
Abstract
To clarify whether heme oxygenase-1 (HO-1) protein plays a protective role against cerebral ischemia, we investigated the effects of an HO inhibitor (tin mesoporphyrin IX [SnMP] three doses of 30 μmol/kg, intraperitoneally) and an HO inducer (hemin, three doses of 30 μmol/kg, intraperitoneally) on the pathologic outcome and on the immunohistochemical reaction for HO-1 after 20-minute transient forebrain ischemia followed by 3-day reperfusion in rats. Hemin significantly increased viable neurons in the cortex (compared to the SnMP-treated group, P<.05) and striatum (compared to the saline-treated group at P<.01 and SnMP-treated group at P<.05), and intense HO-1 immunoreactivity was observed in cortex and striatum, whereas the administration of SnMP tended to decrease viable neurons in the parietal cortex. In contrast, neither hemin nor SnMP affected the pathologic outcome in the CA1 and CA3 hippocampi, in which HO-1 immunoreactivity was weak. These results suggest that induction of HO-1 protein may contribute to cellular defense against ischemic damage in brain regions where potential ability to synthesize HO-1 is retained in ischemia.
While most of protein synthesis is inhibited in cerebral ischemia (Kiessling et al., 1986; Mies et al., 1991), specific stress proteins are induced, including the heat shock proteins (Tomioka et al., 1993), glutathione peroxidase (Takizawa et al., 1994), ornithine decarboxylase (Müller et al., 1991), and others. Heme oxygenase (HO)-1, an inducible form of HO, is well known as the 32-kd heat shock protein which is induced by various stimuli, including heat shock, metals, and oxidative stress (Dwyer et al., 1992; Ewing and Maines, 1991, 1992; Keyse and Tyrrell, 1989). HO catalyzes the rate-limiting step in heme catabolism, generating free iron, carbon monoxide (CO) and biliverdin, which is rapidly reduced to bilirubin by biliverdin reductase (Kikuchi and Yoshida, 1983). Biliverdin and bilirubin have a strong cellular antioxidant activity and may contribute to cellular defenses against oxidative stress (Stocker et al., 1987a, 1987b). CO that activates guanylyl cyclase acts as a putative neural messenger (Verma et al., 1993; Dawson and Snyder, 1994), inhibits nitric oxide synthase (NOS) activity in rat cerebellum (White and Marietta, 1992), and modulates the NO-cGMP signaling system (Ingi et al., 1996). CO has the ability to relax smooth muscle and to block platelet aggregation (Verma et al., 1993). Furthermore, the induction of HO inhibits some of the enzymes involved in arachidonic acid metabolism (Martasek et al., 1991). These findings indicate that HO plays a protective role against ischemia. In contrast, it has been reported that the inhibition of HO activity reduced infarct size and edema accumulation in rat focal ischemia (Kadoya et al., 1995) and blocked the effect of metabotropic glutamate receptor activation (Glaum and Miller, 1993). In addition, an HO inhibitor reduced depolarization-induced glutamate release (Shinomura et al., 1994), although it did not influence glutamate release in transient forebrain ischemia (Takizawa et al., 1996). Thus, an increase in HO activity may produce divergent effects in cerebral ischemia and it has not yet been established whether induction of HO-1 protein plays a protective role against cerebral ischemia. To evaluate the role of HO-1 in cerebral ischemia, we investigated the effects of tin mesoporphyrin IX (SnMP), an inhibitor of HO activity, and ferriprotoporphyrin IX chloride (hemin), an HO inducer, on the pathologic outcome and on the immunohistochemical reaction for HO-1 in rat transient forebrain ischemia.
MATERIALS AND METHODS
Transient forebrain ischemia model
The surgical and anesthetic procedures were approved by the Animal Experimentation Committee, School of Medicine, Tokai University.
The experiments were performed on 26 male Wistar rats (Clea Japan Co. Ltd., Tokyo) weighing approximately 350 g. Animals were fasted overnight but allowed free access to water. Transient forebrain ischemia was achieved by using the four-vessel occlusion method (Pulsinelli and Brierley, 1979) with some modification (Chen et al., 1997). Briefly, on the day before the experiment, after induction of anesthesia with 2.5% halothane, both vertebral arteries were permanently occluded by electrocautery through the alar foramina of the first cervical vertebrae. Both carotid arteries were isolated through a midline neck incision, silk ligatures were placed loosely around them and the wound was closed with surgical clips. All skin wounds were instilled with 1% xylocaine and food was withheld overnight as the animals recovered from anesthesia. On the day of the experiment, under 2.0% halothane anesthesia with a mixture of 30% oxygen and 70% nitrous oxide, the tail artery was exposed and catheterized with polyethylene tubing (PE-50) to allow blood sampling and continuous monitoring of arterial blood pressure. A thermocouple probe (52K/J thermometer; John Fluke Mfg. Co., Everett, WA) was placed in the temporal muscles, and bipolar EEG electrodes were also inserted into the temporal muscles. Three minutes before carotid occlusions, the anesthesia was discontinued. After local infiltration of the neck with xylocaine, both common carotid arteries were lifted using the silk ligatures previously placed around them and occluded for 20 minutes with micro-aneurysm clips (Sugita clip #07-940-12, Mizuho Ika Co., Ltd., Japan). We confirmed that the EEG activity became isoelectric in all rats studied within 10 seconds after carotid occlusions. At the end of 20-minute occlusion, the clips were released and restoration of carotid flow was confirmed by direct observation of the carotid arteries. Just after the start of recirculation, drugs as mentioned below were administered intraperitoneally. During the experiment, temporal muscle and rectal temperatures were controlled at about 37°C with an external heating lamp and a heating pad, respectively. These temperatures and the MABP were continuously monitored. Arterial gases and blood glucose level were examined before ischemia, at 5 minutes after carotid occlusions and 30 minutes after the start of recirculation. Hematocrit was examined before ischemia. At the end of the surgery, all wounds were sutured, the arterial catheter was removed, and rats were allowed free access to water and food.
Three days later, the animals were re-anesthetized with 4% halothane, and killed by transabdominal-arterial perfusion fixation with 100 mL of phosphate-buffered saline, followed by 100 mL of 4% paraformaldehyde in 0.01 mol/L phosphate buffer. The brain was carefully removed from the skull and stored in 4% paraformaldehyde in 0.01 mol/L phosphate buffer (pH 7.4) for 24 hours. Coronal brain sections were obtained at 20-μm intervals and stained with hematoxylin and eosin, as well as HO-1 glial fibrillary acidic protein and tubulin by immunohistochemical techniques, as described below.
Experimental design
Animals were divided into four experimental groups. Groups 1, 2, and 3 were exposed to forebrain ischemia as described above. Group 1 (n=7) received three intraperitoneal doses of 30 μmol/kg SnMP (tin mesoporphyrin IX; M.W., 754.29; Porphyrin Products, UT) dissolved in saline at 0 minutes, 1 and 2 days after carotid occlusions. Group 2 (n=7) received three intraperitoneal doses of 30 μmol/kg hemin (ferriprotoporphyrin IX chloride; M.W., 651.95; Porphyrin Products, UT) dissolved in saline in the same manner as group 1. Group 3 (n=6) received saline alone intraperitoneally in the same manner as group 1. Group 4 (n = 6) consisted of sham-operated control animals, which underwent all surgical procedures except the induction of cerebral ischemia.
Histologic examinations
Hematoxylin and eosin staining was performed in all sections obtained. Simultaneously, using consecutive sections, immunohistochemical staining for HO-1, glial fibrillary acidic protein (GFAP), and tubulin was performed using indirect immunohistochemical staining methods. Briefly, after removal of the paraffin, the coronal brain sections were incubated in a 5-mmol/L solution of hydrogen peroxide for 10 minutes to oxidize pseudoperoxidase compounds and exposed to 5% normal goat serum for 10 minutes. The sections were first incubated with rabbit anti-rat HO-1 (Stress Gen, Victoria, BC, Canada) at 1000-fold dilution, mouse monoclonal anti-GFAP (Amersham, England) at 200-fold dilution, and mouse monoclonal anti-rat tubulin (Amersham, England) at 400-fold dilution for 3 hours in a humidified chamber at 4°C. After having been washed thoroughly with 0.01 mol/L of sodium phosphate-buffered saline (pH 7.2), the sections were incubated with donkey anti-rabbit immunoglobulin G (ab′)2 at 20-fold dilution (for HO-1), or sheep anti-mouse immunoglobulin G F(ab′)2 at 20-fold dilution (for GFAP or tubulin) for 1 hour. The bound antibody was visualized with 3,3′-diaminobenzidine and hydrogen peroxide (Graham and Karnovsky, 1966).
The ischemic damage within the hippocampus CA1 and CA3 regions was quantitated by one observer (H. H.), who was blinded to the experimental protocol, by counting numbers of normal-appearing pyramidal neurons per high-power field (400×) at three levels of the hippocampus, along a predetermined number of grid lengths (~300 μm). Numbers of normal-appearing neurons were also quantitated by counting cells in layers III and IV of the neocortex centered at ~2 mm dorsal to the entorhinal fissure (400×) and in the dorsolateral striatum (400×). Viable neurons were defined as those cells showing a distinct nucleus and nucleolus (Dietrich et al., 1995).
The immunohistochemical staining for HO-1 was evaluated using the semiquantitative scale of Zhang et al. (1993) with some modification. The percentages of HO-1—positive cells among all cells were graded as follows: <2%=0; 2% to 50%=1; >51% = 2. This evaluation was also performed by an observer who was blinded to the experimental protocol.
Statistical analysis
The statistical significance of differences in viable cell count and physiologic parameters among the four groups was analyzed by means of one-way analysis of variance followed by Fisher's protected least significant difference. The statistical significance of differences in the score of HO-1 immunoreactivity among the four groups was analyzed by use of the Kruskal-Wallis test followed by Dunn's procedure. All values were expressed as mean ± SD.
RESULTS
Mean arterial blood pressure, brain and rectal temperatures, arterial blood gases, and blood glucose and hematocrit levels in the four groups are shown in Table 1. There were no statistically significant changes among the four groups.
In sham-operated animals, no ischemic cell damage and no reaction for HO-1 were observed in the CA1 hippocampus (Fig. 1A and Fig. 1B), the CA2 hippocampus (Fig. 2A and Fig. 2B), the parietal cortex (Fig. 3A and Fig. 3B), and the striatum (Fig. 4A and Fig. 4B). None of the tissue sections studied showed any reaction to normal rabbit serum used as an immunologic negative control.
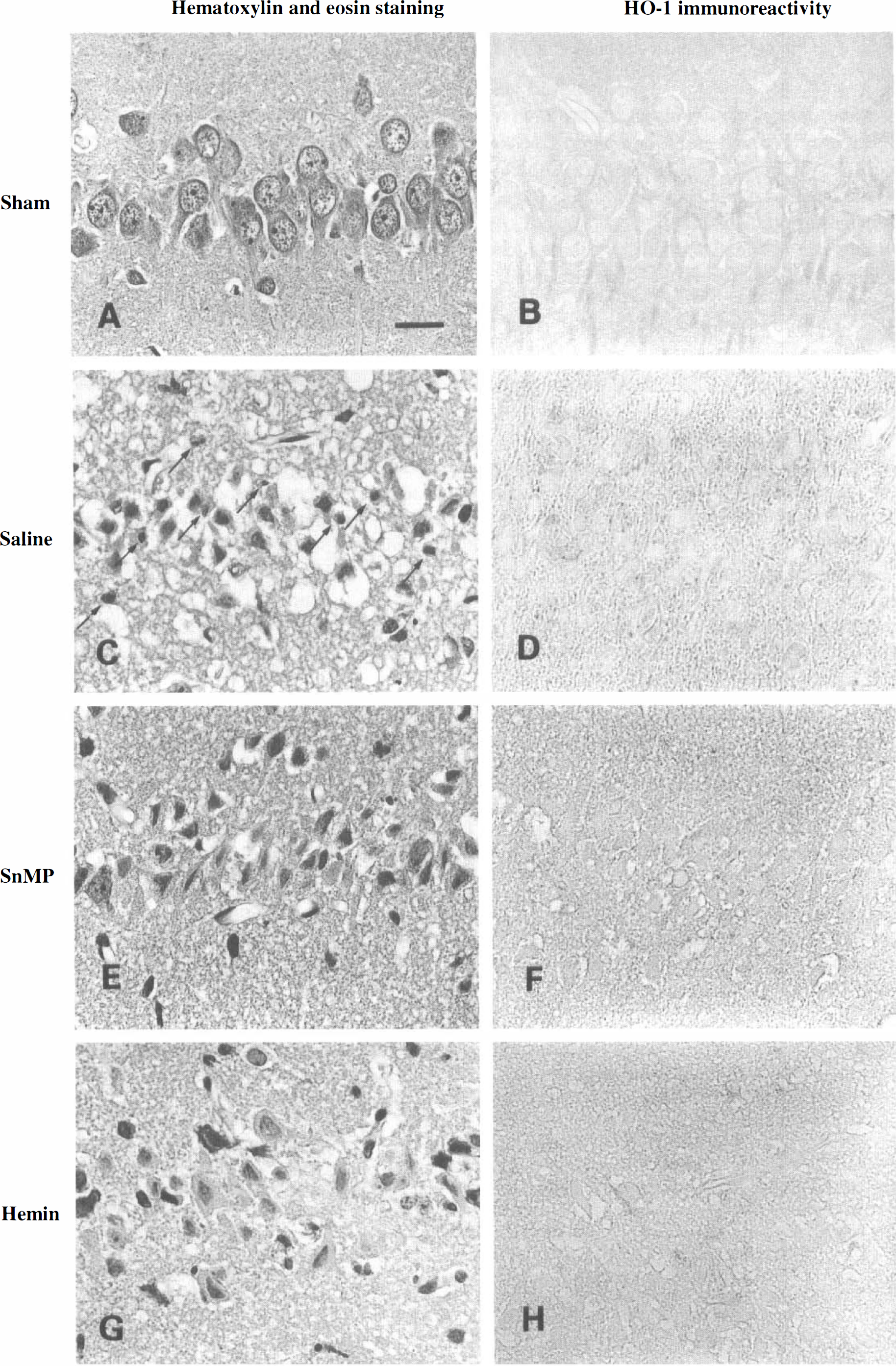
Histopathologic findings in the CA1 hippocampus (600×). In a sham-operated animal, no ischemic cell damage (
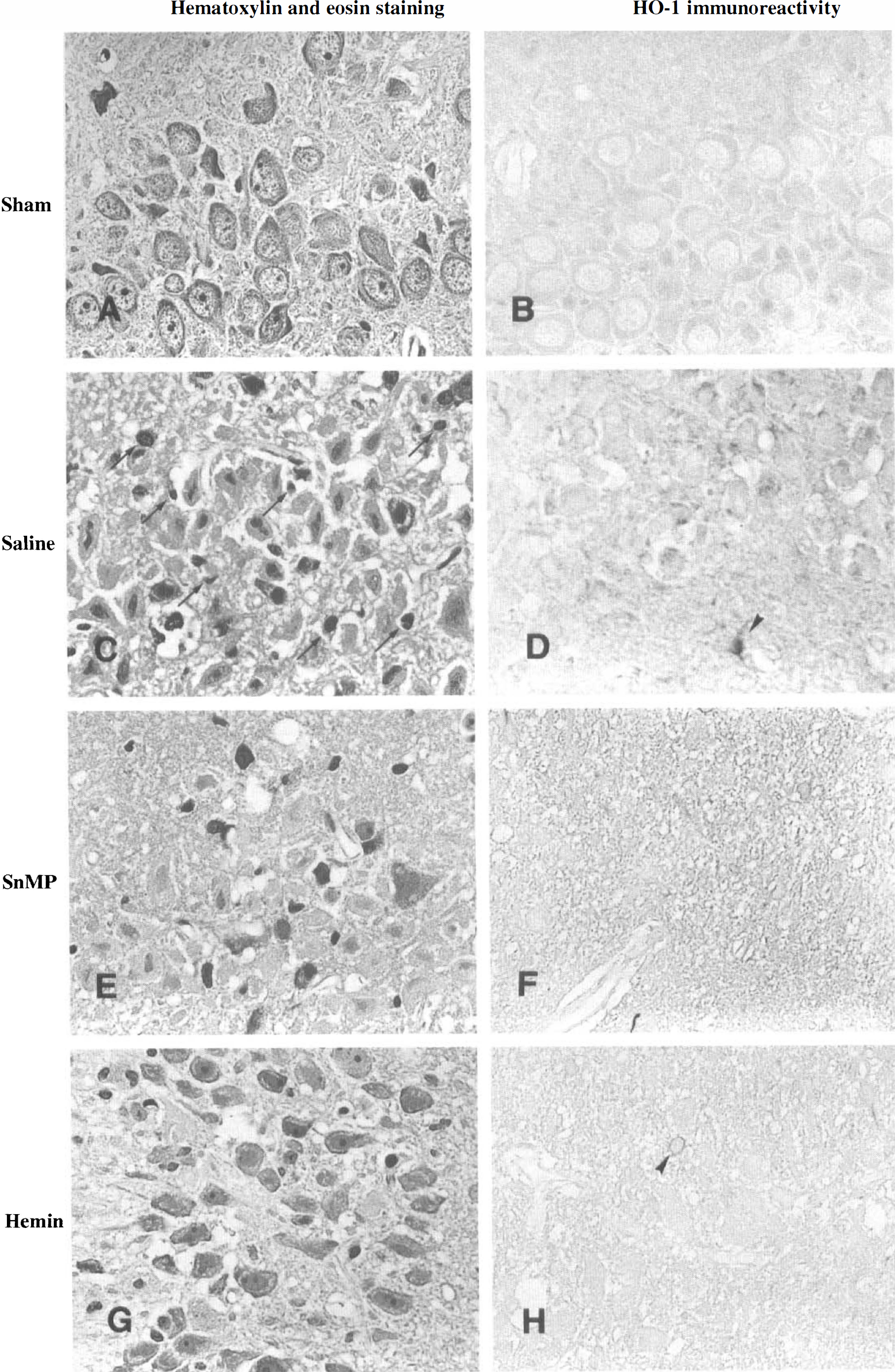
Histopathologic findings in the CA3 hippocampus (600×). In a sham-operated animal, no ischemic cell damage (
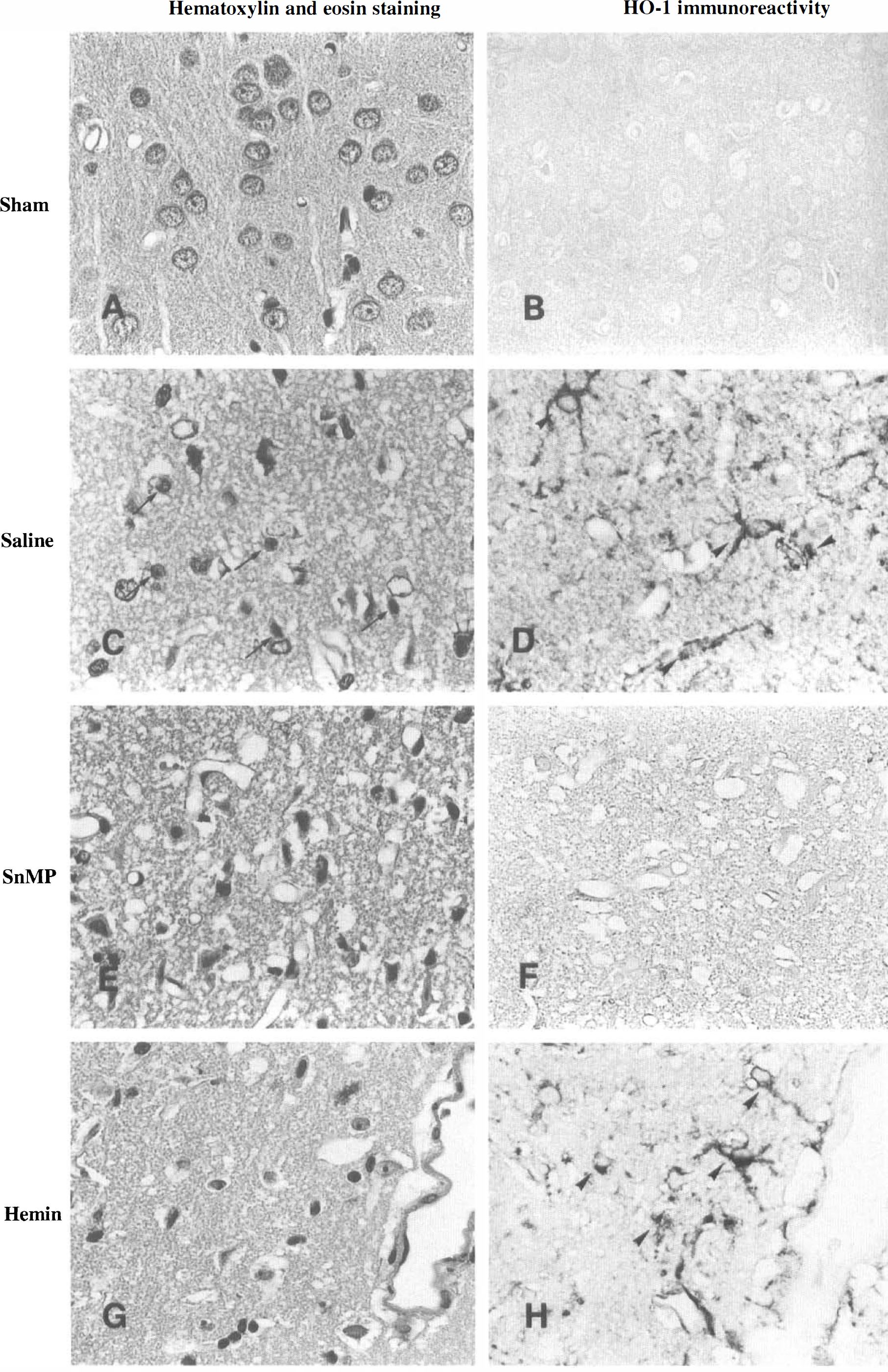
Histopathologic findings in the parietal cortex (600×). In a sham-ope rated animal, no ischemic cell damage (
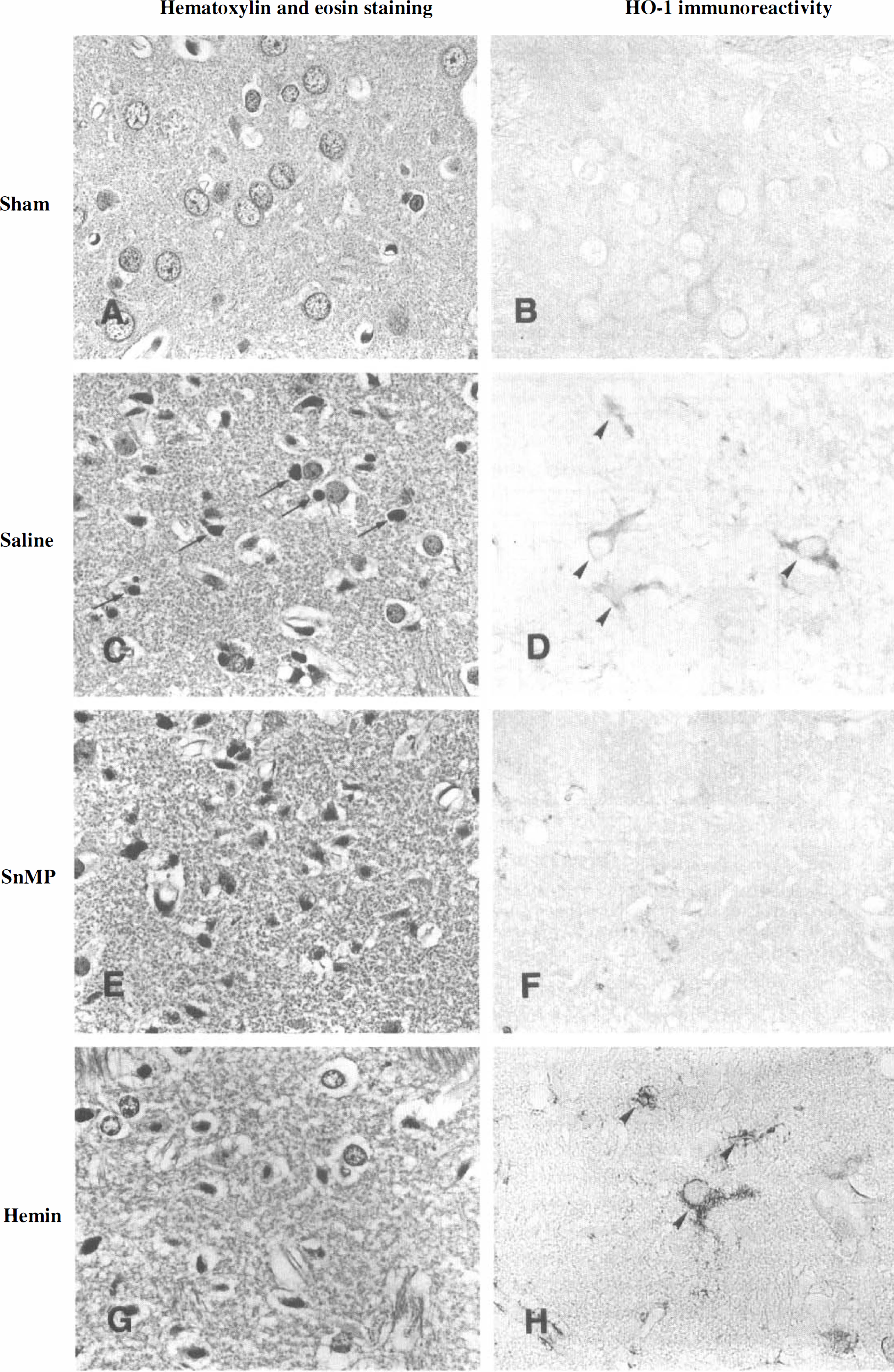
Histopathologic findings in the striatum (600×). In a sham-operated animal, no ischemic cell damage (
Figure 1 shows representative photomicrographs of hematoxylin and eosin staining and immunohistochemical staining for HO-1 in the CA1 hippocampus. In the animals subjected to four-vessel occlusion and treated with saline alone, most of the pyramidal neurons showed ischemic cell damage, characterized by shrunken, darkly stained cytoplasm and pyknotic nuclei, with accumulation of glial cells (Fig. 1C). These findings were confirmed by GFAP and tubulin stainings (not shown). In a consecutive section, no immunohistochemical reaction for HO-1 was seen in neurons or glia (Fig. 1D). In the animals subjected to occlusion and treated with SnMP (Fig. 1E and Fig. 1F) or hemin (Fig. 1G and Fig. 1H), ischemic changes were seen in most of the neurons and no immunohistochemical reaction for HO-1 was observed in the CA1 hippocampus; these findings are similar to those in the saline-treated animals.
Figure 2 shows representative photomicrographs in the CA3 hippocampus. In the animals subjected to occlusion and treated with saline (Fig. 2C) or SnMP (Fig. 2E), the majority of the neurons were damaged and glial cells had proliferated. In consecutive sections, a few HO-1—positive cells were observed (Fig. 2D). In the animals subjected to occlusion and treated with hemin, many neurons were swollen or shrunk, but the destruction of CA3 pyramidal cells was not complete (Fig. 2G). A few of HO-1—positive cells were observed (Fig. 2H).
Figure 3 shows representative photomicrographs in the parietal cortex. In the animals subjected to occlusion and treated with saline alone, most of the neurons were damaged (Fig. 3C) and a strongly positive reaction for HO-1 was observed predominantly in glia and partly in neurons (Fig. 3D). However, the positive reaction for HO-1 was reduced by treatment with SnMP (Fig. 3F), although the degree of ischemic neuronal damage was unchanged (Fig. 3E). Furthermore, treatment with hemin increased surviving neurons and HO-1 immunoreactivity in neurons and glia (Fig. 3G and Fig. 3H).
Physiologic parameters
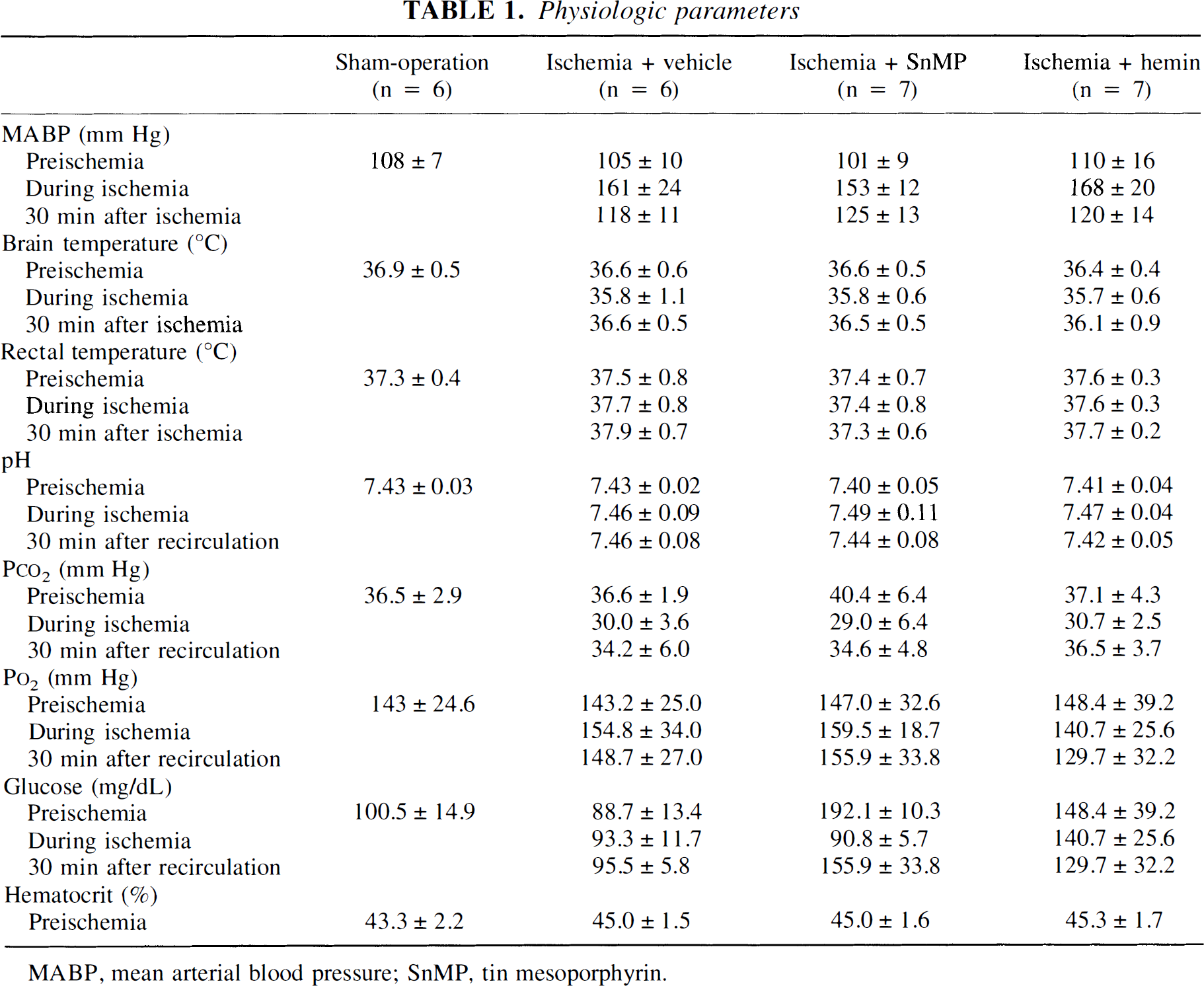
MABP, mean arterial blood pressure; SnMP, tin mesoporphyrin.
Figure 4 shows representative photomicrographs in the striatum. In the animals subjected to occlusion and treated with saline alone, half of the neurons were damaged (Fig. 4C) and a strongly positive reaction for HO-1 was observed predominantly in glia and partly in neurons (Fig. 4D). Treatment with SnMP reduced the number of surviving neurons, although a few HO-1—positive cells were observed (Fig. 4E and Fig. 4F). Treatment with hemin, however, increased both the surviving neurons and HO-1 immunoreactivity in neurons and glia (Fig. 4G and Fig. 4H).
Figure 5 shows quantitative cell counts of normal-appearing neurons and the semiquantitative cell counts of normal-appearing neurons and the semiquantitative scores of immunohistochemical reaction for HO-1 in the CA1 and CA3 hippocampi, the parietal cortex, and the striatum for all experimental groups. In the CA1 hippocampus, forebrain ischemia treated with saline, SnMP, and hemin significantly reduced normal-appearing neurons compared to those in the sham-operated control, but there was no significant difference in cell counts among the former three groups (Fig. 5A). There was no significant difference in the scores of HO-1 immunoreactivity among the four groups, although a weak reactivity was seen in every ischemia group (Fig. 5B). In the CA3 hippocampus, forebrain ischemia treated with saline, SnMP, and hemin reduced viable neurons to 34.8% (P<.05), 50.3% and 73.9% of sham-operated control values, respectively (Fig. 5C), but there was no significant difference in the scores of HO-1 immunoreactivity among the four groups (Fig. 5D). In the parietal cortex (Fig. 5E and Fig. 5F), forebrain ischemia treated with saline and SnMP significantly reduced viable neurons to 50.6% and 42.1% of sham-operated control values (P<.01), and increased the scores of HO-1 immunoreactivity to 1.5 ± 0.5 (P<.05 from the sham-operated control) and 1.1 ± 0.7 (not significant), respectively. However, hemin significantly increased the viable neurons in the cortex compared to the SnMP-treated group (P<.05), and significantly increased the score of HO-1 immunoreactivity compared to the sham-operated group (P<.05). In the striatum (Fig. 5G and Fig. 5H), forebrain ischemia treated with saline and SnMP led to a decrease of viable neurons (P<.01 from the sham-operated group) and increased the scores of HO-1 immunoreactivity to 1.7 ± 0.5 (P<.05 from the sham-operated control) and 1.1 ± 0.7 (not significant), respectively. Hemin, however, significantly increased viable neurons compared to the saline (P<.01)- or SnMP (P<.05)-treated group, and significantly increased the score of HO-1 immunoreactivity to 1.9 ± 0.4 (P<.05 from the sham-operated group).
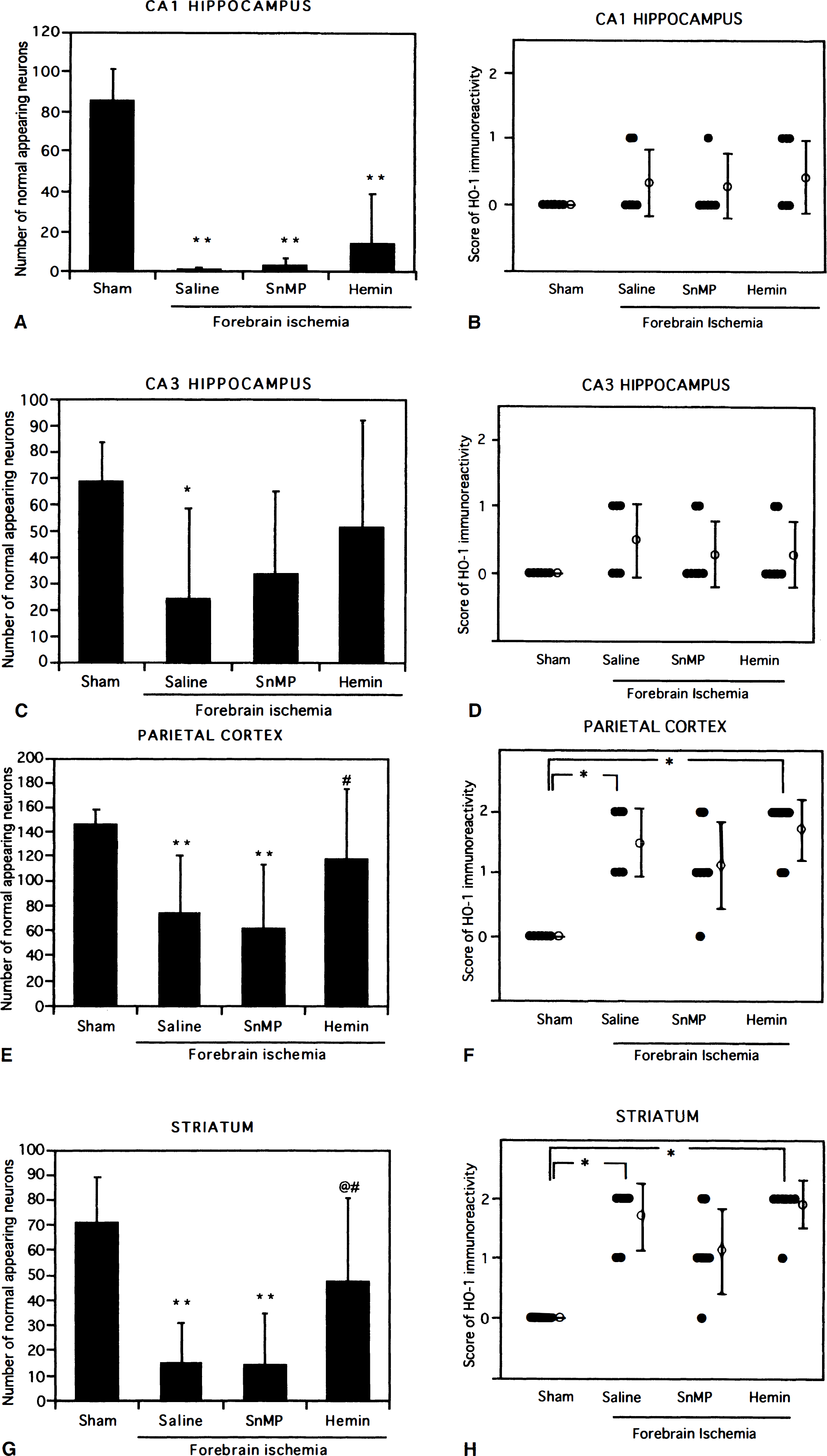
Quantitative cell counts of normal-appearing neurons and semiquantitative scores of immunohistochemical reaction for heme oxygenase-1 in all regions for the four experimental groups (mean ± SD). Significant difference from the sham-operated group at P<.05* and P<.01**; @Significant difference from the saline-treated group at P<.01; #Significant difference from the saline-treated group at P<.05.
DISCUSSION
The role of CO as a neural messenger has been documented (Verma et al., 1993; Dawson and Synder, 1994), but little is known about the influence of CO on cerebral ischemia. The main findings of this study were that hemin, an HO inducer, led to a decrease in neuronal damage in the cortex and striatum, concomitantly with positive HO-1 immunoreactivity, at 3 days after forebrain ischemia, whereas the administration of SnMP, an HO inhibitor, tended to exacerbate the neuronal damage in the parietal cortex. In contrast, neither hemin nor SnMP affected the pathologic outcome in the CA1 and CA3 hippocampi, and HO-1 immunoreactivity in the CA1 and CA3 hippocampi was weak. These results represent the first in vivo evidence that induction of HO-1 protein may contribute to cellular defense against ischemic damage in brain regions where potential ability to synthesize HO-1 is retained in cerebral ischemia. Conversely, if severe ischemia destroys the ability to synthesize HO-1 protein, an HO inducer cannot be expected to have a neuroprotective effect. Similar tissue protection by an HO inducer against glycerol-induced acute renal failure (Nath et al., 1992) and carrageenin-induced pleurisy (Willis et al., 1996) has been reported. These findings, including ours, are consistent with the hypothesis that induction of HO protein may play a considerable role in tissue protection against oxidative stress in some parts of the brain, although the possibility has not been ruled out that HO-1 protein translocation may also be involved.
The mechanism of neuroprotection via an increase in HO activity is unclear. The presently proposed potential role of HO in cellular defense is consistent with its known catalytic action, i.e., production of bilirubin and biliverdin, which act as antioxidants (Stocker et al., 1987a, 1987b), and production of CO, which increases blood flow due to its vasodilating effect (Verma et al., 1993), inhibits some of the enzymes involved in arachidonic acid metabolism (Martasek et al., 1991), and binds heme moieties that can inactivate heme enzymes such as NOS (White and Marietta, 1992). On the other hand, it has been reported that the HO inhibitor zinc protoporphyrin IX (ZnPP) reduced infarct size and edema accumulation in focal ischemia (Kadoya et al., 1995), blocked the effect of metabotropic glutamate receptor activation (Glaum and Miller, 1993), and reduced depolarization-induced glutamate release (Shinomura et al., 1994). However, it has been pointed out that ZnPP inhibits both HO and NOS activities (Meffert et al., 1994; Takizawa et al., 1996); therefore, it is unlikely that the results obtained with ZnPP reflect the inhibition of HO activity alone.
In cerebral ischemia, the expression of HO-1 mRNA and the induction of HO-1 protein immunoreactivity have been reported (Paschen et al., 1994; Takeda et al., 1994; Geddes et al., 1996; Nimura et al., 1996). Interestingly, the distribution and the time course of HO-1 mRNA after forebrain ischemia observed by Paschen et al. (1994) are consistent with those of HO-1 protein in this study. After transient global ischemia followed by recirculation, the HO-1 mRNA level increased consistently up to 24 hours in the cortex and striatum, while it peaked at 4 hours (nine-fold over the control) and thereafter decreased to 4.5-fold over the control at 24 hours in the hippocampus. Because both heat shock and hemin induce HO at the transcriptional level (Shibahara et al., 1987), ischemic insult may also influence HO-1 protein synthesis at the transcriptional level, in parallel with the intense induction of mRNA in the cortex and striatum and the poor induction of mRNA in the hippocampus (Paschen et al., 1994). In our model, HO-1 protein was induced predominantly in glia and partly in neurons in layers III and IV of the neocortex and in the dorsolateral striatum 3 days after transient forebrain ischemia, but not in hippocampus. Geddes et al. (1996) observed that HO-1 immunoreactivity is evident in both neurons and glia in hippocampus, cerebral cortex and thalamus at 24 hours after a 20-minute four-vessel occlusion with hypotension in rats, and predominantly in astrocytes in CA3c and CA1 of the hippocampus at 72 hours after global ischemia. It is conceivable that the degree of glial proliferation and the cell type showing positive HO-1 immunoreactivity may be variable depending on the period of reperfusion after ischemia, and the severity of the ischemia.
HO comprises two isozymes identified as HO-1 and HO-2 (Maines, et al., 1986). HO-1 mRNA and protein are present in selected regions including hilar neurons in the hippocampus under normal conditions (Ewing, et al., 1992), but are increased in response to various insults including oxidative stress (Keyse and Tyrrell, 1989) and hyperthermia (Dawyer et al., 1992; Ewing et al., 1992). On the other hand, HO-2 is a constitutively expressed isoform, which accounts for most of the HO activity in normal brain (Ewing and Maines, 1992), and its immunoreactivity is not altered by transient global ischemia (Geddes et al., 1996). Although SnMP and hemin may modulate both HO-1 and HO-2 activities, our immunohistochemical results may suggest that at least HO-1 contributes to the neuroprotection against ischemic damage. It is unclear whether HO-2 also plays a protective role against ischemia because we did not examine the changes of HO-2 immunoreactivity.
In conclusion, the present results suggest that induction of HO-1 protein by the administration of hemin may contribute to cellular defense against ischemic damage in some brain regions such as the parietal cortex and striatum, where potential ability to synthesize HO-1 may be retained in cerebral ischemia. Our finding, therefore, may suggest a new therapeutic strategy to increase the resistance of the ischemic brain to oxidative stress, although this treatment cannot rescue severely damaged tissue, such as the CA1 hippocampus in global ischemia.
Footnotes
Acknowledgments
The authors thank S. Ogawa (Department of Neurology), Y. Takahari (Laboratories for Experimental Animals and Physiologic Research), and J. Itoh (Laboratories For Structure and Function Research) in Tokai University School of Medicine for their skillful technical assistance.