
Abstract
Systemic inflammation and multi-organ failure represent hallmarks of the post-cardiac arrest syndrome (PCAS) and predict severe neurological injury and often fatal outcomes. Current interventions for cardiac arrest focus on the reversal of precipitating cardiac pathologies and the implementation of supportive measures with the goal of limiting damage to at-risk tissue. Despite the widespread use of targeted temperature management, there remain no proven approaches to manage reperfusion injury in the period following the return of spontaneous circulation. Recent evidence has implicated the lung as a moderator of systemic inflammation following remote somatic injury in part through effects on innate immune priming. In this review, we explore concepts related to lung-dependent innate immune priming and its potential role in PCAS. Specifically, we propose and investigate the conceptual model of lung–brain coupling drawing from the broader literature connecting tissue damage and acute lung injury with cerebral reperfusion injury. Subsequently, we consider the role that interventions designed to short-circuit lung-dependent immune priming might play in improving patient outcomes following cardiac arrest and possibly other acute neurological injuries.
Keywords
Introduction
Despite continued innovation in the prehospital phase of care, the expanded use of induced hypothermia, and advanced ICU level support measures supported by trial data, the morbid and mortal consequences of cardiac arrest (CA) remain a significant challenge. In this review, we discuss both the epidemiology of CA and describe the pathological cascade including selective neuronal vulnerability, neuroinflammation, peripheral immune priming, and neurovascular injury observed after CA. Drawing from the broader stroke and critical care literature, we then investigate how understanding the complex interplay between the brain, gut and lung, and their associated effects on innate immune priming may present a unique opportunity to target a pathological circuit we refer to as lung–brain coupling to improve patient outcomes following CA (Figure 1).
Lung–brain coupling in the post-cardiac arrest syndrome. Acute myocardial ischemia and associated ventricular arrhythmia induce end organ ischemic injury by perturbing systemic perfusion. In the CNS, ischemia induces cellular injury directly through both acute and delayed mechanisms. In the periphery, ischemia damages the physical, biochemical, and immune defenses present within the intestinal mucosae resulting in the translocation of gastrointestinal flora and systemic release of pro-inflammatory molecules and microorganisms. With the return of spontaneous circulation and tissue reperfusion, hematogenous spread of DAMPs from the CNS and PAMPs from the gut trigger acute lung inflammation. These systemic factors, combined with a variety of iatrogenic insults to the lung during the early phase of treatment, contribute to a feed-forward mechanism of reperfusion injury that heightens post-ischemic neuroinflammation.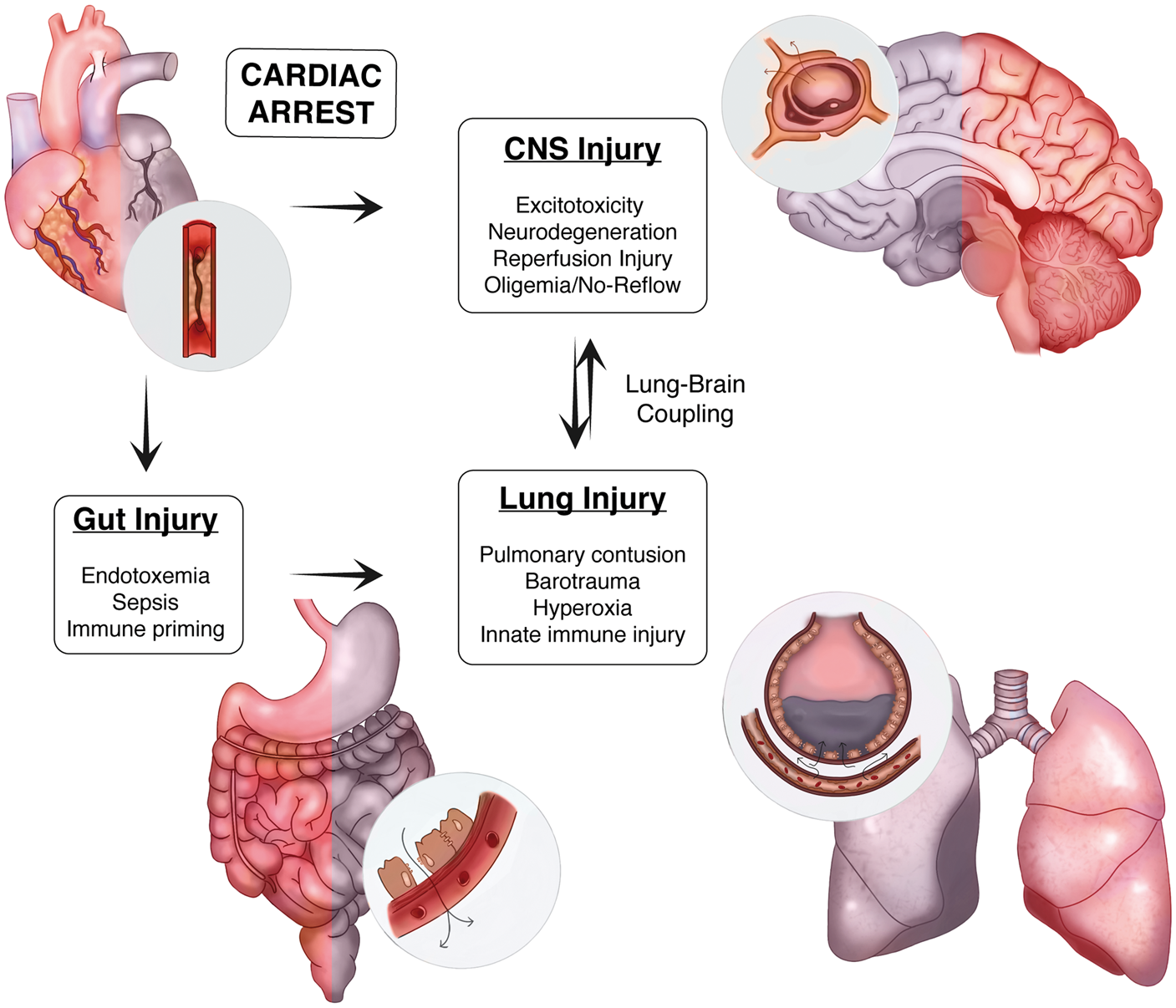
CA: Clinical features
CA is a leading cause of morbidity and mortality in the United States. Ischemic heart disease accounts for the majority (70%) of cases, with cardiac conduction and structural defects, electrolyte disorders, as well as hypoxemia contributing. The mean age of patients experiencing out-of-hospital cardiac arrest (OHCA) is 64, with most subjects judged to be functionally independent (66%) prior to hospitalization. 1 The risk of CA is associated with advanced cardiovascular disease, respiratory disease, end-stage renal disease, smoking, elevated serum CRP levels, and inconsistent patterns of exercise. 2 Ventricular tachycardia (VT) and ventricular fibrillation (VF) are the most common arrhythmias associated with hemodynamic collapse in CA, 3 and patients typically require both ventilator (97%) and hemodynamic support (60%) with one or more vasopressor agents within the first 24 h after admission.
In 2014, there were an estimated 209,000 cases of in-hospital arrests per year compared to 424,000 cases of OHCA. 4 Outcomes also differ considerably between these groups with 37% of in-hospital arrests surviving to discharge compared to rates of 10.7% for OHCA. Although imprecise, time to return of spontaneous circulation (ROSC) is directly proportional to the rate of in-hospital mortality, approaching 62% for patients requiring resuscitation for longer than 20 min. 5 While methodological differences account for some variability in rates of mortality across studies, 6 the poor prognosis associated with OHCA is consistent between countries. 7 Factors associated with poor outcomes include the need for intubation or vasopressors, hypotension at the time of ICU arrival, exposure to 100% O2, persistent coma, and renal failure. 8 Conversely, factors associated with improved survival include VF on presentation, early cardiac catheterization, and a preserved glomerular filtration rate (GFR > 60 mL / min / 1.73 sqm). Notably, antecedent aspirin use and parenteral antibiotic administration within a week of ICU admission are both associated with improved outcomes.9,10 Further gains are expected given ongoing process improvements in the areas of pre-hospital and ICU-phase care, coupled with the expanded use of systemic hypothermia.
Mechanisms of brain injury in cardiac arrest
While multiple organ failure is common with in-hospital arrest (50.9% vs. 9.2%), mortality in patients with OHCA is often attributed to catastrophic brain injury. 11 Relative to focal stroke, the extent of ‘at risk’ brain caused by CA presents a formidable therapeutic target. In addition, pathological changes observed following ROSC are diverse, regionally heterogeneous, and delayed in their maturation. 12 While prolonged arrest is often associated with extensive cortical laminar necrosis, lesser injury triggers post-ischemic neurodegeneration in CA1 hippocampal pyramidal neurons, cortical neurons in layers 3, 5, and 6, cerebellar Purkinje neurons, thalamic reticular neurons, and medium-sized striatal neurons.13–16 These changes give rise to a variety of clinical phenotypes such as memory deficits, movement disorders, and impaired arousal. The kinetics of post-arrest neurodegeneration are also quite delayed with DWI and ADC changes in the parietal lobe and deep grey nuclei with MRI taking up to 36–48 h post-ROSC to become apparent. 17 In studying the utility of DWI in prognostication, Wijman et al. also found that scans obtained between 49 and 108 h after arrest were best at predicting outcomes. 18 These data are consistent with the delayed rise in the serum markers NSE, S100β, and Tau. 19
Given their striking functional diversity and metabolic requirements, it is not surprising that neurons are prime targets for ischemic injury. Relative to other cell types, neurons consume large amounts of ATP to support glutamate-mediated neurotransmission, yet they lack the capacity to either generate or store metabolic precursors in any substantive form. 20 The precipitous decline in neuronal ATP levels during ischemia interferes with the proper function of the energy-dependent sodium-potassium pump causing eventual membrane depolarization. Left unchecked, the rise in net extracellular glutamate, reflecting an increase in release and a decline in re-uptake, promotes regional excitotoxicity caused by uncontrolled spikes in levels of intracellular calcium and zinc.21–23 Neurons failing to recover their electrochemical gradient either succumb acutely to necrosis or succumb to one of several forms of delayed, genetically encoded cell-death.16,24 For example, the pre-treatment of primary and acute slice culture preps with glutamate and AMPA antagonists unmasks cell-autonomous forms of cell death triggered by in vitro oxygen–glucose deprivation that is dependent on de novo transcription and translation. 25 Thus, achieving ischemic neuroprotection at scale will require approaches that mute key sensor-effector pathways involved in parthanatos and necroptosis among other relevant cell death programs. 12
Post-ischemic neuroinflammation
With acute ischemia, IL-6, TNF-α, and interferons generated within the CNS amplify local inflammatory responses and establish chemokine gradients that promote both neuroinflammatory changes and peripheral immune recruitment.26–30 Transcription-dependent mechanisms triggered by hypoxia, hypoglycemia, and endoplasmic reticulum stress activate sensor proteins like AMP-activated protein kinase (AMPK) that in turn stimulate the activity of physiologically responsive transcription factors including HIF-1α and NF-kβ.31,32 The release of damage-associated molecular patterns (DAMPs) from necrotic tissue also triggers local inflammation through a bystander mechanism activating pattern recognition receptors (PRRs) on microglia and macrophages.31,33 Prototypical members of the alarmin pathway include the high mobility group box 1 protein (HMGB1) and S100A8/A9 proteins, which signal through the toll and RAGE receptors to activate microglia, macrophages, and circulating leukocytes. Not surprising, the net effect of post-ischemic inflammation in the nervous system reflects both the magnitude of the response and the balance between the adaptive and maladaptive influence of cytokine signaling in this context. 34
While astrocytes and oligodendroglia participate in ischemic immune signaling, microglia play a particularly interesting role.35,36 After mild ischemic stress, microglia convey an anti-inflammatory bias by reducing net inflammation through phagocytosis of infiltrating neutrophils and dying cells. 37 Microglia can also prune and ingest live neurons through a mechanism described by Brown and Neher as ‘phagoptosis’ that is considered part of the reparative process. 38 However, mice deficient in the phagoptosis regulators MerTK or MFG-E8 also suffered less neuronal injury and fewer functional deficits after ischemia. 39 These results hint at the ability of resident microglia and infiltrating macrophages to undergo a phenotypic switch under permissive conditions. Although details are still being worked out, this dichotomous behavior likely reflects the influence of immune polarization. Like macrophages, microglia tailor their responses in a context-specific manner by assuming either a pro-inflammatory (M1) or anti-inflammatory (M2) posture. In the case of M1 microglia, the typical pattern is characterized by the expression of markers including CD68 and CD86, the production of cytokines, and enhanced phagocytic activity. Conversely, the M2 phenotype promoted by factors including IL-4, IL-10, and TGF-β is characterized by the expression of CD206, arginase 1, and Ym1. 31 Interrogation of the factors governing immune polarization could provide the means to limit tissue damage, hasten the resolution of inflammation, and promote tissue repair. 40
The systemic inflammatory response in PCAS
Systemic inflammation has long been recognized as a central feature of CA. Vladimir Negovsky first coined the term ‘post-resuscitation disease’ in 1972 after observing that patients remained ill and developed a syndromic response in the hours to days after resuscitation from CA. 41 The International Liaison Committee on Resuscitation later formalized the definition for the post-cardiac arrest syndrome (PCAS) to include post-arrest brain injury, persistent precipitating physiology, post-arrest myocardial dysfunction, and systemic ischemia-reperfusion injury (IRI). 42 Notably, the systemic response in PCAS includes many of the core features of both the systemic inflammatory response syndrome (SIRS) and disseminated intravascular coagulation (DIC). Also referred to as a “sepsis-like” syndrome, features of the prototypical SIRS response include changes in core temperature, heart rate, respiratory rate, and the peripheral leukocyte count. DIC involves derangements in the fibrinolytic system and deposition of microvascular thrombi seen as a consequence of elevations in cell-free DNA, DNA-binding proteins, and other DAMPs shed into systemic circulation. In fact, neutrophil-derived myeloperoxidase (MPO) and elastase, as well as proteins associated with neutrophil extracellular traps (NETs), including histones and HMGB1, have recognized procoagulant activity.43,44 Combined with the acute ischemic tissue damage, SIRS and DIC contribute to the development of multi-organ dysfunction syndrome (MODS). MODS reflects a final common pathway involving global microcirculatory compromise, endothelial damage, and ongoing tissue ischemia, coagulopathy, impaired cardiac function, and evolving adrenal insufficiency. 45
While ischemic damage alone is sufficient to induce systemic inflammation, global hypotension after CA contributes in several distinct ways. Arrest-induced damage to the deeper brain structures is associated with hypothalamic-pituitary-adrenal (HPA) axis dysfunction and increased mortality. 46 Interestingly, ischemic damage to central cholinergic pathways induces neuroinflammation that can be suppressed using the selective α7-nicotinic acetylcholine receptor agonist GTS-21. 47 Moreover, destruction of the brain parenchyma also releases DAMPs into the circulation through both venous and lymphatic channels.48,49 These nucleic acids, proteins, and components of the extracellular matrix signal impending danger to downstream tissues stimulating innate immune responses. For example, in a model of focal stroke, Roth et al. 50 demonstrated that alarmin-mediated RAGE receptor activation in monocytes and aortic endothelial cells contributed to remote atherogenesis. And beyond their role as potential therapeutic targets, DAMPs also appear to have value as predictive biomarkers in PCAS. In this regard, the release of the neutrophil-derived heparin-binding protein early after arrest (6–12 h) is associated with longer time to ROSC, poor neurological outcomes, and organ failure. 51 Similarly, levels of HMGB1 correlate with multi-organ damage as measured by the sequential organ function assessment (SOFA). 52 Lastly, elevations of the pyrogen IL-6 predict the need for vasopressor support and 30-day mortality following ROSC.53–55 Other factors associated with poor neurological outcome or mortality include pyrogen receptor antagonist IL-1Ra and neutrophil chemokine IL-8. 55
Neutrophils: The effector limb of CNS reperfusion injury
In contrast to their vital role in defending against invading pathogens, neutrophils play a deleterious role in a broad range of non-infectious etiologies.56–58 Clinical studies support that an elevated neutrophil to lymphocyte ratio at 48 and 72 h after cardiac arrest is associated with poor neurological outcome. 59 Similarly, elevated serum levels of the neutrophil-derived heparin-binding protein is a predictor of multiorgan failure. 60 Using a mouse model of PCAS, we have also shown that neutrophil accumulation in the CNS is directly correlated with both neuronal injury and robust microglial activation. 56 These data are corroborated by similar observations made in preclinical models of focal stroke.61,62 The use of neutrophil margination as a surrogate for tissue damage is supported by mechanistic data regarding their toxic potential. For example, neutrophils amplify the recruitment of inflammatory cells to the site of injury,63,64 and their accumulation within the microvasculature is associated with blood-brain barrier (BBB) compromise and edema formation. 65 Neutrophils also release the contents of different granule subsets in a manner consistent with their degree of activation.66,67 For example, endothelial rolling triggers the exocytosis and fusion of neutrophil secretory vesicles with the cell membrane, resulting in increased surface expression of β integrin Mac1 (CD11b/CD18), reinforcing endothelial attachment. Subsequent activation of secondary granules facilitates neutrophil margination through the release of gelatinase and matrix metalloproteases (MMPs) that degrade type IV and type V collagen, the extracellular matrix, and the endothelial tight junction proteins including occludins, cadherins, and claudins. Consistent with these findings, MMP inhibitors including 1,10-phenanthroline and minocycline effectively reduce BBB permeability, cytokine activation, neutrophil infiltration, and hemorrhagic transformation after focal stroke. 68
Azurophilic granules containing MPO, defensins, cathepsins, and elastase are the last mobilized and contribute to bystander injury under conditions of both sterile inflammation and pyogenic infection. 69 MPO, which produces HOCl through the conversion of Cl− and H2O2 required to kill microorganisms, is commonly used as a marker of neutrophil infiltration after ischemic injury. 70 The relative difficulty of mobilizing azurophilic granules may serve to limit the extent of MPO-related bystander damage. HOCl also extends tissue injury by inducing platelet activation and promoting NETosis, which involves the release of neutrophil extracelluar traps comprising de-condensed chromatin, histones, and degradative enzymes.71–73 NETs are present in thrombi recovered from patients presenting with acute ischemic stroke and in the peripheral blood of patients resuscitated from CA, and may contribute to the no-reflow phenomenon via their pro-coagulant effects.74–76 Based on these observations, co-administration of DNase I with tPA to decrease NET-associated clot formation has been proposed in the treatment of acute ischemic stroke. 74 Neutrophils also undergo reverse migration, which can spread inflammatory-mediated injury to remote locations. Referred to as trans-endothelial migration (TEM), Woodfin et al. 77 found that a portion of neutrophils migrate back into the systemic circulation. Neutrophils that underwent reverse-TEM were phenotypically distinct, possessing higher ICAM-1 levels and ROS-generating abilities compared to those from the marrow or in circulation. Interestingly, after leaving the injured site, these neutrophils preferentially migrate to the lung causing interstitial edema and inflammation.
Post-ischemic neurovascular injury and cerebral edema
The neurovascular bed represents the first point of contact between the activated systemic innate immune system and the CNS. In rodent models, the initial response to transient forebrain ischemia involves transient opening of the BBB within 6 h of injury that closes within 24 h.78,79 This breech effectively short-circuits CNS immune privilege permitting the ingress of serum complement and circulating cytokines and encourages the formation of cerebral edema leading to further microvascular collapse, oligemia, and secondary tissue ischemia. Notably, hypertonic saline has been used experimentally to alleviate post-arrest hippocampal neurodegeneration, further supporting the vasogenic origin of post-arrest cerebral edema. 80 However, other factors contribute as hypertonic saline appears to have variable effects on survival and neurological outcomes in randomized trials for OHCA.
The endothelial glycocalyx serves as the first line of defense protecting the vascular endothelium from reperfusion injury. Extending 200-2000 nm from the endothelial surface, the negatively-charged glycocalyx composed of glycoproteins, proteoglycans, and glycosaminoglycans blocks the penetration of both macromolecules and circulating immune cells into the deeper layers of the vasculature and organ parenchyma.81,82 Under conditions of focal ischemia, the accumulation of free radicals, hyaluronidase, heparinase, and MMPs lead to marked glycocalyx erosion and shedding. 83 With the release of syndecan-1, hyaluronic acid, and heparan sulfate, the exposure of P-selectin and other adhesion factors stimulates the recruitment, activation, and margination of circulating leukocytes into tissues during ischemia/reperfusion.82,84 Shedding of the glycocalyx is also rapid, occurring within 20 min following systemic circulatory arrest. 85 As would be predicted, IV hyaluronidase-induced shedding increases BBB permeability, cerebral edema, and mortality rates after asphyxial CA. Conversely, the use of hydrocortisone interferes with glycocalyx shedding and improves survival after CA. 81
Additional aspects of BBB function provide additional protection against reperfusion-mediated injury. The BBB is a complex structure composed of both cellular elements that include the vascular endothelium, pericytes, astrocytes, and components of the basal lamina (e.g., laminin, collagen IV, and fibronectin), which bond them together. Damage to tight junctions and adherens junctions that interdigitate with the actin cytoskeleton through higher order macromolecular complexes creates discontinuity between the vascular endothelium. Furthermore, loss of α6β4 under ischemic conditions leads to detachment of astrocytic endfeet from the basal lamina.86–88 Much of what we have learned regarding this process have come from elegant studies performed using transient focal models of stroke.78–90 While the aforementioned effects of neutrophils figure prominently in the pathology of IRI, studies have also identified a dual hemostatic and pro-inflammatory role that platelets play in this process. Interactions between platelet-derived sphingolipid sphingosine-1-phosphate (S1P) and the S1P family of receptors expressed on vascular endothelium may be of particular importance. While S1PR is involved in the maintenance of vascular integrity, inhibition of S1PR2 activity with the antagonist JTE013 blocks loss of BBB integrity and other markers of focal IRI. 91
The ‘no-reflow’ phenomenon
Cerebral blood flow exhibits dynamic changes following ROSC in CA. After an initial period of hyperemia approaching 350% of control levels, blood flow drops to 30–50% below control levels continuing for up to 24 h following global cerebral ischemia in the rat. 92 Initially studied using the rabbit model of global cerebral ischemia, 93 the tissue response to localized ischemia described as the ‘no-reflow’ has also been linked to perfusion deficits in the kidney and heart.94,95 Factors contributing to no-reflow in the brain include astrocytic peri-vascular swelling, endothelial damage with increased platelet-neutrophil adherence, and erythrocyte sludging. Increased immune effector transit time induces further inflammation and injury via the release of chemokines and degradative enzymes creating a feed-forward cycle of tissue injury. In focal stroke models, LPS-induced systemic inflammation increases infarct size, 96 while pretreatment with interleukin-1 (IL-1) worsens post-ischemic brain damage via effects on no-reflow. 97 Consistent with these results, studies using derivatives of the IL-1 receptor antagonist Anakinra demonstrate mitigation of reperfusion injury following focal stroke. 98 Taken together, these data support no-reflow as a final common microvascular response to a range of systemic pro-inflammatory stimuli in the setting of cerebral ischemia reperfusion injury.
More recently, there has been great interest in defining the role of pericytes in cerebral blood flow under physiological and pathological conditions. Studies using intravital microscopy indicate that the pial and capillary vascular network regulate 75% of the changes in cerebral blood flow via effects on vascular resistance and mean transit time.99,100 Normally, pericytes support astrocyte polarity and limit endothelial transcytosis required for regulating solute entry to the brain. 101 With physiological stimulation, glutamate, prostaglandin E2, and nitric oxide induce pericyte relaxation allowing for increased blood flow. 100 Conversely, under conditions of ischemic stress, pericyte contraction leads to irreversible capillary constriction and BBB damage. 102 While pericyte death appears to be a rapid response to focal ischemia as well as in acute brain slices, their role in the post-arrest no-reflow phenomenon remains relatively unexplored.
Changes in systemic perfusion pressures are equally important in determining end-organ perfusion. Studies by Kilgannon et al. 103 demonstrate the importance of maintaining a mean arterial pressure above 70 mm Hg in achieving favorable neurological outcomes. 103 Notably, prior trial results indicated that post-ROSC hypotension defined as a systolic pressure below 90 mm Hg within 1 h of ICU arrival was a strong predictor for death (OR 2.7, 95% confidence interval, 2.5-3.0). 104 Others have focused on targeting systemic hypotension as well as inflammation through the combined use of vasopressin and methylprednisolone, followed by stress dose hydrocortisone, and epinephrine (VSE). 105 Results indicate that VSE therapy improves the chances for successful ROSC and lowers post-resuscitation levels of systemic inflammation and organ dysfunction. Specifically, treatment reduced circulating IL-6 within the first seven days of hospitalization and was associated with improvements in neurological function at the time of hospital discharge. 106
Gut injury and systemic immune priming after CA and stroke
Endotoxemia is both a consequence of intestinal ischemia and a contributor to systemic inflammation after CA.107–110 A rise in serum endotoxin levels within 24 to 48 h after ROSC is seen in up to 86% of patients, is associated with post-resuscitation shock, and predicts multiple organ failure.108,111,112 Endotoxemia is associated with non-occlusive mesenteric ischemia in CA and other low-cardiac output states including situations requiring the use of ECMO.113,114 While 60% of CA survivors exhibited early evidence of gut dysfunction, all patients who underwent endoscopic investigation in one study exhibited either hemorrhagic or necrotic lesions. 110 In particular, the colon may also be particularly vulnerable to the damage caused by systemic hypotension due to its propensity to undergo vasospasm. 115 Studies using models of deep hypothermic circulatory arrest reveal that post-arrest disruption of natural intestinal barrier function is marked by hemorrhagic infiltration in the terminal ileum, blunting of the intestinal villi, and the presence of inflammatory infiltrates within the colonic lamina propria. 109 Intestinal ischemia also induces the proliferation of pathogenic intestinal bacterial species. 116 Evidence from models of focal stroke suggests that direct ischemic injury to the gut may not be required to induce alterations in enteric barrier function. 117 In this study, Enterococcus species and Escherichia coli isolated from patients whose strokes were complicated by infection, in fact, originated from a GI source. These results argue that DAMPs released from the post-ischemic brain are alone sufficient to induce bacteremia by altering normal intestinal barrier defenses.
Once within the portal circulation, enteric bacteria and pathogen-associated molecular patterns (PAMPs), including bacterial-derived LPS, trigger robust inflammation in the vascular endothelium as well as circulating neutrophils and platelets by acting upon a family of PRRs including the toll-like receptors (TLRs).118,119 Systemic immune activation can still develop in the absence of detectable bacteremia since gut-associated lymphoid tissue release cytokines and non-microbial pro-inflammatory factors into the lymphatic system in response to mesenteric injury (Figure 2).
120
In fact, this pathway is an important contributor to lung injury since the pulmonary vasculature is exposed to mesenteric lymph via the thoracic duct. Notably, pretreatment of mice with gut-localized antibiotics reduced alveolar macrophage cytokine production and interstitial edema among other markers of acute lung injury (ALI).
121
The lymphatic system and lung–brain immune priming. With the return of spontaneous circulation, both the brain and gut release damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) into the circulation. CNS DAMPs conveyed via cerebral venous return or indirectly via cervical lymphatic channels induce acute lung inflammation. PAMPs generated within the gut are delivered to the pulmonary circulation through analogous venous and lymphatic channels. As shown, the thoracic duct provides lymphatic drainage from the abdominal cavity and lower extremities terminating at the angle of the left subclavian and internal jugular veins. Following the return of spontaneous circulation, these channels contribute to acute lung inflammation, which in turn trigger systemic immune priming and secondary CNS reperfusion injury.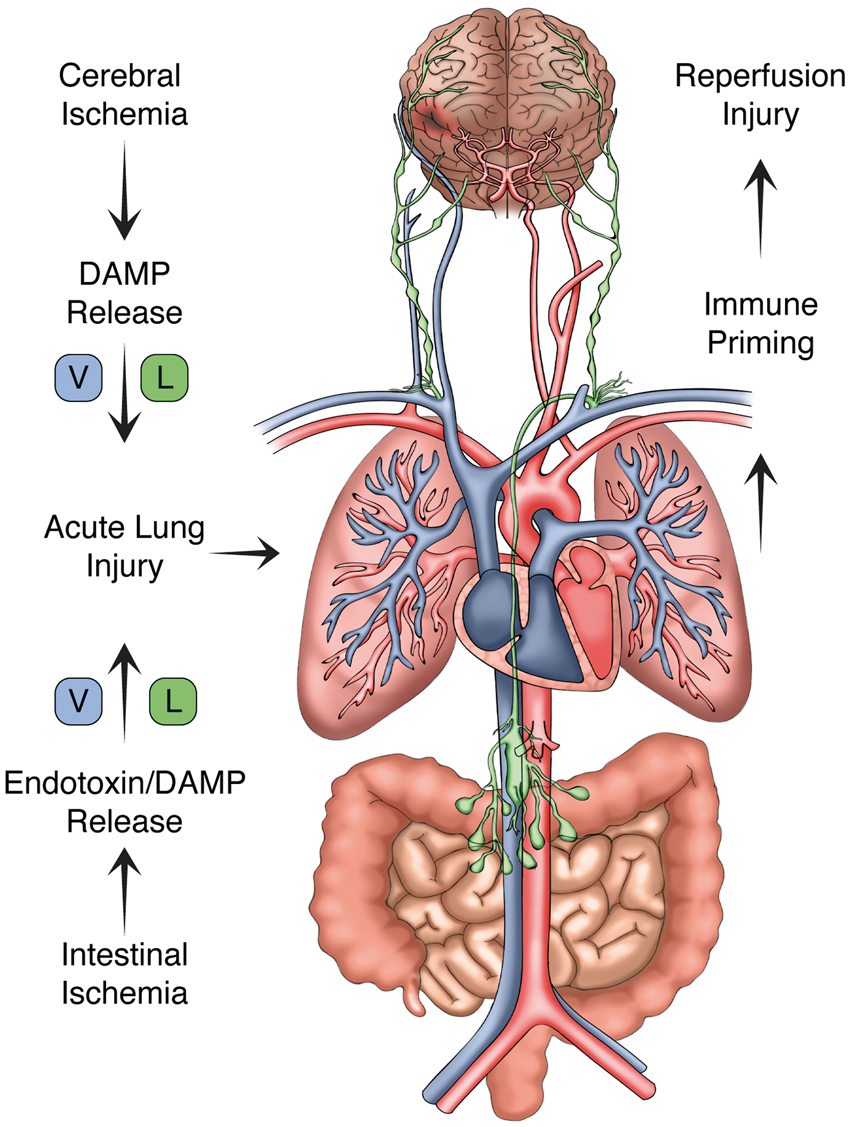
Lung–brain coupling in PCAS
The lung's role in modulating the response to acute organ damage is both underappreciated and incompletely defined. In the setting of CA, lung damage may arise from ischemia, exposure to high-dose oxygen, trauma from chest compressions, and barotrauma during mechanical ventilation. Also, the lung is often a site of secondary infection given the high likelihood for airway compromise and aspiration. Further, neutrophils migrating from sites of ischemic injury preferentially travel to the lung and cause local pulmonary inflammation. 77 In this section, we consider how neurogenic and iatrogenic injury alters normal lung function and promotes pro-inflammatory lung–brain coupling.
Lung involvement in PCAS
The majority of studies investigating the pathological sequelae of CA do so either by inducing cardioplegia or by creating transient global ischemia through the reversible isolation of the cerebral circulation. To model the dynamic interplay between global cerebral ischemia and systemic inflammation observed in PCAS, our group recently studied the effects of transient global brain ischemia induced by three-vessel occlusion (3VO) and simulated low-dose endotoxemia on cumulative reperfusion-related CNS injury separately and in combination. 56 Secondary endpoints in these studies included assessing the extent of innate immune activation as well as the extent to which brain injury and endotoxemia were sufficient to induce an innate immune response in peripheral organs including the lung, kidney, and liver. In comparison to the dose of LPS typically used to model sepsis (25-33 mg/kg), we performed dose-ranging studies and identified a dose (50 µg/kg) that had no measurable effect on serum cytokines but was nonetheless sufficient to induce transient neutrophil activation. We found that when used in combination, ischemia-LPS priming significantly worsened neuro-behavioral scores, doubled the volume of brain injury marked by MAP2 staining, and increased both Iba-1 immunoreactivity and neutrophil accumulation within three days post-reperfusion. Compared with either stimulus, the combination of LPS and cerebral ischemia caused BBB opening within three days as measured by the intraparenchymal accumulation of serum immunoglobulin.
These results were consistent with prior work in focal models of stroke that demonstrated increased BBB permeability, neuroinflammation, and cerebral damage.97,122,123 However, an unexpected finding in our study and the basis for this review was the extent to which low-grade systemic endotoxemia contributed to ALI following three-vessel occlusion. While the dose of LPS selected enhanced neutrophil activation in the peripheral circulation, it did not cause the degree of interstitial edema and immune infiltration typically seen in sepsis-induced ALI. Transient global cerebral ischemia alone was associated with increases in interstitial lung edema and neutrophil accumulation beyond what is typically observed using intraperitoneal (IP) LPS administration. Presumably, this effect was provoked in part by central nervous system (CNS) DAMPs delivered to the lung via lymphatic and venous channels. There is a precedent for focal brain injury causing reactive inflammation. For example, the introduction of the glycosaminoglycan hyaluronan (HA) to endothelial cultures enhanced neutrophil recruitment by facilitating endothelial interactions, 124 while macrophage clearance of HA promoted the resolution of lung inflammation. 125 Wu et al. also reported that the timing of ICAM-1 upregulation in the brain and lung (6-24 h) following intracerebral hemorrhage (ICH) correlated with the onset of neutrophil infiltration and alveolar destruction. 126 Taken together, these observations suggest that brain-lung immunologic coupling could have important effects on both lung function and secondary CNS injury in the sub-acute period. We next deconstruct aspects of the proposed lung-brain axis and discuss the lung's response to acute brain injury, its effect on peripheral immune priming, and its potential as a therapeutic target in PCAS.
The lung: Immuno-homeostatic mechanisms
The lungs receive the entirety of the cardiac output and possess an immense vascular surface area related to the millions of branching capillaries measuring 2-15 µm.127,128 Barring contributions from either intrapulmonary or intracardiac shunts, the lung receives 100% of cardiac return, placing it in prime position to sense and respond to circulating inflammogens. Cellular constituents of the lung consist of type I and type II alveolar epithelial cells, capillary endothelium, and interstitial fibroblasts. The capillary endothelium performs important gatekeeper functions controlling permeability and edema formation, coagulation, platelet aggregation, as well as neutrophil recruitment. Recent studies also indicate that the lung is host to a substantial proportion of marrow-derived megakaryocytes responsible for producing upwards of 50% of the total fraction of circulating platelets. 129 In addition, the lung contains a significant number of alveolar and interstitial macrophages that sense and respond to both respiratory and hematogenous infection.
The lung possesses intrinsic antioxidant defenses to protect against inhaled toxicants and damage caused by acquired respiratory infections and inflammatory conditions. Relevant factors include members of the superoxide dismutase (SOD) family of proteins as well as catalase present in peroxisomes. Typically, 98% of O2 is converted to H2O through oxidative phosphorylation, while roughly 2% results in the generation of the superoxide radical (O2-). As implied by their name, each of the SOD enzymes is capable of converting this potent toxicant into H2O2, which is subsequently converted to water via catalase. The gene Sod3 produces the extracellular form of superoxide dismutase (EC-SOD) that performs a critical function in the lung by stabilizing the extracellular matrix and inhibiting both inflammation and fibrosis under stress conditions. 130 The lung redox system also buffers against neutrophil-mediated damage conveyed by the highly reactive HOCl molecule generated by MPO. In the critical care setting, the increased burden placed upon endogenous lung antioxidant systems caused by aspiration, neutrophil toxicity, and gut-associated xanthine oxidase activity challenge endogenous SOD function and promote a milieu of chronic inflammation. However, it is important to note that maintenance of homeostatic lung function requires a balance between endogenous lung antioxidant effects and pro-oxidant signaling mediated by neutrophils. Importantly, while Sod3 deletion leads to ALI in response to ambient air, deletion of Mpo predisposes mice to develop pneumonia.131,132
Lung injury in CA
Neurogenic lung injury is a well-recognized complication of acute neurological injuries ranging from ischemia damage to traumatic brain injury and recurrent seizure.133,134 Verein et al. 135 described the evolution of neurogenic pulmonary edema (NPE) between the fifth and seventh day after stroke correlating the extent of brainstem dysfunction with extravascular lung fluid volume. 135 The effects of neuroinflammation on the posterior hypothalamus, caudal ventrolateral medulla, nucleus tractus solitarius, and area postrema are thought to underlie NPE by driving sympathetically mediated surges in catecholamines. 136 Increases in sympathetic tone also raise pulmonary capillary pressures, further influenced by sympathetic-mediated pulmonary venoconstriction. 137 Lastly, sympathetic stimulation produces interstitial edema by increasing pulmonary vascular permeability independent of effects on vascular pressure. Importantly, NPE responds to treatment with hypertonic saline or mannitol by increasing plasma colloidal pressure. 136
It has become increasingly clear that iatrogenic factors affecting the lung may also adversely influence patient outcomes after CA (Figure 3). Examples include the mechanical trauma caused by CPR, the effects of mechanical ventilation, as well as the biological effects of both hyperventilation and the use of supplemental oxygen. Rib fractures are common, occurring in up to 32% of patients resuscitated after an arrest,
138
and may be associated with lung contusions reported to resolve within seven days.
139
Ventilator-associated lung injury is caused by three primary forces that include barotrauma, volutrauma, and atelectrauma. This is particularly relevant since ventilator-induced barotrauma has been associated with hippocampal neuron apoptosis as well as possible long-term cognitive deficits.
140
Mechanical ventilation may also contribute to the formation of lung edema and worsening gas exchange by increasing intrathoracic pressure and disturbing normal lymphatic outflow.
141
Mechanisms of acute lung injury in PCAS. The mechanisms leading to acute lung injury in the post-cardiac arrest syndrome are divided into neuro-humoral, immune-mediated and iatrogenic etiologies. Neurogenic pulmonary edema (NPE) reflects an increase in pulmonary interstitial and alveolar fluid that stems from sympathetically mediated pathways. The lung is also subject to immune priming caused by the ischemia-induced release of both DAMPs and PAMPs from post-ischemic tissues combined with the inflammatory effects of aspiration pneumonitis. Equally important, iatrogenic factors encountered in the course of cardiopulmonary resuscitation (CPR), as well as those associated with ventilator-induced lung injury (VILI), are shown.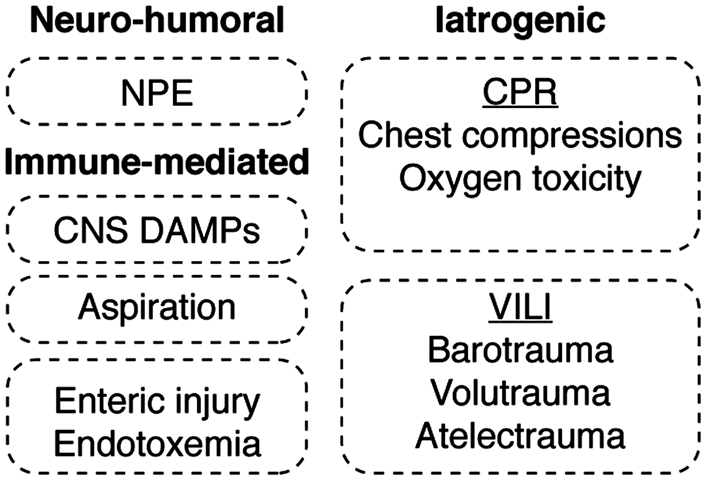
The delivery of high fractional O2 in the acute care setting is also recognized as a risk factor for both lung and brain injury. A multicenter observational study of over 6300 patients conducted by the Emergency Medicine Shock Research Network Investigators found that hyperoxia exposure observed in 18% of subjects was associated with an odds ratio for death of 1.8. 1 In preclinical models of global cerebral ischemia, the use of supplemental oxygen induces CA1 neuronal injury, neuroinflammation, and memory deficits. 142 While it stands to reason that hypoxemia should be avoided in the post-arrest patient, establishing protocols that limit lung damage from high flow oxygen therapy remain an area of active investigation. 143 And while considered a temporizing measure in the management of evolving cerebral edema, hyperventilation-induced hypocapnia leads to cerebrovascular constriction which may exacerbate tissue ischemia. This mechanism is thought to underlie the improved neurological outcomes observed with permissive hypercapnia.144,145
As described, the lung also receives lymphatic flow from the thoracic duct that serves as the primary conduit for returning abdominal lymph to the general circulation. Duct ligation has been shown to blunt acute lung inflammation in several models. For example, intestinal ischemia–reperfusion resulted in IL-1β-dependent bronchial inflammation that was disrupted by prophylactic thoracic duct ligation. 146 An analogous function is provided by the meningeal lymphatic vessels which function as a final common drainage pathway for brain interstitial fluid and serve an essential role in regulating immune surveillance under homeostatic and diseased states. 147
The lung as a site of neutrophil priming
Because CA induces both systemic ischemia and inflammation, neutrophils are primed in the periphery long before reaching the CNS. 148 Yet, while genetic models of neutrophil deficiency support their role in exacerbating cerebral reperfusion injury, 149 anti-neutrophil strategies have failed in clinical trials for acute cerebral ischemia. 150 This apparent contradiction may reflect the fact that like microglia, neutrophils are also capable of adopting either pro- (N1) or anti- (N2) inflammatory phenotypes.151,152 Notably, evidence from the literature suggests that reversal of neutrophil priming may be a target for therapeutic intervention.153–155
Neutrophils are released from the marrow in a state of quiescence and exhibit a circulatory half-life of 6-8 h. 156 Neutrophils are 6-8 µm in diameter, and in humans, they are wider than 38% of pulmonary capillary segments. 157 Their transit time through the pulmonary vasculature is slower than that of red blood cells and other leukocytes, and they must cross 40-100 capillary segments positioned between pre-capillary arterioles and post-capillary venules. 158 Neutrophils can roll in capillary segments of equal diameter and deform to fit narrower microvessels. Under standard physiological conditions, a large ‘marginated’ pool of neutrophils resides within the pulmonary vasculature, maintained by mechanical retention in capillaries and by interactions with P-selectin and ICAM-1 in arterioles and venules. 159 In humans, this pool is approximately 4 × 108 cells/kg, 160 and some studies suggest it may exceed the circulating neutrophil pool. 161 Upon activation with endotoxin, TNF-α, or DAMPs, actin reorganization results in increased neutrophil stiffening and reduced compliance resulting in their increased transit time through the lung where they are removed from circulation and retained.64,162,163 In addition to these structural changes, immune priming increases surface expression of TLRs on both neutrophils and endothelial cells rendering them more responsive to circulating alarmins. Notably, TLR4-dependent neutrophil activation has been linked to lung injury caused by high tidal volume ventilation. 164
Given the kinetics of neutrophil trafficking through the lung and their established role in ALI, several groups have suggested that the healthy pulmonary endothelium may also play a role in neutrophil de-priming. Under this model, unprimed neutrophils enter and pass through the pulmonary circulation, surveilled by the endothelium.153,165 In the case of low-grade neutrophil activation, an otherwise healthy lung serves a “catch and release” function by depriming and releasing them or by removing them from circulation, relying on the actions of pulmonary reticuloendothelial cells through the process of efferocytosis required for the resolution of lung inflammation. 166 However, under conditions where the lung is also injured and can no longer serve as a site of neutrophil depriming and clearance, neutrophils pass through the lung in a heightened state of activation. By way of example, Nahum et al. 167 used the production of H2O2 as a surrogate marker of neutrophil activation, comparing arterial and venous blood from septic patients with and without lung infiltrates. Levels of arterial H2O2 were reduced in patients without lung injury, suggesting that the lung either de-primed neutrophils or removed them from circulation. Conversely, arterial H2O2 levels were higher in patients with pulmonary infiltrates, suggesting that the injured lung may promote neutrophil activation. Summers et al. also examined trans-pulmonary neutrophil migration in patients receiving 111-indium-tropolonate-labeled autologous neutrophils exposed to the priming agents GM-CSF or PAF. Relative to controls, less than 5% of unprimed neutrophils were retained in the pulmonary vasculature after the first pass, though neutrophil transit was slower than that of erythrocytes. Priming with GM-CSF caused first-pass neutrophil retention, with 48% still retained after 40 min. Analyses of neutrophil activation have also been performed on blood drawn from the internal jugular vein and radial artery of patients with sepsis, acute respiratory distress syndrome (ARDS), and perioperative controls. Results indicate that arterial neutrophils from patients with ARDS expressed less CD62L on their surface suggesting lung-dependent activation. 155 These studies suggest that injured lungs not only failed to de-prime neutrophils but that they can also serve as a priming site.
What then are the implications of the lung's involvement in delayed post-ischemic neurodegeneration during PCAS? The extent to which DAMPs and PAMPs released from the brain and intestinal tract contribute in PCAS remains unexplored. In concept, however, these factors likely induce endothelial activation, increase capillary permeability, and heighten levels of inflammation in the lung, driving local innate immune activation and recruitment of circulating immune cells. As noted, these changes will worsen gas exchange with repercussions on cerebral perfusion and metabolism by altering the ratio of delivered oxygen and carbon dioxide. The loss of intrinsic pulmonary de-priming will also favor the polarization of circulating neutrophils and macrophages toward pro-inflammatory phenotypes. And, given the prominent role that pulmonary megakaryocytes play in systemic platelet production, it is reasonable to expect that lung injury may also cause both quantitative and qualitative changes in platelet function with marked effects on the neurovascular unit, no reflow, and overall neuroinflammatory responses. While iatrogenic lung injury may be a modifiable target, it is worth noting that many patients presenting after OHCA harbor pre-existing intrinsic lung disease that also compromises the organ's ability to adapt to acute injury. Thus, future work geared towards understanding how co-morbid disease and early immunological signals conveyed by the brain and gut conspire to cause post-ischemic brain injury may provide options to improve long-term patient outcomes.
Lung–brain coupling: A new therapeutic target in cerebrovascular disease?
At present, post-resuscitation care provided in the ICU setting focuses on (1) reversal of the precipitating pathology and achieving coronary reperfusion, (2) maintaining serum pH neutrality as well as electrolyte balance and euglycemia, (3) monitoring for and treating seizures, (4) maximizing cerebral perfusion with fluids and pressors, and (5) maintaining normothermia while treating infection when suspected. 168 While work to define best practices continues, these measures alone have not addressed the holy grail of post-arrest care, namely the ability to induce clinically meaningful neuroprotection. In the pre-hypothermic cooling era, trials focusing on excitotoxicity, post-anoxic seizures, and improving cellular bioenergetics failed to improve patient outcomes. This was indeed the intended consequence of induced hypothermia, thought to limit tissue metabolic demands and suppress post-resuscitation fever and inflammation associated with poor neurological outcomes.169,170 While targeted temperature management has changed the landscape of post-arrest survival, 171 studies indicate equivalency in outcomes when comparing cooling to 33 versus 36 ℃. 172 Data from acid- as well as ventilator-induced ALI suggest that hypothermia may confer some benefit on systemic inflammation by inhibiting the expression of iNOS and MPO, among other markers of acute lung inflammation, which would in turn potentially reduce pathological lung–brain coupling.173,174 That said, lower temperatures do not appear to reduce systemic expression of IL-6 or related cytokines. 175 In fact, rewarming after induced systemic hypothermia enhances immune cell endothelial transmigration through the induction of endothelial adhesion proteins including Junctional Adhesion-Molecules A&B. 176 Notably, neither cooling in the pre-hospital phase of care nor prolonged cooling beyond 24 h provide additional benefit.177,178 These data underscore the fact that the mechanism underlying the protective effects of targeted temperature management remains elusive. Considering that this is the largest randomized trial yet for cooling after CA, there remains a pressing need for complementary therapies for PCAS.
In contrast to the rapid evolution of tissue damage associated with acute ischemic stroke, the window of opportunity to intervene in CA is far less constrained. Considering the multiple potential points for therapeutic intervention within the conceptual framework of lung–brain coupling (Figure 4), we can now consider the issue of how best to protect the brain from PCAS. These include targeted delivery of agents via the intranasal or enteral routes to reduce early-phase organ injury, administration of inhaled agents to prevent ALI, and the IV administration of small molecules capable of stabilizing the neurovascular unit and reducing microvascular complications (Figure 5).
Neurotherapeutic targets in PCAS. The identification of lung-brain coupling in the post-cardiac arrest syndrome provides an additional point of intervention to reduce post-ischemic neurodegeneration. These include shortening the time to ROSC and optimization of supportive measures during the prehospital phase of care (1) and devising acute interventions (2) that can be combined with targeted temperature management to induce early neuroprotection (3). The delayed administration of agents that block required ligand-receptor immune interactions in the periphery could prevent systemic priming. Finally, interventions that reduce acute lung injury (4) or preserve blood-brain barrier (BBB) integrity (5) would, in theory, reduce systemic leukocyte activation, reduce cerebral edema, maintain CNS immune privilege, and hasten the resolution of neuroinflammation.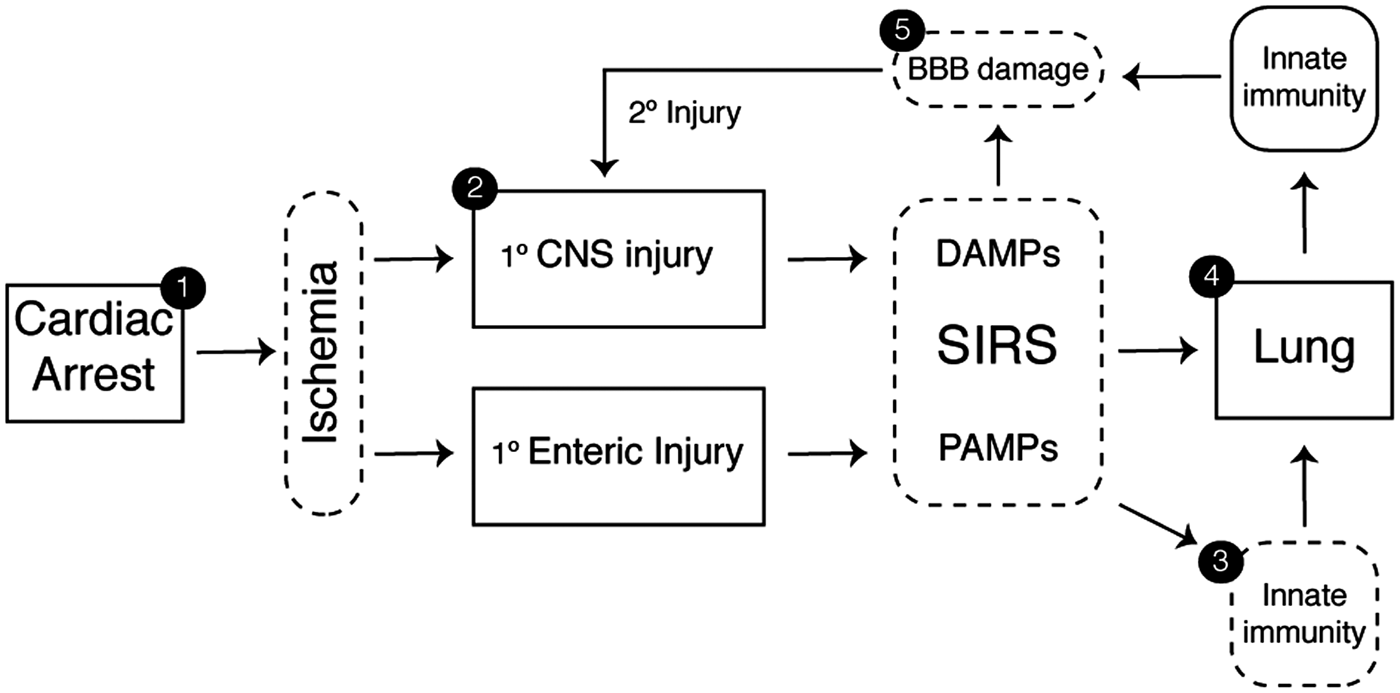
Intranasal therapeutic delivery
Intranasal administration has proven an effective approach to deliver anti-inflammatory and neuroprotective agents in rodent models of disease. Studies have shown that the intranasal route bypasses the BBB targeting the olfactory and trigeminal neural pathways resulting in CNS delivery comparable to that achieved with IV administration in a relatively rapid time frame.
179
This route has been used to deliver the anti-oxidant agent pyrrolidine dithiocarbamate (PDTC), which produces neuroprotection and reduces neuroinflammatory responses in a model of neonatal hypoxia–ischemia.
180
Similar results have been obtained using intranasal delivery of (+)-naloxone, which suppresses the neuroinflammatory response to focal stroke.
181
Others have shown that delivery of orexin-A influences both the state of arousal and levels of neuroinflammation after CA.
182
Moreover, intranasal delivery of caspase-1 inhibitors blocks both apoptotic signaling and neuroinflammation induced by transient global cerebral ischemia.
183
Lastly, intranasal delivery of fusogenic peptides like the NF-kB fusion Tat-NBD reduces neuroinflammation after hypoxia–ischemic injury.
184
Considering that nasopharyngeal evaporative cooling is being developed as a putative therapy to achieve cerebral hypothermia, it is interesting to speculate whether the use of cooling in combination with the delivery of one or more therapeutic agents may exhibit synergistic protective effects after acute brain injury.
185
Thus, targeting the brain through intranasal administration of a therapeutic would, in theory, provide direct protection by limiting early ischemic-neurodegeneration and neuroinflammation while limiting the shedding of CNS DAMPs and secondary systemic inflammatory responses.
Therapeutic approaches to target lung–brain coupling in PCAS. Developing effective neuroprotective strategies for the post-cardiac arrest syndrome may require utilizing unconventional delivery routes. While traditional IV delivery of BBB-penetrant small molecules remains a viable approach, intranasal administration of both peptides and small compounds has shown promise in conveying anti-inflammatory and neuroprotective effects on the CNS. Also, targeting the alimentary tract through either oral or IP delivery of a therapeutic could mitigate against mesenteric injury and the production and release of priming antigens produced by the enteric flora. To reduce the effects of acute lung injury on CNS reperfusion, inhalation of therapeutic compounds and biologics during the pre-hospital phase of care could modulate lung–brain coupling early in the course of disease.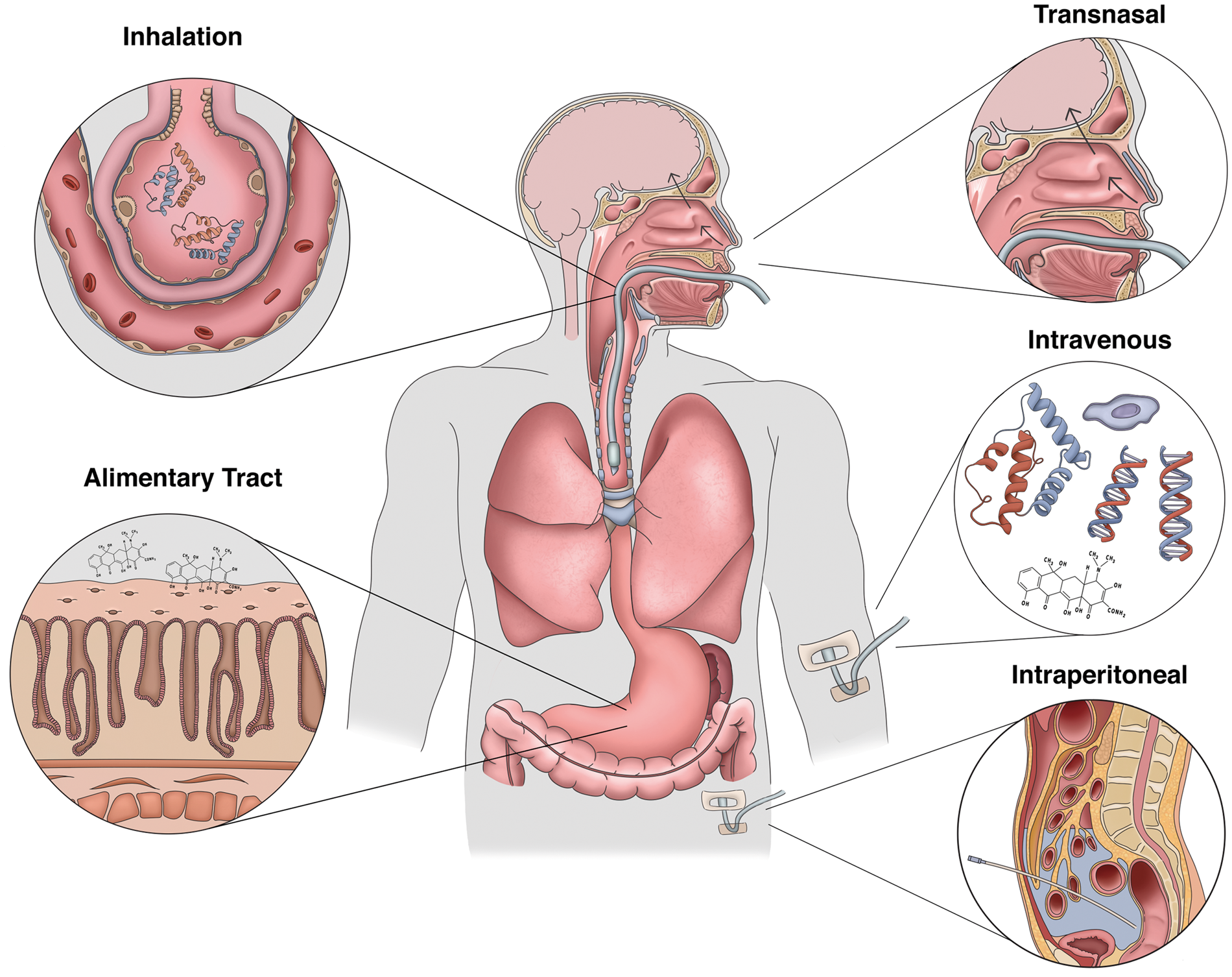
Neutralizing the gut microbiome
As noted, the gut represents an important source of immunological priming and cause of ALI after CA. In the context of CA, the potential therapeutic benefit of reducing enteric bacterial growth that would otherwise contribute to systemic endotoxemia has been investigated. Work by Davies et al. 10 suggests that antibiotic treatment is associated with improved survival in OHCA with a number needed to treat of five; however, randomized trials are required in order to confirm this effect. While there is limited evidence to suggest that prophylactic antibiotics play a significant role in determining outcomes after PCAS in patients without signs of infection by tracheobronchial aspiration, 186 selective decontamination of the digestive tract (SDD) could prove to be an effective alternative. In randomized controlled trials of patients admitted to the ICU, SDD was shown to reduce mortality in critically ill patients with a reduction in MODS by 50% (OR 0.5, CI 0.34-0.74). The goal of SDD is to prevent the emergence of pathogenic microorganisms, and treatment involves the combined use of IV and enteral antibiotics, strict hygiene, and the use of surveillance cultures to ensure response to treatment. In a similar respect, it may be advantageous to consider whether IP delivery of one or more agents (commonly performed preclinical protocols) may be therapeutically beneficial. IP delivery could have multiple beneficial effects including suppressing gut barrier decay, promoting anti-inflammatory polarization of innate immune cells, as well as inducing bacteriostatic or bactericidal effects on the resident gut flora. To this end, IP delivery of the macrolide class of antibiotics has been shown to be protective in models of focal stroke. 187 In this work, Amantea et al. demonstrated that IP injection of azithromycin reduced BBB damage and the infiltration of myeloid cells into the cerebrum of mice following ischemic stroke. Moreover, neuroprotection in their model correlated with increases in the M2 markers arginase and Ym1.
Inhaled therapies to short-circuit lung–brain coupling
Prior work has established the respiratory system as a tractable therapeutic target using gasses, small molecules, and biologics to treat both acquired and inherited disorders. In the case of CA, drug-delivery to the lung could be achieved readily during resuscitation in the pre-hospital phase of care and after admission as most patients require intubation. In the context of CA, therapeutic uses for formulated gas mixtures have been studied. For example, Geng et al. 188 demonstrated that hydrogen sulfide inhalation reduces early BBB permeability and brain edema in a model of CA. These experiments were based on prior work demonstrating that the use of hydrogen sulfide (H2S) is protective in models of reperfusion injury through the detoxification of oxygen radicals. Animals in this protocol were treated post-ROSC for 1 h followed by free ranging in a chamber enriched in 50% oxygen with or without 80 ppm H2S for an additional hour. Relative to controls, exposure to H2S was associated with improvements in neurological function, survival, timed tape removal testing, and the extent of CA1 neuroprotection. Similarly, in addition to improving oxygen delivery in up to 50% of subjects, inhaled nitric oxide (NO) increases the likelihood for resuscitation after experimental arrest and is associated with improved survival one week after injury. 189 At the biochemical level, NO treatment is associated with lower serum lactate and inflammatory cytokine levels. Recruitment is currently underway for a phase II, double-blind randomized controlled trial of inhaled NO (20 ppm) initiated within 4 h of ROSC following OHCA (Trial NCT03079102). Finally, given its role as a scavenger of oxygen radicals and therapeutic antioxidant, molecular hydrogen (H2) has also shown benefit in one small, randomized clinical trial as an acute treatment in acute cerebral infarction with improvements on imaging and reduced scores on both the NIH stroke scale and Barthel Index. 190
Advances in methods to create aerosols from small molecules and peptides among other preparations have expanded the range of options to manipulate lung biology. As mentioned, cleaved fragments of hyaluronic acid (HA) are highly immunogenic acting through TLRs 2 and 4, 191 suggesting a potential role for the delivery of the hyaluronidase inhibitor L-Ascorbic acid 6-hexadecanoate to reduce CNS IRI by limiting HA degradation. A similar approach involving delivery of the endogenous antioxidant SOD has been studied in neonatal models of hyperoxic lung injury. Likewise, CuSOD reduced markers of inflammation and injury and reduced rates of CNS complications including intraventricular hemorrhage and periventricular leukomalacia. 192 Use of aerosolized catalase and SOD mimetics has shown similar protection in models of ARDS. 193 Lastly, inhaled antisense oligonucleotides are effective in suppressing pulmonary inflammation comparable to effects seen using monoclonal antibodies directed towards allergen-specific T-cells. 194
It is intriguing to consider scenarios in which the above interventions might be used in combination with total liquid ventilation (TLV). Studies indicate ultrafast cooling with TLV provides both cardio- and neuro-protective benefits after asphyxial CA in the rabbit with survival rates of 58% in the TLV group compared to 0% for controls and 8% in groups receiving conventional cooling. 195 The concurrent administration of small molecules, dissolved gas mixtures, and the removal of pro-inflammatory intermediates and transmigrating immune effectors by liquid recirculation could all ostensibly limit collateral damage.
Intravenous delivery
Of course, the intravenous route has been widely used to deliver small molecules, recombinant proteins, antisense, and viral vectors in preclinical models of CA. Moreover, the IV route is required for some molecules due to the challenges posed by stability and absorption across the pulmonary or gastric endothelium. Over the past two decades, there has been widespread interest in leveraging the non-antibiotic properties of tetracycline (TC) derivatives in acute models of CNS ischemia. Minocycline, perhaps the best studied among the TCs, exhibits potent anti-inflammatory effects acting in part by limiting the polarization of microglia and other innate immune effectors.196,197 TCs have also been shown to blunt neutrophil chemotaxis, reduce MMP-mediated tissue injury, and limit the production of HMGB1 and other DAMPs from the brain.198–201 Using a porcine model of post-perfusion ARDS, Carney et al. 202 showed that treatment with the TC derivative CMT-3 (6-demythyl-6-deoxy-4dedimentylamino-TC) prevents lung dysfunction, reducing elastase and gelatinase activity, lavage fluid protein content, and neutrophil infiltration. Similar protective effects were seen with the use of a non-antimicrobial TC after mesenteric ischemia–reperfusion and peritoneal sepsis. 203 And while experience regarding the neuroprotective effects of TCs in models of CA is limited, the data support their potential therapeutic benefit via reductions in the production of neuronal TNFα, neuronal loss, and post-arrest anxiety.204,205
Conclusions
In this review, we present the salient clinical aspects of CA providing context regarding the notable pathological features that contribute to the post-CA syndrome. Based on the established link between pathological and iatrogenic events that alter pulmonary physiology after ROSC in CA, we propose a theoretical model referred to as lung–brain coupling that may play a critical role in regulating systemic inflammation and reperfusion injury. We hope that validation of the model and further study regarding essential sensor-effector pathways involved will provide novel therapeutic approaches that can be used to reduce the morbidity and mortality associated with this devastating condition.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants to MWH from the National Institute Neurological Disorders and Stroke (NINDS) (NS092455) and the Department of Defense (W81XWH-16-1-0191).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article. The authors followed criteria for authorship as set forth by the International Committee of Medical Journal Editors. Authorship credit was based on substantial contributions to the conception and design of the review, as well as the drafting of the article (NM and MWH), as well as the critical revision and contributions regarding to intellectual content (NM, SK, KG, and MWH). Digital illustrations in this review were created by Olivia Mackay (Figure 1), Emily Hochstedler (Figure 2), and Alyssa Marsh (![]() ). All authors have provided final approval for the final version to be published.
). All authors have provided final approval for the final version to be published.