.10,12 By contrast, the nNOS inhibitor L-NG-nitroarginine methyl ester (L-NAME) had no effect on superoxide production. The inhibitory effect of Tat-NR2B9c on NMDA-induced superoxide production was further confirmed by imaging formation of the lipid peroxidation product, 4-hydroxynonenal (Figures 1D and 1E). Additional evidence that Tat-NR2B9c prevents NOX2 activation was obtained by assessing phosphorylation of the p47phox serine 328 residue, which is required for assembly of the functional NOX2 complex. 11 NMDA induced a marked increase in phospho-ser328 p47phox immunoreactivity in cultured neurons, and this increase was negated by co-incubation with the Tat-NR2B9c (Figures 2A and 2B).
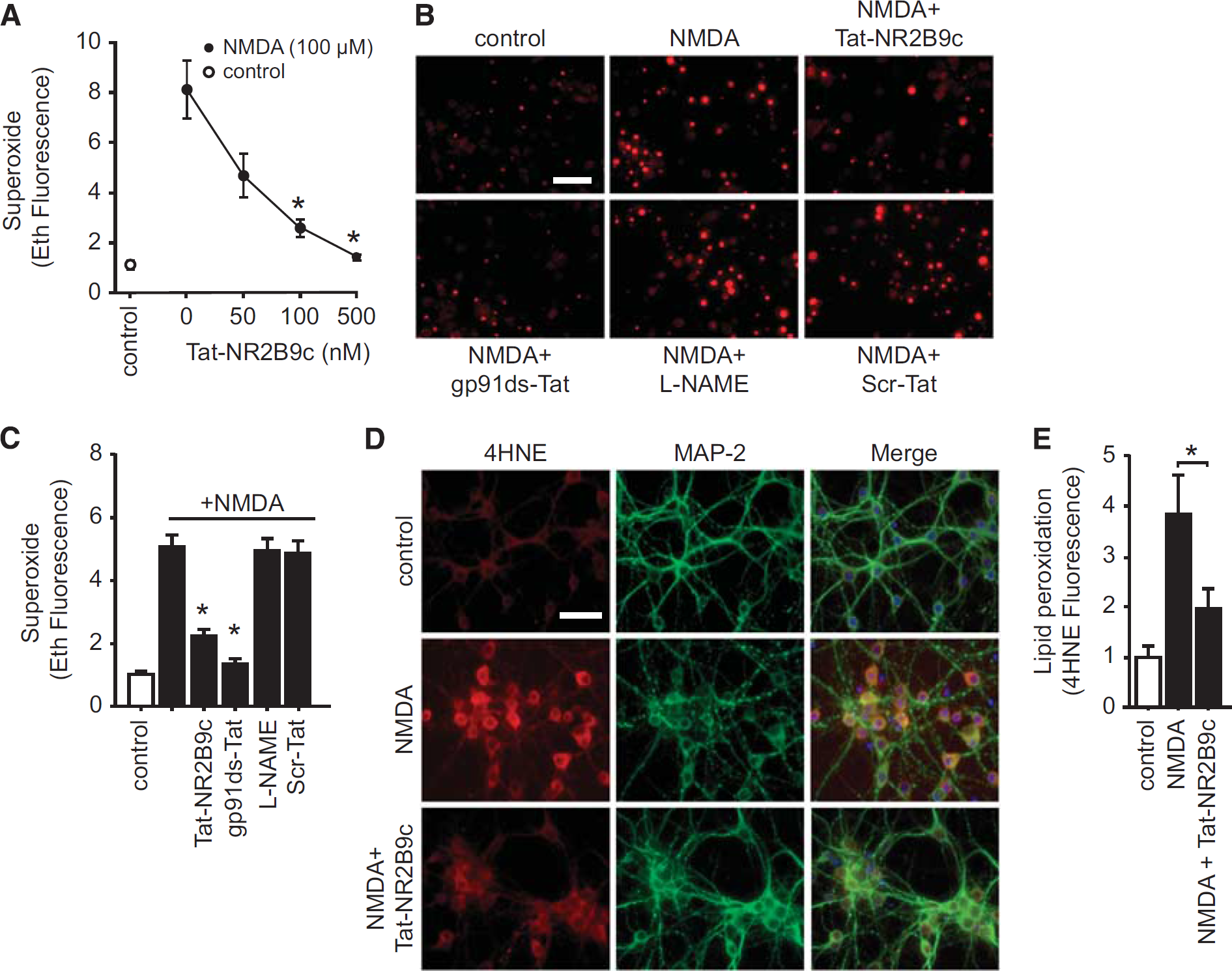
Tat-NR2B9c blocks
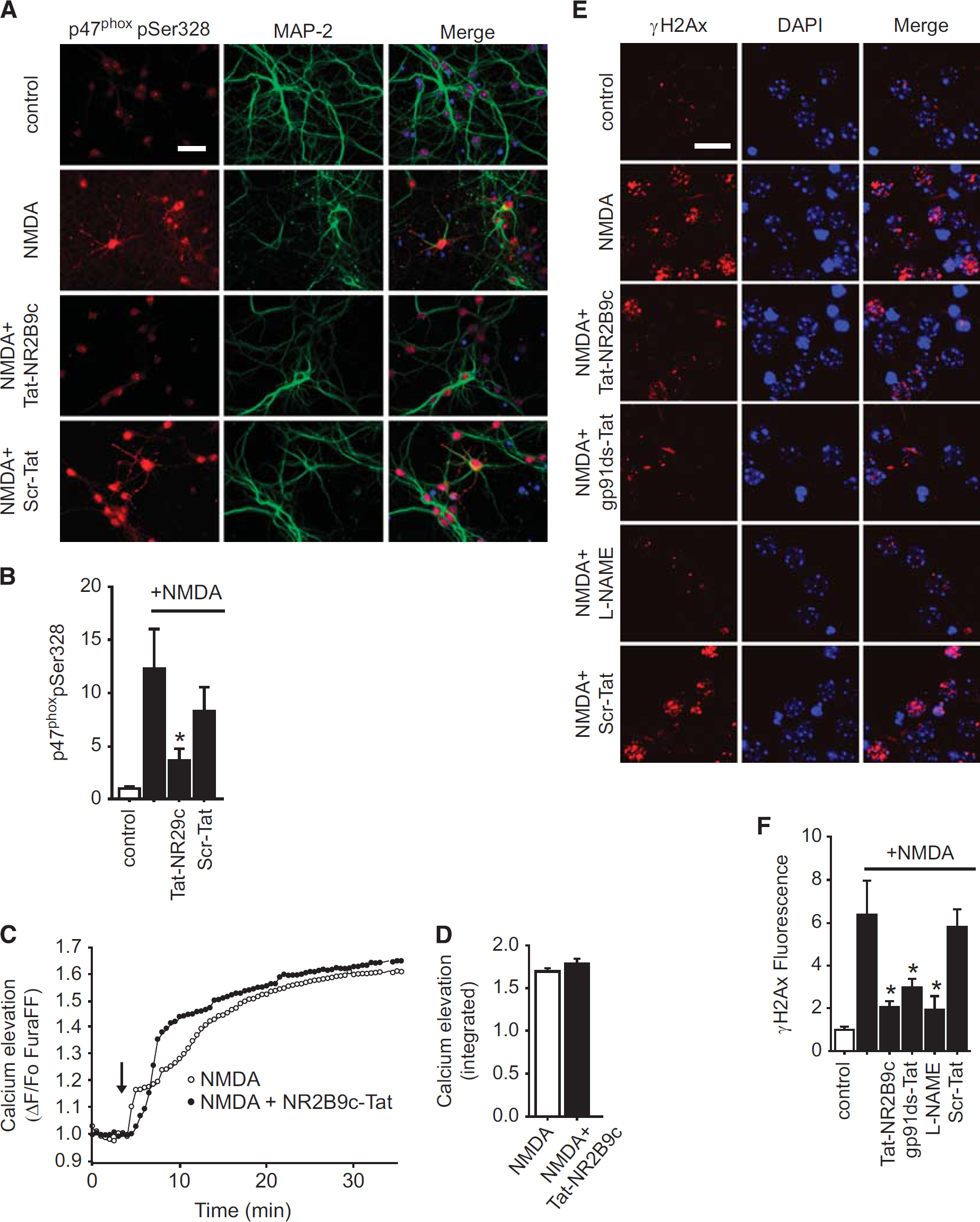
Tat-NR2B9c prevents
These results suggest that NR2B binding to PSD-95 is an essential step linking NMDAr activation to NOX2 activation. However, Tat-NR2B9c might alternatively impair NOX2 activation by reducing NMDA-induced calcium influx. This possibility was evaluated using the low-affinity calcium indicator Fura-FF. Tat-NR2B9c was found to have no measurable effect on the rate or magnitude of NMDAr-induced calcium influx (Figures 2C and 2D), consistent with prior reports. 5
NMDA-induced cell death is caused primarily by the formation of peroxynitrite and related reactive oxygen species, which induce DNA strand breaks and resultant activation of the poly(ADP-ribose) polymerase cell death cascade.1,2 DNA strand breaks thus provide a link between oxidative stress and cell death. Immunostaining for phosphorylated histone H2A.X (γ-H2A.X), which accumulates at DNA strand breaks, showed that the NOX2 inhibitor gp91ds-Tat prevented NMDA-induced DNA breaks, and that Tat-NR2B9c had comparable efficacy (Figures 2E and 2F). Importantly, NMDA-induced DNA strand breaks were also prevented by the nNOS inhibitor L-NAME, confirming that NO production was suppressed by L-NAME under the conditions of this study. Assessment of NMDA-induced cell death under these same treatment conditions gave a parallel result; neuronal death was significantly reduced by Tat-NR2B9c, gp91ds-Tat, or L-NAME (
DISCUSSION
The Tat-NR2B9c peptide is one of the first neuroprotectant agents to show efficacy in patients with acute stroke. Tat-NR2B9c is designed to uncouple NO production from NMDAr activation by blocking PSD-95 binding to NMDAr and nNOS. The present studies show that Tat-NR2B9c also prevents NMDA-induced superoxide formation by blocking p47phox phosphorylation, which is required for assembly of the functional NOX2 complex. 14 These findings indicate that PSD-95 couples NMDAr to the signaling pathway activating NOX2. Given that superoxide production is required for excitotoxic cell death,1,2,8,9 the findings also suggest that the neuroprotective effect of Tat-NR2B9c may be partly or wholly attributable to its suppression of NOX2 activation.
The signaling pathway linking NR2B to NOX2 involves activation of the atypical protein kinase PKCζ by phosphoinositide 3-kinase, and subsequent phosphorylation of p47phox by PKCζ. 10 This process is regulated at multiple steps, and requires a conformational change in p47phox to expose the critical phosphorylation sites. 14 It has not yet been established how the binding of NMDA (or glutamate) to NR2B activates phosphoinositide 3-kinase, but prior reports indicate that PSD-95 provides a structural framework for this interaction. 15 Our results support this idea by showing that the Tat-NR2B9c, which targets the PDZ domain of PSD-95, disrupts the functional coupling between NR2B and NOX2. Although PDZ domains are present on proteins other than PSD-95, specifically the PDZ domains on PSD-95 are essential for the neuroprotective effect of Tat-NR2B9c. 4
The oxidation of dihydroethidium to fluorescent ethidium species is specific for superoxide under cell-free conditions, but metals and peroxidases present in cells can enable dihydroethidium oxidation by other reactive oxygen species. Here, the observation that NMDA-induced oxidation of dihydroethidium was prevented by NOX2 inhibition confirmed that the observed ethidium signal was attributable to superoxide.
Given that Tat-NR2B9c is known to uncouple NMDA receptors from nNOS, we also considered the possibility that the inhibitory effect of Tat-NR2B9c on NOX2 activation might in some way be secondary to this inhibitory effect on nNOS. Indeed, the NOS inhibitor L-N
The present findings do not identify the mechanism by which Tat-NR2B9c prevents superoxide production, but the effect of Tat-NR2B9c on p47phox phosphorylation suggests an effect upstream of this event. We propose that Tat-NR2B9c blocks p47phox phosphorylation through its disruption of PSD-95/NR2B binding, given that NMDA-induced phosphorylation of p47phox requires activation of the PI3K pathway9,10 and that PI3K is coupled to NR2B by PSD-95. 15 Additional study will be required to confirm this proposal; however, regardless of the mechanism, the present results suggest that the inhibitory effect of Tat-NR2B9c on NMDA-induced superoxide production may contribute to the neuroprotective effect of this promising therapeutic agent.
DISCLOSURE/CONFLICT OF INTEREST
The authors declare no conflict of interest.