
Abstract
Proinflammatory cytokines are important mediators of neuroinflammation after traumatic brain injury. The role of interleukin (IL)-18, a new member of the IL-1 family, in brain trauma has not been reported to date. The authors investigated the posttraumatic release of IL-18 in murine brains following experimental closed head injury (CHI) and in CSF of CHI patients. In the mouse model, intracerebral IL-18 was induced within 24 hours by ether anesthesia and sham operation. Significantly elevated levels of IL-18 were detected at 7 days after CHI and in human CSF up to 10 days after trauma. Published data imply that IL-18 may play a pathophysiological role in inflammatory CNS diseases; therefore its inhibition may ameliorate outcome after CHI. To evaluate the functional aspects of IL-18 in the injured brain, mice were injected systemically with IL-18–binding protein (IL-18BP), a specific inhibitor of IL-18, 1 hour after trauma. IL-18BP—treated mice showed a significantly improved neurological recovery by 7 days, accompanied by attenuated intracerebral IL-18 levels. This demonstrates that inhibition of IL-18 is associated with improved recovery. However, brain edema at 24 hours was not influenced by IL-18BP, suggesting that inflammatory mediators other than IL-18 induce the early detrimental effects of intracerebral inflammation.
The pathology of traumatic brain injury is very complex and still poorly understood. Research efforts in the past decade have highlighted an important role for cytokines released systemically as well as locally within the intrathecal compartment after brain injury. A dual effect of proinflammatory cytokines, such as tumor necrosis factor (TNF), interleukin (IL)-6, or IL-8, was demonstrated based on findings of a time-dependent beneficial or adverse effect of these cytokines (Gruol and Nelson, 1997; Morganti-Kossmann et al., 1997; Kossmann et al., 1997, Shohami et al., 1999; Scherbel et al., 1999; Stahel et al., 2000; Whalen et al., 2000). IL-18 is a proinflammatory cytokine structurally related to the IL-1 family, previously known as interferon-γ–inducing factor (Okamura et al., 1995), mediating a broad array of effector functions of both the innate and acquired immune system (Dinarello, 1999; McInnes et al., 2000; Akira, 2000; Nakanishi et al., 2001). IL-18 is constitutively expressed in human blood monocytes, and recent studies have demonstrated that it is also expressed in the normal CNS of mice, rats, and humans (Culhane et al., 1998; Jander and Stoll, 1998; Prinz and Hanisch, 1999; Fassbender et al., 1999; Wheeler et al., 2000; Hodge et al., 2001; Menge et al., 2001). Furthermore, IL-18 expression was detected in primary cultures of astrocytes and microglia, but not neurons, in vitro (Conti et al., 1999). In clinical studies, increased IL-18 levels have been reported in the CSF of patients with inflammatory CNS diseases, such as bacterial meningitis and viral meningoencephalitis (Fassbender et al., 1999), and in the CSF of patients with multiple sclerosis (MS) (Losy and Niezgoda, 2001). Consistent with the finding of elevated intrathecal IL-18 levels in patients with MS, increased IL-18 mRNA expression was demonstrated in spinal cords of Lewis rats with experimental autoimmune encephalomyelitis, the animal model for MS (Jander and Stoll, 1998). In addition, a recent study demonstrated an increase of IL-18 protein, but not mRNA expression, after experimental crush injury to optic and sciatic nerves in rats (Menge et al., 2001). Although these data imply that IL-18 may play a pathophysiological role in inflammatory CNS diseases, the expression and functional significance of IL-18 after traumatic brain injury has not been investigated to date. IL-18–binding protein (IL-18BP) is a naturally occurring, specific inhibitor of IL-18 (Aizawa et al., 1999; Kim et al., 2000). IL-18BP has been shown to bind IL-18 with high affinity and effectively inhibit its biological activities, such as the IL-18–mediated early Th1 cytokine response (Novick et al., 1999, 2001; Reznikov et al., 2000). Interestingly, recombinant IL-18BP does not exhibit species specificity, and both human and murine IL-18BP have been shown to block the binding of IL-18 to its receptor cross-species (Aizawa et al., 1999; Kim et al., 2000; Novick et al., 1999).
In the present study, we analyzed the expression of IL-18 at the protein level both in brain homogenates from mice with experimental closed head injury (CHI) and in CSF and matching serum samples from patients with CHI. In addition, we assessed the functional role of IL-18 inhibition by treatment of mice with IL-18BP 1 hour after experimental CHI. This protocol was designed with the aim to create a new therapeutic approach with relevance to clinical situations. We hypothesized that the antiinflammatory effect of IL-18BP would result in an improved clinical recovery and attenuated extent of brain damage as compared to vehicle-treated control animals.
MATERIALS AND METHODS
Trauma model
The mice used in this study were 8-to 16-week-old males weighing 30 to 35 g. They were bred in a specific pathogen-free environment, kept under standard conditions of temperature and light in cages of 4 to 6 animals, and fed with food and water ad libitum. The study was performed according to the guidelines of the Institutional Animal Care Committee of the Hebrew University of Jerusalem, Israel. Experimental CHI was performed using a modified weight-drop device previously developed in our laboratory (Chen et al., 1996). Briefly, after induction of ether anesthesia, a midline longitudinal incision was performed, the skin was retracted, and the skull was exposed. The left anterior frontal area was identified and a tipped Teflon cone was placed 1 mm lateral to the midline, in the midcoronal plane. The head was fixed and a 75-g weight was dropped on the cone from a height of 18 cm, resulting in a focal injury to the left hemisphere. After trauma, the mice received supporting oxygenation with 100% O2 for no longer than 2 minutes and were then brought back to their cages.
Assessment of interleukin-18 levels in mouse brains
For quantification of intracranial IL-18 levels, mice of the C57BL/6 (B6) strain (total n = 81) were assigned to six distinct groups: (1) “normal controls”—untreated B6 mice (n = 10); (2) “ether anesthesia”—mice were anesthetized with ether for 10 minutes and decapitated after 24 hours (n = 10) or 7 days (n = 8); (3) “sham operation”—these mice underwent anesthesia and longitudinal scalp incision and were killed after 24 hours (n = 10) or 7 days (n = 8); (4) “trauma group”—experimental CHI was performed as described above, and the animals were decapitated in ether anesthesia at 4 hours (n = 7), 24 hours (n = 7), and 7 days (n = 7) after trauma; (5) “IL-18BP group”—in order to evaluate whether the systemic injection of IL-18BP (see protocol that follows) results in attenuated intracranial IL-18 levels, an additional group of B6 mice (n = 10) were killed 7 days after CHI for analysis of IL-18; (6) “vehicle control group”—four additional mice were treated as described for the “IL-18BP group,” but were injected with vehicle solution only, as negative control. In all animals, the brains were immediately removed after decapitation, snap-frozen in liquid nitrogen, and stored at −70°C until analyzed. Brains from the trauma group were separated into left (ipsilateral) and right (contralateral) hemisphere, in order to allow a comparison of IL-18 levels in the injured versus noninjured hemisphere. Tissue homogenization was performed with a Polytron (Kinematica, Kriens, Switzerland) using a dilution of 1:4 in ice cold extraction buffer (W/W) containing Tris 50 mmol/L (pH 7.2), NaCl 150 mmol/L, Triton-X-100 1% (Boehringer Mannheim, Rotkreuz, Switzerland), and protease inhibitor cocktail (Boehringer Mannheim). The homogenate was shaken on ice for 90 minutes and then centrifuged for 15 minutes at 3,000 g and 4°C. The supernatants were aliquoted and stored at −70°C until analysis. The concentrations of total protein in the brain extracts were measured by Bradford assay (Bio Rad Laboratories, Munich, Germany) and were found to be very consistent in all mice assessed (12.1 ± 2.1 mg/mL; mean ± SD). Quantification of intracerebral cytokine levels was performed by enzyme-linked immunosorbent assay (ELISA) specific for both mature and precursor form of murine IL-18 according to the manufacturer's instructions (R&D Systems, Abingdon, U.K.). The sensitivity of the assay was 5 pg/mL. For comparison of the intracerebral IL-18 levels between the different animal groups, all concentrations below the detection limit of 5 pg/mL were assigned a value of 4.9 pg/mL. The samples were run undiluted in duplicate wells, and the final concentration was calculated from the mean OD of duplicate samples. The OD was determined by spectrophotometer (Dynatech Laboratories Inc., Chantilly, VA, U.S.A.) at an extinction wavelength of 405 nm. The final IL-18 concentrations were divided by the total protein concentration of the according brain hemisphere, and the data are presented as picograms per milligram tissue protein. Since IL-18 is produced and stored as a biologically inactive precursor (pro-IL-18), biologically active IL-18 can be accurately detected in supernatants and CSF, whereas brain homogenates consist of a mixture of pro-IL-18 and mature IL-18. Thus, the use of ELISA on brain homogenates results in measurement of a nondistinguishable form of IL-18, “immunoreactive IL-18” (irIL-18). In this study, the levels derived from brain homogenates (“irIL-18”) are denoted as “IL-18” for simplicity throughout the article.
IL-18BP treatment protocol
Male Sabra mice of the Hebrew University strain (n = 51) were used for the IL-18BP studies. Anesthesia and experimental CHI were performed as described above. IL-18BP was provided by Serono Intl., Switzerland. For the treatment protocol, the animals were stratified into two groups. In the control group (n = 25), mice were subjected to experimental CHI, and 1 hour later they were injected with vehicle alone (0.5 mL sterile phosphate-buffered saline [PBS] intraperitoneally) and were observed for 7 days for neurological assessment (n = 16) or sacrificed after 24 hours for evaluation of posttraumatic brain edema (n = 9). In the treatment group (n = 26), the mice were injected 1 hour after CHI with 50 μg human recombinant IL-18BP given intraperitoneally diluted in 0.5 mL sterile PBS. Human IL-18BP was previously shown to cross-react between humans and mice (Novick et al., 1999). Since the blood—brain barrier permeability is increased five-to sixfold between 1 to 4 hours after CHI, as previously determined in the same experimental model (Chen et al., 1996), a time-point was selected where the inhibitor should be available to the intrathecal compartment after systemic administration. The IL-18BP—treated mice were either assessed for neurological impairment for 7 days (n = 18) or killed after 24 hours for quantification of brain edema (n = 8), as described below.
Evaluation of neurological impairment
For assessment of posttraumatic neurological impairment, a Neurological Severity Score (NSS) has been previously developed and validated in our laboratory (Beni-Adani et al., 2001). The score consists of 10 individual clinical tasks on motor function, alertness, and physiological behavior, whereby one point is given for failure of task performance and zero points for success. A maximal NSS of 10 points indicates severe neurological dysfunction, with failure of all tasks, whereas a score of zero is achieved by healthy uninjured mice. The NSS at 1 hour after trauma reflects the initial severity of injury and is inversely correlated with clinical outcome (Beni-Adani et al., 2001). Evaluation of task performance was done by two investigators who were blinded about the study groups at the time-points 1 hour, 24 hours, 72 hours, and 7 days after experimental CHI. The ΔNSS, calculated as the difference between NSS at 1 hour and the NSS at any later time-point, is a parameter that reflects the degree of spontaneous recovery following brain injury, as described earlier (Chen et al., 1996).
Assessment of brain edema
The extent of cerebral edema was evaluated by determining the tissue water content in the injured hemisphere, as previously described (Chen et al., 1996). Briefly, mice were anesthetized as described above at 24 hours after trauma, which corresponds to a time-point at which edema is maximal in this model system. After decapitation, a cortical segment of approximately 20 mg was prepared from an area bordering the trauma site and from the contralateral hemisphere. The right (noninjured) hemisphere was used as an internal control. The tissue slices were weighed and dried for 24 hours at 95°C. After weighing the “dried” sections, the percentage of brain water content was calculated as %H2O = [(wet weight − dry weight) × 100] / wet weight.
Brain injury patients
Ten patients with isolated CHI (mean age ± SD: 37 ± 10 years; range 24 to 57 years; 9 men and 1 woman) and a Glasgow Coma Scale score less than or equal to 8 (Teasdale and Jennett, 1974) after cardiopulmonary resuscitation were included in this study. Following computed tomography scan evaluation, the patients received intraventricular catheters for therapeutic CSF drainage when the intracranial pressure (ICP) exceeded 15 mm Hg. No patient was treated with steroids. Patients with multiple injuries requiring interventions for concomitant thoracic, abdominal, pelvic, spinal injuries, or long bone fractures were excluded from the study. The individual outcome was assessed using the Glasgow Outcome Scale (Jennett and Bond, 1975). The protocol for CSF and serum collection was approved by the Ethics Board Committee of the University Hospital, Zurich.
Sample collection and interleukin-18 analysis
The CSF and matched serum samples of CHI patients (n = 10) were collected daily at a fixed time-point. Control CSF was collected from patients undergoing diagnostic spinal tap (n = 5). These patients did not have signs of inflammatory CNS disease, based on normal CSF values of protein, glucose, and cell count (data not shown). In the CHI group, sample collection was performed for 10 days after trauma, unless the ventricular catheter was removed earlier, for example, in cases where the ICP remained in a normal range (≤15 mm Hg) for more than 24 hours. A total of 106 matched CSF and serum samples were collected in the trauma patients analyzed in this study. All samples were immediately centrifuged after collection, aliquoted, and frozen at −70°C until analysis. Quantification of IL-18 levels in CSF and serum was performed by ELISA specific for both the mature and the pro-form of human IL-18 (R&D Systems). For comparison of the IL-18 CSF levels between CHI patients and controls, all concentrations less than the detection limit of 5 pg/mL were assigned a value of 4.9 pg/mL.
Data analysis
Statistical analysis of the data was performed on commercially available software (SPSS 9.0 for Windows). The nonparametric Mann-Whitney U test was used for analysis of data that were not normally distributed, such as the neurological scores (NSS and ΔNSS). The unpaired Student's t-test was used for comparison of intracerebral IL-18 concentrations in the different mouse groups and for analysis of differences in brain water content in the IL-18BP—treated versus vehicle-injected mice. The comparison of human IL-18 levels, either in daily CSF versus matched serum samples in CHI patients, or in trauma versus control CSF, were determined using the general linear model for repeated measures analysis of variance (ANOVA). A P value less than 0.05 was considered to be statistically significant.
RESULTS
Intracerebral interleukin-18 levels in mice
As shown in Fig. 1, IL-18 was detectable by ELISA in brain homogenates of untreated (“normal”) control mice of the B6 strain (n = 10), with a mean level of 2,105 ± 375 (± SD) pg/mg protein. In the experimental groups, the induction of ether anesthesia alone or in combination with “sham” operation (i.e., ether anesthesia and longitudinal scalp incision) resulted in significantly elevated intracranial IL-18 levels of 3,575 ± 390 pg/mg protein (“ether” group, n = 8) and 3,650 ± 780 pg/mg protein (“sham” group, n = 13) within 24 hours (P < 0.01 versus “normal” mice, unpaired Student's t-test;Fig. 1). In contrast, the difference between the “ether” and “sham”-treated animals was not statistically significant (P = 0.24). In the trauma group (n = 21), induction of CHI resulted in elevated IL-18 levels both in the injured and in the contralateral hemispheres within 4 hours (4,810 ± 405 and 4,560 ± 590 pg/mg protein) to 24 hours (5,060 ± 220 and 4,895 ± 600 pg/mg protein) after trauma. However, the levels were not significantly higher than in the “ether” or “sham” groups sacrificed after 24 hours (P > 0.05). In contrast, by 7 days after CHI, a significant increase of intracerebral IL-18 was detected in both hemispheres (6,475 ± 890 and 5,870 ± 785 for left and right hemispheres, respectively), compared to ether-anesthetized or sham-operated animals sacrificed after 7 days (3,280 ± 215 and 3,220 ± 395 pg/mg protein, respectively;P < 0.01).
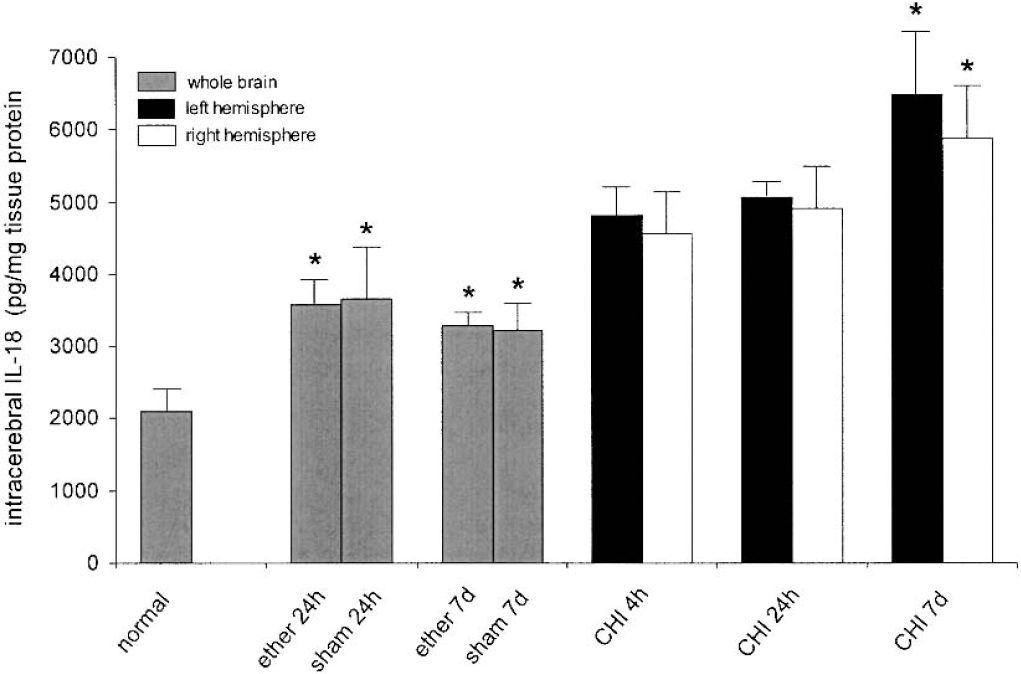
Intracerebral interleukin-18 levels in mice of the B6 strain (n = 67), as determined by enzyme-linked immunosorbent assay in brain homogenates. The animals were either untreated (“normal”), or subjected to ether anesthesia, “sham” operation (ether anesthesia and scalp incision), or to experimental closed head injury (CHI), and killed after 4 hours, 24 hours, or 7 days. The data are presented as mean ± SD. * P < 0.01 for “ether”/“sham” groups versus normal mice at 24 hours and 7 days, and for CHI versus “ether” or “sham” groups at 7 days.
Effect of posttraumatic IL-18BP treatment on the neurological recovery
In order to investigate our hypothesis that inhibition of IL-18 might facilitate recovery after brain injury, we compared the clinical state of the traumatized mice at different time points after a single injection of recombinant IL-18BP. In order to achieve groups of animals with comparable trauma, mice were assigned to different treatment groups after their initial NSS was evaluated 1 hour after CHI. As shown in Fig. 2A, both groups had a similar initial NSS 1 hour after trauma, with a mean score of 7.69 ± 1.25 (± SD) for the control group (n = 16) and 7.44 ± 1.62 for the IL-18BP group (n = 18), indicating a comparable severity of injury. Evaluation of NSS at later time-points revealed that the IL-18BP group exhibits a tendency toward less neurological impairment at 24 hours and 72 hours after CHI, as shown by lower mean NSS values compared to the vehicle-treated control group. After 7 days, the IL-18BP—treated mice showed a significantly attenuated neurological impairment compared to the control group, as evident by a significantly lower mean NSS in the IL-18BP group (3.89 ± 2.30 versus 5.94 ± 1.73, respectively, P < 0.05, Mann-Whitney U test). In addition, the extent of neurological recovery after trauma, as expressed by the ΔNSS, revealed that IL-18BP—treated mice had a significantly higher ΔNSS after 24 hours and 7 days compared to the control group, demonstrating a better neurological recovery. As shown in Fig. 2B, the ΔNSS was significantly higher in the IL-18BP group compared to controls after 24 hours (2.0 ± 1.46 versus 1.06 ± 1.12;P < 0.05) and after 7 days (3.56 ± 1.69 versus 1.75 ± 1.77, P < 0.01), whereas the difference in ΔNSS at the time-point 72 hours was not statistically significant (P > 0.05).
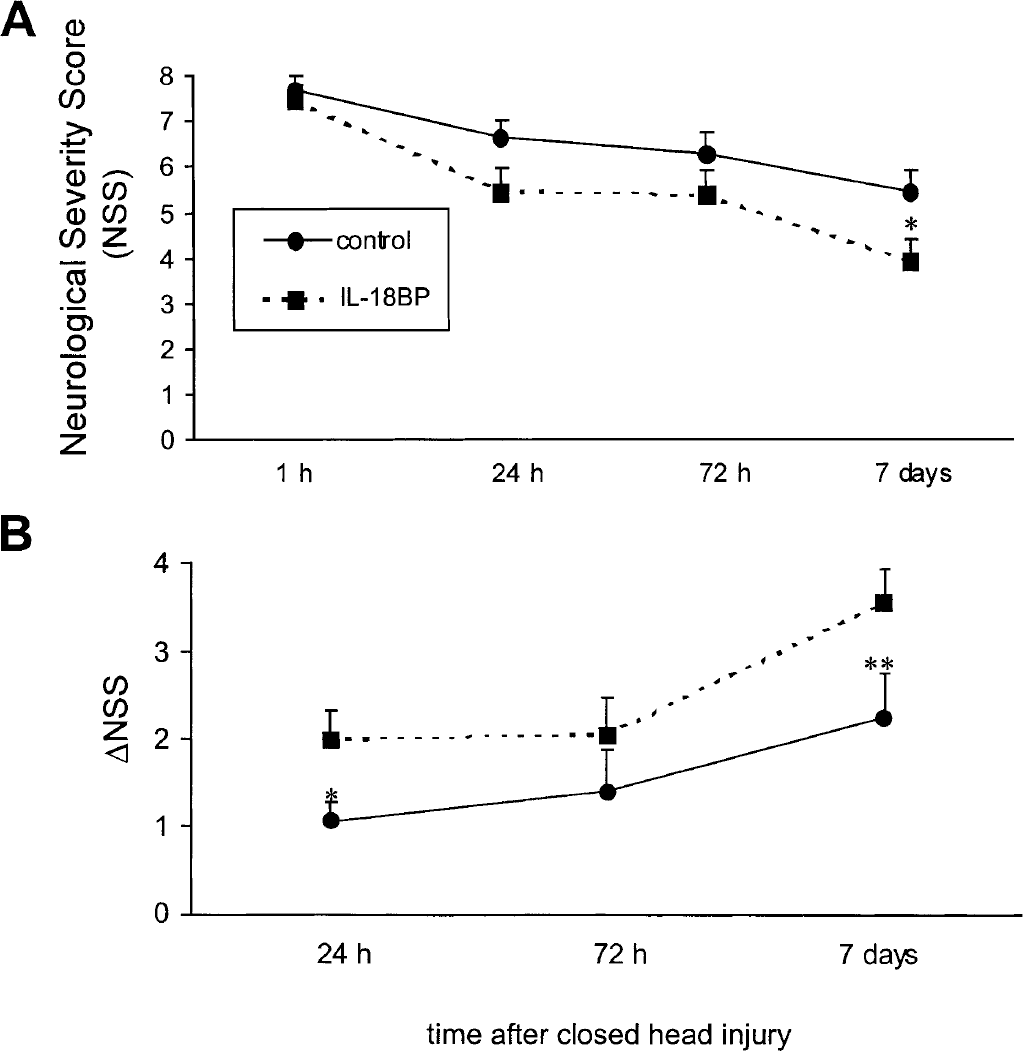
Improvement of posttraumatic neurological recovery after administration of interleukin-18–binding protein (IL-18BP), as determined by the Neurological Severity Score (NSS)
Assessment of posttraumatic brain edema
Brain water content measured 24 hours post CHI was significantly higher in the contused (79.77 ± 1.5%) than in the contralateral hemisphere (77.40 ± 0.57%) of the brain-injured mice. No effect of IL-18BP was found in either hemisphere (79.57 ± 1.64% versus 77.05 ± 0.91%). Although the water content was significantly elevated in the injured compared to the contralateral hemisphere (P < 0.001, unpaired Student's t-test), the extent of posttraumatic cerebral edema was not different between the IL-18BP—treated mice and the vehicle control group (data not shown).
IL-18BP reduces intracerebral interleukin-18 levels
In order to determine whether the administration of IL-18BP results in attenuated intracerebral IL-18 levels after trauma, IL-18 was measured in injured brains of IL-18BP—treated mice after 7 days (n = 10). This time-point was selected based on the observation that both the maximal elevation of IL-18 in the injured brain as well as the best functional recovery in the IL-18BP group was seen at 7 days after CHI (Figs. 1 and 2). As shown in Fig. 3, the intraperitoneal injection of 50 μg IL-18BP at 1 hour after trauma resulted in significantly attenuated intracranial IL-18 levels by 7 days, both in the injured left hemisphere (3,730 ± 470 pg/mg protein) and in the contralateral hemisphere (3,485 ± 530 pg/mg protein), compared to untreated brain-injured mice (6,475 ± 890 and 5,870 ± 785 pg/mg protein for left and right hemispheres) and to vehicle-injected mice (6,610 ± 415 and 6,200 ± 260 pg/mg protein for left and right hemispheres) 7 days after trauma (P < 0.001).
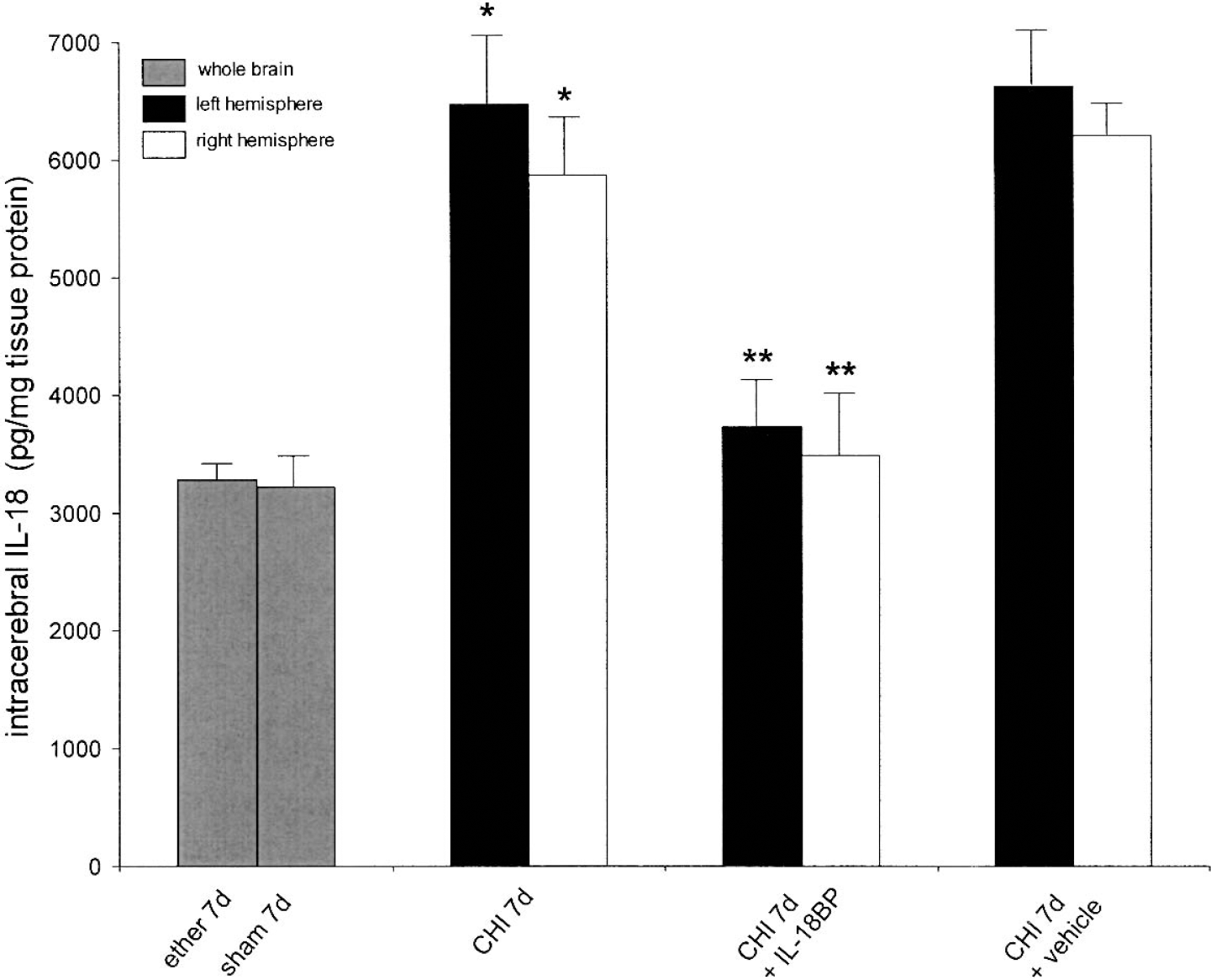
Interleukin-18–binding protein (IL-18BP) reduces intracerebral IL-18 levels after closed head injury (CHI) in B6 mice (n = 37). One hour after CHI, a group of mice was injected with 50 μg human recombinant IL-18BP intraperitoneally or with vehicle solution only and IL-18 levels were assessed by enzyme-linked immunosorbent assay in brain homogenates after 7 days. Data are shown as mean ± SD. * P < 0.01 for CHI and for vehicle-treated CHI versus “ether” and “sham” controls, and ** P < 0.001 for IL-18BP—injected versus untreated or vehicle-injected mice 7 days after CHI (unpaired Student's t-test).
Elevated interleukin-18 levels in human cerebrospinal fluid after brain injury
IL-18 levels were measured in daily CSF and serum samples from 10 patients with severe CHI for up to 10 days after trauma. The patients' demographic and clinical data and the median IL-18 levels with individual ranges in CSF and serum are presented in Table 1. The median levels of IL-18 in CSF were significantly elevated in 9 of 10 CHI patients compared to control CSF from 5 patients without trauma or inflammatory neurological disease (P < 0.05; repeated measures ANOVA). One CHI patient (patient 10) had elevated daily peak levels of up to 169 pg/mL in the CSF; however, the median IL-18 value in CSF was not significantly elevated above control (P = 0.31;Table 1). Intrathecal IL-18 was detectable by ELISA in 90% of all CSF samples in the trauma group, whereas only 40% of control CSF samples had detectable IL-18 in CSF (i.e., >4.9 ng/mL). Notably, the maximal IL-18 concentrations in CSF (966 pg/mL) were almost 200-fold higher in head-injured patients than in controls. In 8 of 10 CHI patients, the median IL-18 concentrations were significantly higher in CSF than in serum (P < 0.05). However, in two patients (patients 9 and 10) the median IL-18 levels in serum exceeded the corresponding concentrations in CSF, as shown in Table 1.
Demographic and clinical data of patients with closed head injury
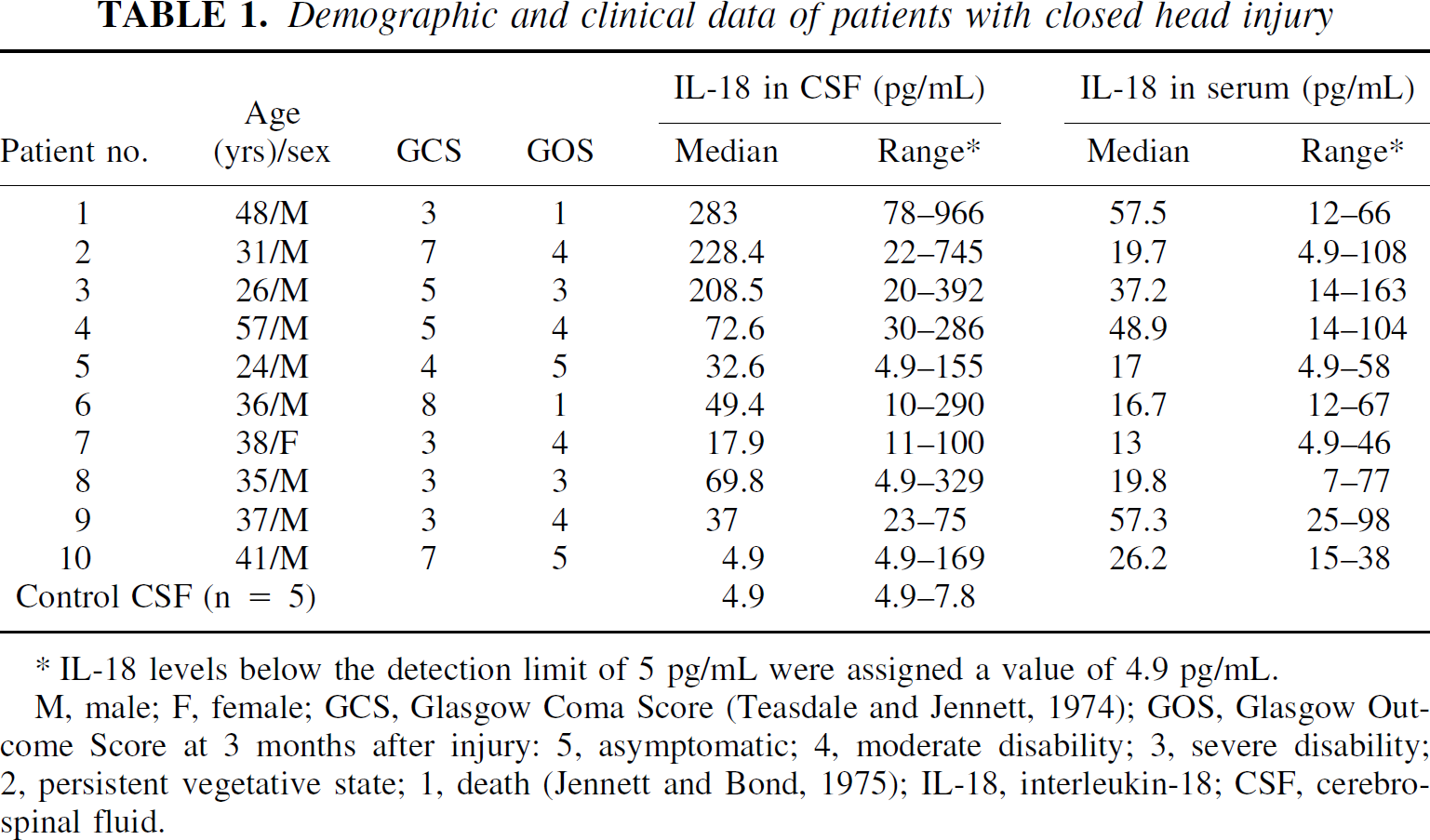
M, male; F, female; GCS, Glasgow Coma Score (Teasdale and Jennett, 1974); GOS, Glasgow Out-come Score at 3 months after injury: 5, symptomatic; 4, moderate disability; 3, severe disability; 2, persistent vegetative state; 1, death (Jennett and Bond, 1975); IL-18, interleukin-18; CSF, cerebrospinal fluid.
IL-18 levels below the detection limit of 5 pg/mL were assigned a value of 4.9 pg/mL.
DISCUSSION
In this study, we demonstrate for the first time elevated intracranial levels of IL-18 after traumatic brain injury, both in a clinical setting, by analysis of human ventricular CSF samples, and in a standardized experimental model of CHI, by analysis of murine brain homogenates. Previous studies have revealed a constitutive expression of IL-18 in the CNS, based on studies in humans and rodents (Culhane et al., 1998; Jander and Stoll, 1998; Prinz and Hanisch, 1999; Fassbender et al., 1999; Wheeler et al., 2000; Conti et al., 1999). Furthermore, IL-18 was shown to be upregulated under inflammatory conditions in the CNS, such as bacterial or viral infection or autoimmune-mediated inflammation, both at the protein and mRNA level (Jander and Stoll, 1998; Fassbender et al., 1999; Losy and Niezgoda, 2001). However, the intracranial expression and regulation of IL-18 after CNS trauma has not been previously investigated. In the experimental part of our study, we confirm previous findings of constitutive IL-18 expression in normal murine brains (Prinz and Hanisch, 1999; Hodges et al., 2001). We also demonstrate for the first time a significant induction of intracerebral IL-18 within hours following trivial stimuli in control groups, such as ether anesthesia or “sham” operation (a combination of ether anesthesia and scalp incision). These control groups exhibited a significant elevation of intracerebral IL-18 levels at 24 hours and 7 days, compared to untreated normal mice (Fig. 1), suggesting that the induction of ether anesthesia represents a potent mediator of intracerebral IL-18 expression.
In “sham”-operated mice, the additional incision of the scalp did not result in a significant further induction of IL-18, suggesting that the additional surgical measure does not influence IL-18 production in the CNS. Although epidermal cells, such as keratinocytes, represent a cellular source of IL-18 (Kampfer et al., 2000), it would not seem reasonable to hypothesize that the peripheral release of IL-18 through these cell types after scalp incision may contribute to elevated intracerebral cytokine levels. Interestingly, the experimental trauma itself did not induce significantly higher IL-18 levels above enhanced levels in “ether” or “sham” control groups during the early posttraumatic time period up to 24 hours. In addition, the observation of elevated IL-18 levels to a similar extent both in the injured (left) and the contralateral (right) hemispheres is furthermore surprising and suggests a diffuse mechanism, e.g., the induction of general anesthesia, to be responsible for the intracerebral upregulation of IL-18 in brain-injured mice at early time-points, rather than the focal impact itself. We have previously demonstrated that other inflammatory mediators, such as TNF and IL-1β (Shohami et al., 1994) as well as CC- and CXC-chemokines (Otto et al., 2001), are significantly upregulated in the injured hemisphere, but not in the contralateral side, within 1 to 24 hours after trauma, using the same experimental model system. However, other experimental studies using the same model system have brought evidence that the contralateral hemisphere is not completely unaffected after trauma. For example, markers of oxidative stress indicate that early after CHI, the contralateral hemisphere is also under stress (Beit-Yannai et al., 1997). In addition, Holmin et al. demonstrated that after mild brain injury, late cytokine production occurred not only in the contused hemisphere, but in the contralateral corpus callosum as well (Holmin et al., 1997). Moreover, recently it was found that presenilins are upregulated at 3 to 7 days after CHI also in the contralateral corpus callosum (R. Stein, personal communication). Thus, the diffuse induction of IL-18 seems to represent a phenomenon specific to this cytokine, in contrast to mediators of the early inflammatory response after trauma, such as other proinflammatory cytokines (TNF, IL-1β, IL-6) and chemokines. The exact mechanisms of anesthesia-induced expression of IL-18 remain to be further elucidated. Seven days after trauma, IL-18 levels were significantly elevated in both hemispheres compared with corresponding control groups analyzed at the same time-point (Fig. 1). These data demonstrate that the induction of CHI leads to an exacerbation of IL-18 production above anesthesia-induced levels only at later time-points. This is in contrast to the temporal changes in other cytokines that are upregulated only at very early times after CHI (Shohami et al., 1994, 1997). Our present findings are compatible with a recent report describing the dynamic changes of brain IL-18 after cerebral ischemia. In this article, Jander et al. also found that the increase of intracerebral IL-18 is delayed, starting at 48 hours and peaking at 7 to 14 days, and proposed that this is part of the delayed inflammatory response (Jander et al., 2002). A late peak (4 to 6 days) of brain cytokine production was also reported by Holmin et al. (1997), using a mild contusion model in the rat.
The further design of the study was aimed at assessing the potential IL-18 expression in the CNS in vivo. We have assessed the functional significance of intracranial IL-18 after brain injury by pharmacological inhibition with IL-18BP, a recently described specific inhibitor of IL-18 (Novick et al., 1999; Dinarello, 2000). Since we found that IL-18 is elevated in murine brains within a week after experimental CHI, we hypothesized that inhibition of IL-18 may result in a reduced inflammatory response and improved recovery after trauma. In order to achieve a potential clinical relevance for therapeutic intervention, we selected a time-point within the “window of opportunity” at 1 hour after CHI for administration of IL-18BP. The extent of posttraumatic neurological recovery was determined using a previously characterized score (NSS) that correlates with the severity of the injury and with the volume of injured brain tissue, as determined by magnetic resonance imaging and histological analyses (Beni-Adani et al., 2001). The systemic application of 50 μg IL-18BP 1 hour after CHI resulted in a significantly improved neurological outcome of brain-injured mice within 7 days, as reflected by a reduced NSS and elevated ΔNSS (Fig. 2). It is interesting that contrary to other cytokines that are elevated within the intrathecal compartment for a shorter period after CHI, such as TNF or IL-6 (Shohami et al., 1994), IL-18 reached significant peaks within the injured brain only after 7 days. It may be assumed that in the first few days after CHI other cytokines play a more significant role than IL-18 in determining the extent of intracerebral inflammation and that the inhibitory effect of IL-18BP becomes evident only after peak levels of other cytokines decline. This hypothesis may explain the lack of influence of IL-18BP on the extent of posttraumatic brain edema within 24 hours after trauma. In order to further investigate whether the beneficial effects of IL-18BP after 7 days are accompanied by a reduction of intracerebral IL-18, we assessed IL-18 levels in brain homogenates of IL-18BP—treated mice 7 days after trauma. In these studies, IL-18 was significantly attenuated within 7 days in mice treated with IL-18BP 1 hour after trauma, as compared to untreated or to vehicle-injected brain-injured mice sacrificed at the same time-point (Fig. 3), demonstrating that the neuroprotective effects of IL-18BP are associated with attenuated IL-18 levels in the injured brain. It should be noted that pro-IL-18 binds to IL-18BP with about 10 times lower affinity as compared to the mature IL-18 (Novick et al., 2001) thus, the decrease of irIL-18 shown after treatment with IL-18BP implies that indeed the mature form of the cytokine was assayed. In the last part of our project, the results derived from the experimental studies were extrapolated to a clinical setting, in order to determine whether IL-18 is elevated in the intrathecal compartment during the posttraumatic course after CHI in humans. The analysis of serial daily CSF samples revealed that IL-18 is significantly elevated for up to 10 days after trauma in severely brain-injured patients, as compared to matched serum samples or to healthy control subjects (Table 1). However, it has to be taken into account that assessment of ventricular CSF has limitations in that “true” brain tissue levels of IL-18 may not be accurately reflected. This notion is supported by the observation of approximately 1,000-fold elevated IL-18 levels in murine brain homogenates compared to human CSF samples. Further studies are required for assessing the exact pathophysiological role of IL-18 in the posttraumatic course after brain injury, e.g., by the use of IL-18–deficient mice (Wei et al., 1999).