
Abstract
Activating transcription factor 3 (ATF3) is one of the most important transcription factors that respond to and exert dual effects on inflammatory responses. Recently, the involvement of ATF3 in the neuroinflammatory response to acute brain injury (ABI) has been highlighted. It functions by regulating neuroimmune activation and the production of neuroinflammatory mediators. Notably, recent clinical evidence suggests that ATF3 may serve as a potential ideal biomarker of the long-term prognosis of ABI patients. This mini-review describes the essential inflammation modulatory roles of ATF3 in different disease contexts and summarizes the regulatory mechanisms of ATF3 in the ABI-induced neuroinflammation.
Introduction
Activating transcription factor 3 (ATF3), a transcription factor with a basic leucine zipper, has long been recognized as a member of the ATF/cAMP response element binding (CREB) protein family. 1 The expression of ATF3 is at low levels in quiescent cells, but increases in response to stressors such as oxidative damage and ischemia/reperfusion.2–4 ATF3 is emerging as a master regulator of inflammation in multiple pathophysiological processes such as cardiovascular disease, dementia, and ischemia/reperfusion-induced damage.1,5–15
Neuroinflammation is one of the key pathological features of a wide range of acute brain injuries (ABIs),16–19 which mainly include stroke and traumatic brain injury (TBI).20,21 Neuroinflammation plays a pivotal role in ischemic brain injury during the acute phase of ABI, with excessive immune cell activation and the release of inflammatory mediators.22–24 A persistent neuroinflammatory response will further lead to secondary neurodegenerative processes such as cognitive impairment and dementia.18,25–27 Clinical evidence also supports that unresolved neuroinflammation continuously shapes the evolving brain pathology and affects the long-term neurological outcome of patients.16,28–30 Therefore, it’s urgently needed to understand the putative mechanisms for dysregulated brain inflammatory responses in the pursuit of improving long-term neurological outcomes after ABI. Regarding ABI, recent studies suggested that ATF3 could be an ideal predictor of outcomes in stroke and TBI,31,32 and ATF3 failure to regulate inflammatory responses account for poorer ABI outcomes by exacerbating neuroinflammation.33,34
In this mini review, we describe the regulation of ATF3 in various inflammatory responses, scrutinize the latest research findings, and highlight new insights into the potential involvement of ATF3 in neuroinflammation and several common ABIs.
ATF3, is a stress-associated transcription factor that regulates local and systemic inflammation
ATF3 is a stress-associated transcription factor and a key regulator of local and systemic inflammation. Taking type 2 diabetes mellitus as an example, ATF3 is necessary for the complete induction of matrix metalloproteinase (MMP12) expression, a negative regulator of glucose metabolism, in macrophages, which connects microbiota-dependent inflammation to insulin resistance.35–37 In addition to proinflammatory effects, ATF3 deficiency is associated with exacerbated inflammation and inflammation-related disease processes, such as neuronal apoptosis and, cellular metabolic dysfunction of macrophages and endothelial cells (ECs) in ischemia/reperfusion injury and cancer therapy.2,5,14,38,39 Protein-protein interaction network analysis further revealed that ATF3 interacts with 48 important stress-relevant proteins including other transcriptional complexes involved in inflammatory responses (Rela, Jun, Junb, Jund, Fos), indicating the core position of ATF3 in cellular adaptive-response networks. 40
ATF3 exerts double-sided effects on inflammatory response
As a multifaceted and controversial transcription factor, ATF3 exerts both protective and deleterious effects on inflammatory responses. Under stress, ATF3 may play a protective role against inflammation-associated damage in diseases as a compensatory factor. 35 It functions as an immunomodulator by interacting with nuclear factor kappa B (NF-κB) and suppressing proinflammatory cytokines such as interleukin(IL)-6-, IL-12, and toll-like receptor (TLR) 4 in mice exposed to lipopolysaccharide (LPS).41–45 Furthermore, ATF3 interacts with BRG1-Associated Factor 60 A (BAF60a), a regulator of obesity-induced adipose tissue macrophages, to inhibit proinflammatory cytokine production in obese people. 46 In addition to cytokines, ATF3 links to the regulation of chemokine C-C motif chemokine 4 (CCL4) in macrophages and the attenuation of inflammatory responses. 47 In the early stress response, acute hypoxia-induced activation of tumor necrosis factor-alpha (TNF-α) enhances ATF3 expression in ECs, and ATF3 regulates the activation of the c-Jun NH2-terminal protein kinase pathway, which protects cardiomyocytes from doxorubicin-induced death. 48 Thus, ATF3 can engage in a compensatory or adaptive homeostatic response to alleviate inflammatory responses during inflammation–related pathological processes.
By contrast, ATF3 has also proinflammation properties and can detect molecular patterns generated by necrotic cells to induce inflammation. 49 In respiratory infections caused by Staphylococcus aureus (S. aureus), ATF3 promotes inflammation by upregulating IL-6 and IL-12p40. 50 The amplified inflammatory response supports the antimicrobial effects, and ATF3 stimulates the migration of F4/80 macrophages for pathogen elimination in S. aureus-induced pneumonia. 50 Moreover, the ATF3-dependent upregulation of IL-6R and IL-6 may increase drug resistance to sorafenib and regorafenib in hepatocellular carcinoma. 39 In the tumor microenvironment of an immune-competent mouse model, shATF3 significantly decreased the PD-L1 level, thereby boosting the activated CD8+ T cell population to increase its anti-tumor efficacy. 51 Notably, the ATF3/c-Jun/Lgals3 axis may enhance microglial activation in central diabetes insipidus following hypothalamic damage. 52
ATF3 mediates inflammatory responses by interacting with a complex metabolic-immune response network
Transcription and metabolic regulation have been recognized as powerful principles guiding inflammation.53–56 As an important transcription factor, ATF3 interacts with a complex metabolic-immune response network in various cells, such as macrophages and ECs. 57 Several studies have suggested that the ATF3 function in metabolic, tumor, and cardiovascular diseases in part originates from its influence on cell metabolism.1,58–60
Abnormal metabolism induces stress reactions and subsequently influences ATF3-mediated inflammatory responses.36,46,53 Guided by symptoms of lipodystrophy and fever among patients with proteasome-associated autoinflammatory syndromes, scientists discovered that proteasome dysfunction blocks adipogenesis and induces inflammation by activating ATF3. 61 It is of note that ATF3 negatively regulates the acquisition of a pro-inflammatory phenotype induced by high-density lipoprotein (HDL) in macrophages. 20 ChIP-sequencing revealed that several key pro-inflammatory genes were directly targeted by ATF3 following its induction by HDL. 20 As an HDL-inducible target gene in macrophages, ATF3 down-regulates the expression of TLR-induced proinflammatory cytokines and alleviates cellular stress. 20 Another disease that involves ATF3 and lipid metabolism is atherosclerosis, a progressive disease characterized by the accumulation of lipids and inflammatory responses in arteries. The synthetic c-type lectin receptor 4e (Clec4e) ligand induces nuclear translocation of ATF3 in a Clec4e-dependent manner in both peritoneal macrophages and bone marrow-derived macrophages.21,62–64 On the other hand, ATF3 can promote cholesterol efflux by regulating Clec4e signaling in macrophages, alleviate Clec4e-induced endoplasmic reticulum (ER) stress response, and reduce the production of pro-inflammatory mediators and growth factors. 65 Furthermore, it limits macrophage foam cell formation and inflammation in granuloma remodeling and prevents the progression of atherosclerosis. 65 Another way to prevent macrophage foam cell formation by ATF3 relies on regulating the intracellular neutral fat accumulation by inhibiting the cholesterol 25-hydroxylase in macrophages and, modulating cytokine production and ER stress. 66 Therefore, ATF3 can be initially activated by metabolic stressors, then works as a modulator and inhibitor in response to a variety of inflammatory stimuli.7,12,50,67
Apart from the anti-inflammatory properties mentioned above, persistent ATF3 expression induced by excessive reactive oxygen species (ROS) or ER stress may have detrimental effects, overwhelming its initial compensatory property. For instance, triglyceride-rich lipoprotein (TL) lipolysis products promote nuclear ATF3 accumulation in carotid artery ECs of mice and increase vascular apoptosis, 68 whereas ATF3 promotes lipolysis products-induced transcription of E-selectin and IL-8 in human aortic Ecs. 68 Hence, ATF3 deletion can prevent vascular apoptosis by decreasing triglyceride-rich lipoproteins and lipolysis products-induced inflammation. 68 Additionally, decreased ATF3 levels were necessary for HDL-induced inhibition of saturated fatty acid-mediated enhancement of LPS-induced inflammatory responses in ECs. 69 In response to lipotoxic brain microvascular damage, ATF3 mainly governs TL-induced inflammation and TNF signaling in the cerebrovascular system and increases EC apoptosis. 67 The ATF3 effects on inflammation depends on the various cellular environments under stress (Figure 1).
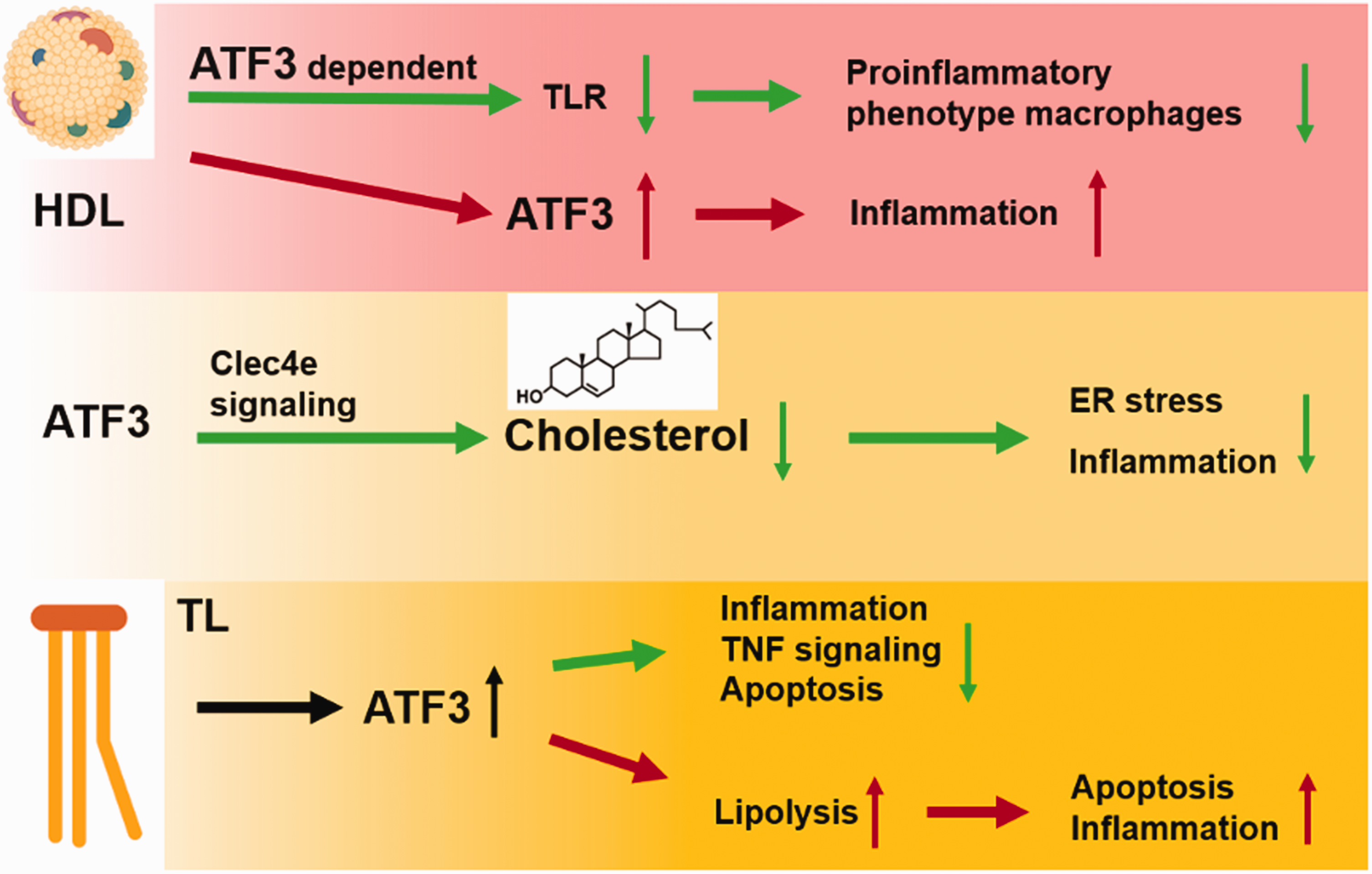
The interplay of ATF3 with metabolism on inflammation. HDL mediates anti-inflammatory reprogramming of macrophages via ATF3 in macrophages, but also induced ATF3 activity in endothelial cells then promote inflammation. ATF3 regulates Clec4e signaling to increase cholesterol efflux and reduce ER stress and inflammation. TL increases nuclear ATF3 accumulation. The accumulation of ATF3 can down-regulate TNF signaling and alleviate inflammation and apoptosis. ATF3 can also increases lipolysis, which exacerbates apoptosis and inflammation in carotid artery endothelial cells in the cerebrovascular system.
In summary, ATF3 may be therapeutically targeted to combat excessive inflammatory responses, or conversely, ATF3 can be repressed to strengthen an insufficient immune system.
Neuroinflammation is an emerging therapeutic target for improving neurological function following ABI
ABI triggers robust inflammatory- responses,28,70–72 then peripheral and brain immune dysregulation initiate a cascade of inflammatory signaling events, ultimately resulting in neuroinflammation. 73 The resulting intense neuroinflammation can last for years and trigger secondary neurodegeneration due to the excessive neuroimmune reaction induced by both resident brain cells and infiltrating peripheral inflammatory cells.26,74–77 Thus, controlling disproportionate neuroinflammatory responses represents a promising therapeutic strategy to improve neurological outcomes after ABIs.25,78,79
Both innate and adaptive immune cells are highly involved in ABI–induced neuroinflammation. Neutrophils are the first leukocyte subtype to infiltrate the lesion site within 30 min after stroke and release excessive ROS, proinflammatory cytokines (IL-1β, IL-6, TNF-α) and chemokines (MCP-1, MIP-1α, IL-8), MMPs and adhesion molecules (PSGL-1, L-selectin), which may exacerbate brain injury. 80 Microglia are primary innate immune cells within the central nervous system (CNS) and can produce pro-inflammatory factors in response to noxious stimuli, such as IL-1β, interferon γ, TNF-α, nitric oxide, and ROS.81,82 Representative adaptive immune cells T and B lymphocytes migrate into the brain at a later stage than neutrophils after brain injury, they can significantly influence microglial phenotype and function.75,83 Neuroinflammation is also driven by neuroimmune–related inflammatory mediators, for instance, complement activation promotes neuroinflammation and secondary brain injury after ABI, such as stroke, TBI, and intracerebral hemorrhage.84–86 Inhibition of complement can reverse neuroinflammatory transcriptome changes and significantly improve cognitive performance after TBI.76,84,87,88 TLR represent another recently emerging target for modulation of neuroinflammation after ABI89–92 Inhibition of TLR4 signaling pathways has been suggested to reduce microglia/macrophages activation and neutrophil infiltration, thereby ameliorating neuroinflammation and improving neurological function in animal models of ABI.93,94
In addition to basic research, clinical studies also emphasize the significance of neuroinflammation in patients with ABI. 95 Imitating the pathogenesis of ischemic stroke, neonates receiving cardiac surgery with deep hypothermic circulatory arrest showed immediate inflammatory responses based on the analysis of their vena cava superior blood samples, showing increased immune cell counts and cytokine concentrations. 96 The neuroinflammatory biomarker MMP-9 was highly correlated with poor prognosis of patients with ischemic stroke.97,98 Moreover, atorvastatin and rosuvastatin exhibited in TBI patients neuroprotective effects with better functional outcomes due to the anti-inflammatory properties of statins.99,100
The efforts of limiting neuroinflammation after ABI are rewarded by improved neurological recovery, therefore, attracting increasing attention and interest in the exploration of novel neuroinflammatory modulators.
The emerging role of ATF3 in ABI associated neuroinflammation
Stroke, divided into cerebral ischemic and hemorrhagic strokes, is, together with TBI the most typical ABI which is characterized byneuroinflammation.101,102 Subsequently, the activation of cerebral resident neural and peripheral immune cells amplifies neuroinflammatory responses due to the compromised permeability of the blood brain barrier (BBB) and the proinflammatory cytokine storm.103,104
Evidence shows that ATF3 can be expressed in microglia,105,106 brain ECs,67,107 and neurons9,108 in the CNS and can exert effects on neuroinflammatory modulators to affect brain physiologic function or development.31,32 Similarly, the expression of ATF3 in peripheral immune cells, such as macrophages, is involved in peripheral inflammation. 57 Thus, the underlying regulatory mechanism of ATF3 in inflammation is highly correlated with its participation in neuroinflammation.
ATF3 inhibits neuronal apoptosis following ischemic stroke
ATF3 has been implicated in the cellular response to ischemia/reperfusion of the brain, 5 which features by inflammatory responses109–111 (Figure 2). Accumulating evidence suggests that ATF3 is activated in the ischemic brain.32,112,113 Recently, ATF3 as an immediate early gene was proposed as a potential biomarker for diagnosis and prognosis in elderly women with focal ischemia.1,32,112,113 Studies discovered that gene transcript and protein levels of ATF3 were increased at the acute stage after ischemia/reperfusion, inhibiting glutamate-induced apoptosis. 114 Reasonably, knockout of ATF3 significantly increased the infarct volume, worsening neurological function, as well as eliciting the cellular inflammatory response 3 days after transient middle cerebral artery occlusion (MCAO). 33 Although it was reported that ATF3 expression was decreased 3 days after MCAO, 32 ATF3 overexpression attenuated neuronal caspase-dependent apoptosis, microglial activation, and pro-inflammatory cytokine production to alleviate brain injury.32,113 For example, ATF3 can block CCL2 transcription and TLR4/NF-κB signal activation to reduce the activation of microglia and prevent neuronal apoptosis. Another is to prevent neuronal apoptosis induced by c-Jun N-terminal kinase by promoting the expression of the anti-apoptotic neuronal survival factor HSP27 and activation of Akt.32,113 As mentioned above, ATF3 can be therapeutically targeted to modulate ischemic stroke. However, existing evidence of ATF3 regulation is currently described only for ischemic, not hemorrhagic stroke. No research has investigated the role of ATF3 during the chronic phase of inflammation. Thus, further research is required in the future.
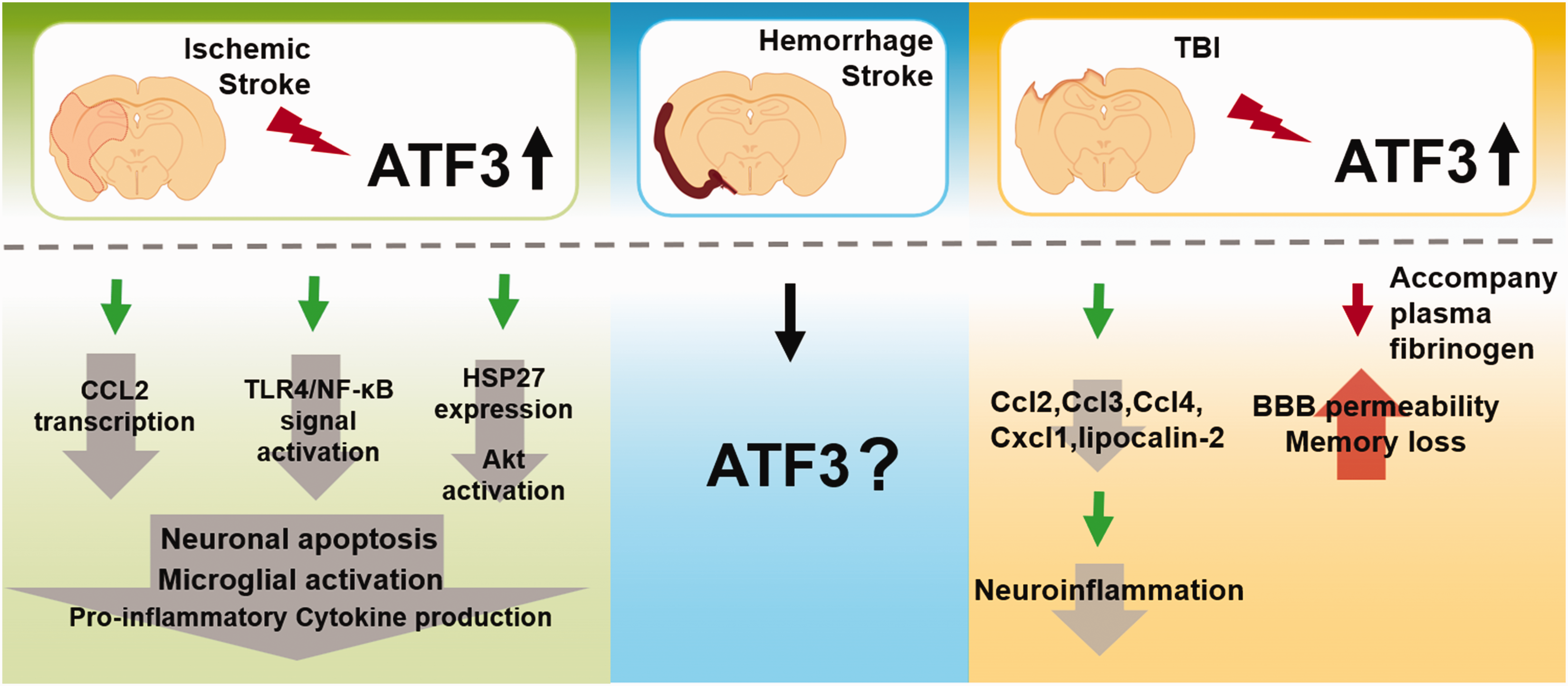
The function of ATF3 on acute brain injury. In the ischemic stroke, ATF3 overexpression attenuated neuronal caspase-dependent apoptosis, microglial activation, and pro-inflammatory cytokine production to alleviate brain injury. ATF3 can block CCL2 transcription and TLR4/NF-κB signal activation to reduce the activation of microglia and prevent neuronal apoptosis. Another is to prevent neuronal apoptosis induced by c-Jun N-terminal kinase by promoting the expression of the anti-apoptotic neuronal survival factor HSP27 and activation of Akt. There is a lack of knowledge regarding the regulation of ATF3 in hemorrhagic stroke. In TBI, upregulation of ATF3 in the brain negatively regulates CCL and CXCL chemokines (Ccl2, Ccl3, Ccl4, and Cxcl1) and lipocalin-2 expression, which then alleviates neuroinflammation after TBI. But increased BBB permeability and loss of memory after TBI are both caused by Fg-induced overexpression of ATF3.
TBI induces upregulation of ATF3 and exerts dualistic roles in neuroinflammation and BBB damage
TBI triggers an inflammatory response in the CNS and poses a substantial risk for neurodegenerative diseases.115,116 Previous studies shown that ATF3 is induced 2 hours after TBI31,117,118 and rapidly up-regulated in neuronal and immune cells, thereby regulating neuroinflammation 34 (Figure 2). Similar to ischemic stroke models, the upregulation of ATF3 occurs within 1–2 hours after TBI and decreases 2–3 days later. 34 Functionally, ATF3 is necessary to inhibit the production of CCL and CXCL chemokines (CCL2, CCL3, CCL4, and CXCL1), as well as lipocalin-2, subsequently reducing neuroinflammation in the subacute phase of TBI. 34 However, ATF3 overexpression following TBI caused by plasma fibrinogen is associated with increased BBB permeability 119 and memory loss. The fibrinogen inhibitor fibrinogen antisense oligonucleotide can decrease inflammatory responses and BBB permeability, thereby ameliorating neurological diseases. 120 Further studies are warranted to illustrate more detailed regulatory mechanisms of ATF3 in TBI owing to its dual effects in inflammatory responses.
Conclusion
Neuroinflammation is one of the key pathological features of a wide range of ABIs. Unresolved neuroinflammation continuously shapes the evolving brain pathology and affects the long-term neurological outcomes of affected patients.16,30 Neuroinflammation is an emerging therapeutic target for improving neurological function following ABI.
As an intermediate modulator in various pathological contexts, ATF3 plays a pivotal role in the regulation of inflammatory responses. In this mini review, we described that ATF3, a stress-associated transcription factor, can be rapidly induced in the brain after ABI and detail multiple ways by which ATF3 can participate in neuroimmune reactions after ABI, e.g., modulating the production of inflammatory mediators and altering cerebral vascular integrity. ATF3 might be therapeutically targeted to combat excessive inflammatory responses to modulate brain damage. However, there is a lack of knowledge regarding the regulation of ATF3 in hemorrhagic stroke and subarachnoid hemorrhage, in which neuroinflammation plays a critical role in determining neurological functions. Further studies are highly warranted to expand our knowledge of ATF3 regulation following hemorrhagic stroke and subarachnoid hemorrhage. Moreover, the differential role of ATF3 in acute versus chronic inflammation after brain injury should be explored in the pursuit of providing a framework to develop novel therapies for ABI.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: W.Y is supported by the National Natural Science Foundation of China (32030043,81971223), Shanghai Municipal Key Clinical Specialty (shslczdzk03601), Shanghai Engineering Research Center of Peri-operative Organ Support and Function Preservation (20DZ2254200), Area Municipal Commission of Health and Family Planning Funding (PWZxq2017-06), National Key R&D Program (2018YFA0108204), the Shanghai Municipal Education Commission (Grant number 2019-01-07-00-01-E00074). P.L. is supported by the National Natural Science Foundation of China (NSFC 91957111, 81971096, 82061130224, M-0671), New Frontier Technology Joint Research sponsored by Shanghai Shenkang Hospital Development Center (SHDC12019102), Shanghai Municipal Education Commission-Gaofeng Clinical Medical Grant Support (20181805), Shuguang Program supported by Shanghai Education Development Foundation and Shanghai Municipal Education Commission (20SG17), and Shanghai Outstanding Academic Leaders’ Program from Shanghai Municipal Science and Technology Committee (20XD1422400), Newton Advanced Fellowship grant provided by the UK Academy of Medical Sciences (NAF\R11\1010) P.L. is also supported by SHSMU-ZLCX20211602, Innovative Research Team of High-level Local Universities in Shanghai. W.X. is supported by the National Natural Science Foundation of China (NSFC, 81901985). X. W. is supported by the National Natural Science Foundation of China (NSFC, 82001381) and Shanghai Sailing Program (20YF1425400). This work is also supported by Clinical Research Plan of SHDC.
Acknowledgements
Thank the authors of the references
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.