
Abstract
The cerebrovascular effects of the heme oxygenase–carbon monoxide pathway were studied in the rat hypothalamus. Intraperitoneal administration of the heme oxygenase inhibitor zinc deuteroporphyrin 2,4-bis glycol (ZnDPBG, 45 μmol/kg) had no significant effect on the resting cerebral blood flow, but increased hypothalamic nitric oxide synthase activity by 67% without changing the CSF cyclic GMP concentration. After pharmacologic inhibition of nitric oxide synthase, the diminished cerebral blood flow was further reduced by 22% after administration of ZnDPBG, and the effect showed direct correlation with the baseline perfusion level. Therefore, endogenous carbon monoxide may significantly contribute to the cerebral vasoregulation under resting conditions and in pathophysiologic states associated with diminished nitric oxide synthesis.
Heme oxygenase (HO)-mediated heme degradation is the primary cellular mechanism for production of endogenous carbon monoxide (CO). Analogous to nitric oxide synthase (NOS), the HO enzyme has different constitutive and inducible isoforms (HO-1, HO-2, and HO-3). The physiologic roles of nitric oxide (NO) and CO also display notable similarities, as both molecules appear to be neurotransmitters in the brain and peripheral autonomic nervous system. Furthermore, both NO and CO are endothelial-derived relaxing factors for blood vessels (Barañano and Snyder, 2001).
The participation of CO in the in regulation of the cerebral circulation has received little attention (Koehler and Traystman, 2002), although several studies have found that HO is expressed in the cerebral vasculature (Parfenova et al., 2001; Vigne et al., 1995; Zakhary et al., 1996). Freshly isolated cerebral microvessels produce CO from endogenous heme under basal conditions, and increased CO production was reported after stimulation of ionotropic glutamate receptors (Leffler et al., 2003a, b ; Parfenova et al., 2003). Both exogenous CO and the HO substrate heme-L-lysinate induced pial arterial dilatation in piglets, and the latter could be inhibited by the HO blocker chromium mesoporphyrin. Furthermore, hypoxia-induced vasodilatation was inhibited and vasoconstriction in response to hypercapnia was increased by application of chromium mesoporphyrin (Leffler et al., 1999, 2001; Winestone et al., 2003). During seizures, pial arteriolar dilation in piglets and an increase in CBF in adult rats were attenuated by HO inhibitors (Montécot et al., 1998; Pourcyrous et al., 2002). Recently, it was shown that endogenous CO dilates cerebral arterioles by augmenting the coupling of Ca2+ sparks to KCa channels in smooth muscle cells (Jaggar et al., 2002).
The aim of the current study was twofold: (1) to investigate the involvement of endogenous CO in the regulation of the resting cerebral circulation, and (2) to clarify its possible interaction with the L-arginine–NO pathway. The cerebrovascular effects of HO blockade were studied in the rat hypothalamus, because in this brain region both HO-1 and HO-2 are expressed at a relatively high level (Ewing and Maines, 1992).
MATERIALS AND METHODS
In the first part of the study, the effects of HO blockade on the systemic circulation and hypothalamic blood flow (HBF) were studied under physiological conditions. Animals in group 1 (n = 6) served as vehicle-treated controls and received an intraperitoneal injection of saline. In group 2 (n = 8), zinc deuteroporphyrin 2,4-bis glycol (ZnDPBG, 45 μmol/kg, Frontier Scientific Europe Ltd, Carnforth, Lancashire, UK), was applied intraperitoneally to inhibit the HO pathway (Appleton et al., 1999; Chernick et al., 1989). HBF was measured before and 15, 30, and 45 minutes after the administration of saline or ZnDPBG. ZnDPBG was preferred to other metalloporphyrins because it has been shown that when this dose is intraperitoneally injected, it crosses the blood–brain barrier and markedly inhibits cerebral HO activity (Johnson et al., 1997).
The experiments were carried out in adult (300–400 g) male Wistar rats anesthetized with and intraperitoneal injection of Urethan (1.3 g/kg; Sigma, St. Louis, MO, U.S.A.) and spontaneously breathing via the cannulated trachea. Catheters were inserted into both femoral arteries (to measure blood pressure and for blood sampling) and into the left femoral vein for drug administration. The skull was fixed in a stereotaxic headholder. Body temperature was kept constant between 36°C and 38°C with a controlled heating lamp.
Systemic arterial pressure was continuously recorded on a polygraph (Model 7E; Grass, Quincy, MA, U.S.A.). CBF in the ventromedial hypothalamic area was determined using the H2-gas clearance method of Aukland et al. (1964) as described elsewhere (Benyó et al., 1995). Briefly, a 100-μm-diameter Teflon-coated Pt electrode with a 1-mm bare tip was introduced stereotactically into the ventromedial hypothalamic area. H2-washout curves were produced by H2-gas inhalation, and were recorded on the polygraph. HBF was calculated from the washout curves by using the initial slope technique, omitting the first 0.5 minutes, blood gas values (PaCO2, PaO2, O2 saturation) and pH in femoral arterial samples were measured by a Radiometer Blood Gas Analyzer (ABL-300, Copenhagen, Denmark), and MABP, heart rate, and ventilation rate were determined at the same time. After the last measurements were taken, CSF was obtained from the cisterna magna for cyclic GMP (cGMP) content determination. Thereafter the anesthetized animals were rapidly exsanguinated and hypothalamic tissue samples were excised for the measurement of NOS activity. CSF and tissue samples were rapidly frozen and kept at −25 °C or −75 °C until further analysis. The cGMP content of the CSF was determined by using an enzyme immunoassay kit according to the instructions of the manufacturer (Assay Designs Inc., Ann Arbor, MI, U.S.A.). The hypothalamic tissue NOS activity was measured on the basis of the formation of labeled citrulline from labeled L-arginine (Benyó et al., 1995; Nagy et al., 2000). Hypothalamic tissue samples were homogenized in brainhomogenizing solution containing 50-mmol/L TrisHCl (pH 7.4), 0.3-mol/L sucrose, 0.1-mmol/L EDTA, 1-mmol/L dithioerythritol, and 1-mmol/L phenyl-methyl-sulfonylfluorid (a protease inhibitor). Homogenates were added to the samples containing 5-mmol/L HEPES, 1-mmol/L NADPH, 10-μmol/L tetrahydrobiopterin, 30-μmol/L calmodulin, and 2.5-mmol/L CaCl2 at pH 7.4, and the reaction was initiated by adding 20-μmol/L 3H-arginine (final concentrations). After a 30-minute incubation, the reaction was stopped, samples were put onto 2-cm Dowex 50 × 8 resin columns, and eluates were mixed with 5 mL dioxane-based scintillation fluid and measured in a Beckman TriKarb liquid scintillation spectrometer. Protein contents were determined by the biuret reaction, and NOS activities were calculated in picomoles citrulline formed per minute per milligram protein units.
In the second part of the study, the effects of HO blockade on the systemic and hypothalamic circulatory parameters were studied in the absence of NO. Therefore, in these experimental groups (groups 3 and 4), NOS blockade was introduced by intravenous administration of 50-mg/kg NG-nitro-L-arginine methyl ester (L-NAME, Sigma). Animals in group 3 (n = 9) served as vehicle-treated controls and received an intraperitoneal injection of saline 30 minutes after the onset of NOS blockade. In group 4 (n = 8), ZnDPBG was administered at the same time. HBF and the corresponding physiological parameters were determined with the same protocol as described for groups 1 and 2.
All values are presented as mean ± SD. Statistical analysis was performed using analysis of variance for repeated measurements or one-way analysis of variance followed by Fisher protected LSD post hoc test. For comparison of two groups, the unpaired t-test was used. A P value of less than 0.05 was considered to represent a statistically significant difference.
RESULTS
In the experimental groups without L-NAME pretreatment (groups 1 and 2), the physiologic variables were within the normal range at the time of the first HBF determination before administration of saline or ZnDPBG (Table 1). These cardiovascular, respiratory, and acid–base parameters did not change significantly during the whole experimental period. Furthermore, there were no differences with respect to the corresponding values between the two groups at any time point.
Hemodynamic, respiratory, and acid-base parameters at the time of the first CBF determination in the four experimental groups
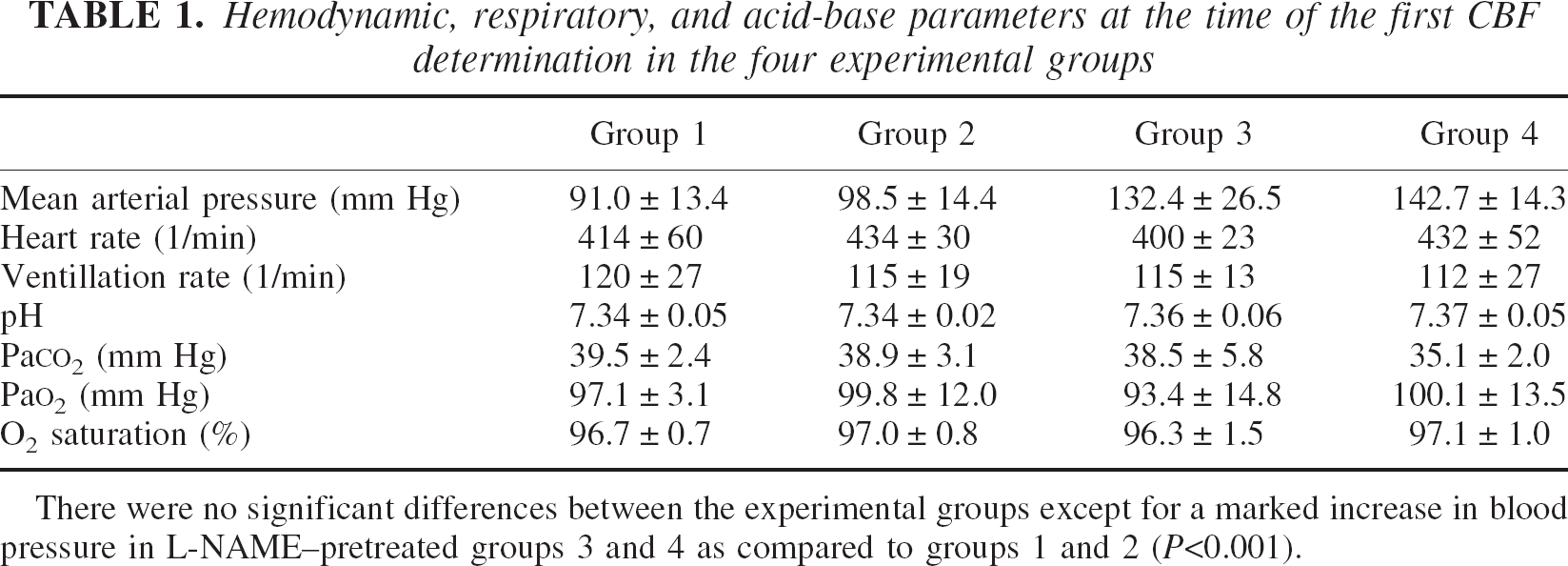
There were no significant differences between the experimental groups except for a marked increase in blood pressure in L-NAME–pretreated groups 3 and 4 as compared to groups 1 and 2 (P<0.001).
HBF was similar in groups 1 and 2 during the first determination (103.1 ± 6.7 vs. 108.2 ± 25.0 mL · 100 g−1 · min−1 in groups 1 and 2, respectively). Neither saline (group 1) nor ZnDPBG (group 2) induced any significant changes in HBF (data not shown). Furthermore, there was no significant difference in the CSF cGMP concentration at the end of the experiments (450.7 ± 165.2 vs. 568.8 ± 148.0 pmol/L in groups 1 and 2, respectively). However, we observed a marked increase (P = 0.001) in hypothalamic NOS activity in the ZnDPBG-treated animals (4.17 ± 0.87 pmol citrulline·mg protein−1 · min−1) as compared to the vehicle-treated group (2.49 ± 0.38 pmol citrulline·mg protein−1 · min−1).
Since HBF remained unchanged in ZnDPBG-treated animals despite an increase in NOS activity, we hypothesized that the HO blockade had a CBF-decreasing effect, which counteracted the action of NO. In order to study this possible direct effect of the HO blockade on the hypothalamic circulation in two additional experimental groups (groups 3 and 4), we repeated our measurements after NOS blockade induced by L-NAME.
The respiratory and acid–base parameters after NOS blockade in groups 3 and 4 were not significantly different from those observed in groups 1 and 2 (Table 1). Although the initial MABP was significantly higher in these groups (Table 1), it did not change further during the experiments (data not shown). Moreover, despite an increased MABP, initial HBF was significantly (P<0.001) lower in group 3 (65.7 ± 20.0 mL · 100 g−1 · min−1) and group 4 (65.9 ± 12.1 mL · 100 g−1 · min−1) when compared to the groups without NOS blockade. The already diminished HBF was further reduced after administration of ZnDPBG in group 4, but did not change significantly after saline administration in group 3 (Fig. 1A). Moreover, there was a significant linear correlation between the HBF values after NOS blockade and the reduction of HBF in response to HO inhibition (Fig. 1B), indicating that HBF was better preserved in animals that expressed higher HO activity levels in the hypothalamus. The physiologic parameters presented in Table 1 did not change throughout the experiments, and no differences were observed between groups 3 and 4 (data not shown).
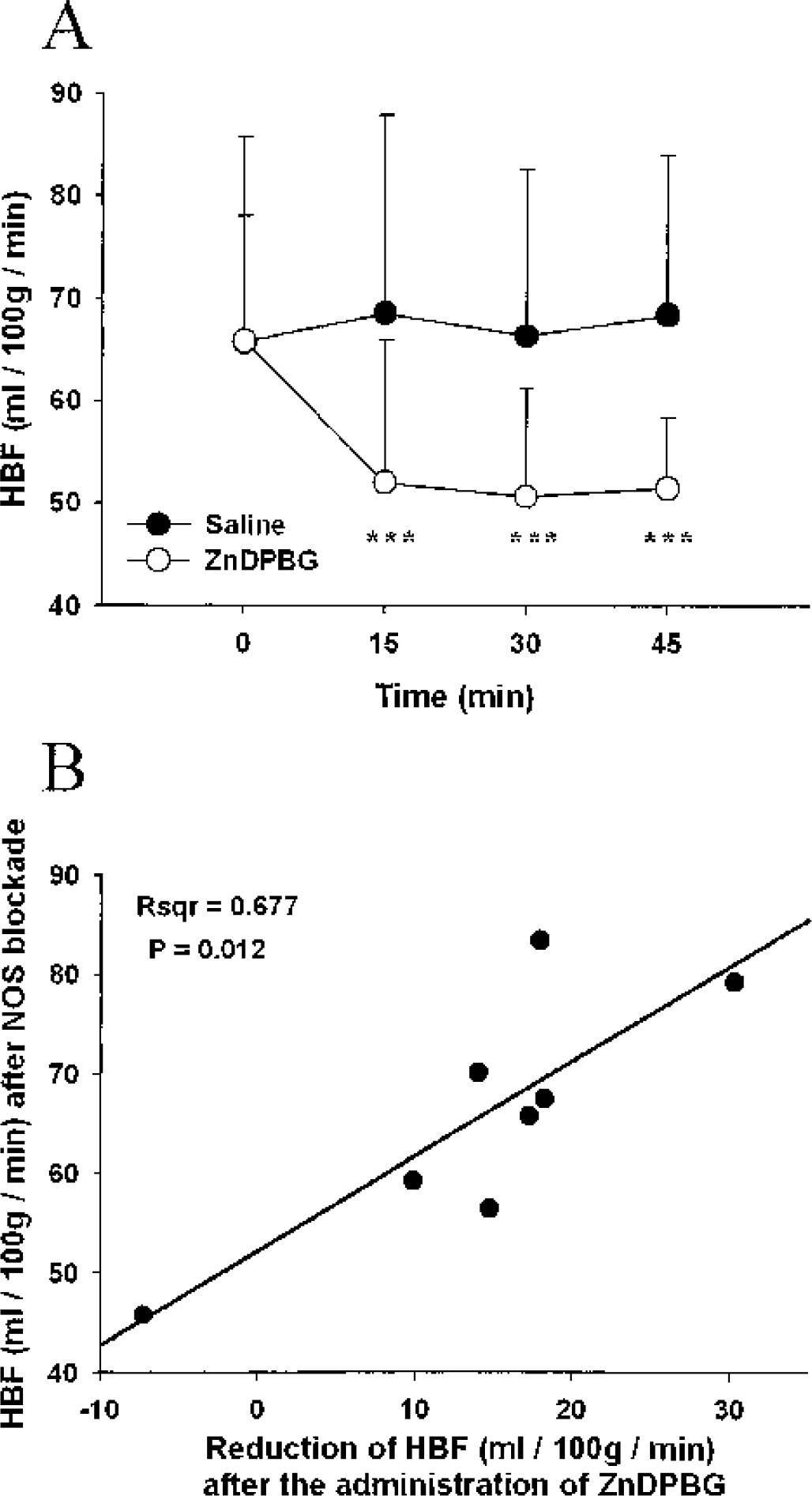
(
DISCUSSION
The current study found that under physiologic conditions, HO blockade increased hypothalamic NOS activity without changing HBF. This finding indicates that endogenous CO has a dual effect on the hypothalamic circulation—a direct vasodilatory action that is compensated by the inhibition of NOS. Therefore, endogenous CO appears to exert a dilatory influence in the hypothalamus under resting conditions, but inhibition of CO production does not cause a decrease in HBF because HO blockade results in increased NO production. After NOS blockade, however, inhibition of HO induced a further significant reduction of the HBF, indicating that in the absence of NO, CO becomes responsible for the maintenance of cerebral blood perfusion. Furthermore, a direct correlation between HBF in NOS-blocked animals and its reduction after HO inhibition was observed. In other words, the HO blockade induced most marked flow reduction in animals that showed relatively high blood flow in the absence of NO. This observation indicates that after NOS blockade the preservation of HBF is dependent on the vasodilatatory action of CO.
The increased NOS activity in group 2 after administration of ZnDPBG indicates that constitutive CO release may tonically suppress NO production in the hypothalamus. A similar interaction between CO and NOS has been proposed by others using different experimental approaches. Leffler et al. (1999) reported that inhibition of the HO pathway with chromium mesoporphyrin results in a NO-mediated dilatation of the pial arterioles in piglets. In studies by Mayevsky et al., administration of exogenous CO resulted in cerebrocortical blood flow oscillations very similar to those observed after inhibition of NO synthesis (Lacza et al., 2000; Mayevsky et al., 1995; Meilin et al., 1999). Furthermore, the HO inhibitor zinc protoporphyrin IX reportedly upregulates NOS-III messenger RNA in rat aortic endothelial cells (Seki et al., 1997) and increases the NO release from myenteric neurons of the rabbit (Chadker et al., 1996). Conversely, increased HO activity due to induction of HO-1 was reported to suppress NOS activity in the rat brain (Maines et al., 1993).
The molecular mechanism by which CO may suppress hypothalamic NOS activity remains to be elucidated. CO binds to NOS and inhibit NO production (McMillan et al., 1992; White and Marletta, 1992), but whether this interaction could influence NOS activity in ex vivo hypothalamic tissue samples is questionable. Therefore, changes in the turnover rate or cofactor availability of NOS could explain the increased NO-synthesizing capacity observed after HO blockade (Maines, 1997). Regardless of its mechanism, the increased NOS activity after HO blockade explains the unchanged CSF cGMP content in group 2 despite the loss of CO-mediated stimulation of soluble guanylate cyclase (sGC). The observations obtained by microdialysis in the frontal cortex of the rat (Laitinen et al., 1997) strongly support this view. In that study, the cGMP release significantly decreased 30 minutes after the onset of zinc protoporphyrin-IX, but returned to the normal level at 60 minutes, when in our study the CSF was sampled. The transient decrease in the cGMP production may reflect that while HO blockade results in an acute loss of CO-mediated sGC activity, the secondary activation of the NO–sGC pathway is a slower process that is probably induced by a change in the NOS turnover rate. Maines et al. (1993) demonstrated the dual effect of CO on cerebral cGMP production, and reported a 36% reduction in cerebellar NOS activity after experimentally increasing HO activity by 65%; however, these two influences on the sGC activity compensated each other, and the cGMP level remained unchanged.
Although the HO pathway apparently has no significant influence on the resting HBF under physiologic conditions, one may not underestimate its potential importance in pathophysiologic states associated with diminished NO synthesis. It is noteworthy that in the second part of our study (i.e., in animals subjected to NOS blockade), the already compromised HBF further diminished after inhibition of endogenous CO production. Therefore, in the absence of NO, CO functions as a backup vasodilator. Although CO alone is unable to completely restore the blood flow in the hypothalamus, the partial improvement in perfusion may prevent the development of irreversible neurologic deficits. This role of the HO pathway is obviously important in circulatory shock or global cerebral ischemia, where the hypothalamus plays a central role in the coordination of defense mechanisms activated to prevent the collapse of the systemic circulation. Our observations provide a possible explanation for the relative preservation of the HBF during ischemia/reperfusion (Liachenko et al., 2001).
Of course, other endothelial vasodilators such as prostacyclin or EDHF may also be involved in the compensatory mechanism activated after the loss of NO (Faraci and Heistad, 1998). However, CO is unique in that it can also be synthesized by smooth muscle cells to relax vessels in case of sustained damage of the endothelium (Cristodoulides et al, 1995; Johnson et al, 2002).
In conclusion, our study indicates a dual role of endogenous CO in the hypothalamic circulation: a direct vasodilatory effect but at the same time suppression of NO release. Therefore, the HO pathway represents a silent mechanism of the vasoregulation under physiologic conditions, but significantly contributes to the preservation of the hypothalamic circulation when NO production is diminished.
Footnotes
Acknowledgments:
The authors thank Professor Stefan Offermanns for critically reading the manuscript, Professors Nader G. Abraham and Hendrik J. Vreman for their valuable suggestions during the preparation of the final version of the manuscript, Dr. Zsombor Lacza for his advice regarding statistical analysis, and Maria H. Velkei, Judit Szabó, and Antal Holly for their expert technical assistance.