
Abstract
Alzheimer’s disease (AD) is a very common neurodegenerative disorder. Although a majority of the AD cases are sporadic, most of the studies are conducted using transgenic models. Intracerebroventricular (ICV) administered streptozotocin (STZ) animals have been used to explore mechanisms in sporadic AD. In this study, we have investigated memory and neurometabolism of ICV-STZ-administered C57BL6/J mice. The neuronal and astroglial metabolic activity was measured in 1H-[13C]-NMR spectrum of cortical and hippocampal tissue extracts of mice infused with [1,6-13C2]glucose and [2-13C]acetate, respectively. STZ-administered mice exhibited reduced (p = 0.00002) recognition index for memory. The levels of creatine, GABA, glutamate and NAA were reduced (p ≤ 0.04), while that of myo-inositol was increased (p < 0.05) in STZ-treated mice. There was a significant (p ≤ 0.014) reduction in aspartate-C3, glutamate-C4/C3, GABA-C2 and glutamine-C4 labeling from [1,6-13C2]glucose. This resulted in decreased rate of glucose oxidation in the cerebral cortex (0.64 ± 0.05 vs. 0.77 ± 0.05 µmol/g/min, p = 0.0008) and hippocampus (0.60 ± 0.04 vs. 0.73 ± 0.07 µmol/g/min, p = 0.001) of STZ-treated mice, due to similar reductions of glucose oxidation in glutamatergic and GABAergic neurons. Additionally, reduced glutamine-C4 labeling points towards compromised synaptic neurotransmission in STZ-treated mice. These data suggest that the ICV-STZ model exhibits neurometabolic deficits typically observed in AD, and its utility in understanding the mechanism of sporadic AD.
Introduction
Alzheimer’s disease (AD) is the most common neurodegenerative disorder, and a leading cause of dementia worldwide. It is characterized by a progressive loss of memory, compromised intellectual abilities and cognitive functions. 1 According to the popular amyloid cascade hypothesis, genetic or environmental factors are considered as the primary cause of AD. These factors induce amyloid-beta (Aβ) aggregation and tau hyper-phosphorylation followed by the formation of toxic Aβ oligomers, plaques and neurofibrillary tangles (NFTs), ultimately leading to neurodegeneration and dementia. 2
AD has been broadly categorized into familial and sporadic (sAD) 3 based on the genetic and non-genetic cause of its origin. Although sAD accounts for a majority of patients (95%), 4 the underlying mechanism in its onset and progression is least explored. Hence, there is a need for detailed investigations of different aspects of pathophysiology and mechanism of sAD using non-transgenic models. 5 Several chemical interventions have been used in rats and mice to develop sAD models. 6 These include administration of Aβ peptides, scopolamine, streptozotocin (STZ), L-methionine and heavy metals. Intracerebroventricular (ICV) STZ administration is widely used to model sAD in rodents. 7 It induces an insulin-resistant brain state 8 that arises due to impaired insulin response signaling, 9 ultimately leading to decreased glucose transport and metabolism, overproduction of Aβ peptides, tau hyperphosphorylation and neurofilaments in the brain.8–11 The reduction in cerebral glucose metabolism has been considered as a key event in inducing AD-like phenotypes in ICV-STZ-treated animals. These features strengthen the relevance of the ICV-STZ model in understanding the pathophysiology of sporadic AD.
Glutamate and γ-aminobutyric acid (GABA) are the major excitatory and inhibitory neurotransmitters, respectively, in the matured mammalian central nervous system. 12 These neurotransmitters play major roles in cortical excitability and cognitive functions including learning and memory. The rates of neurotransmitter cycling and cerebral metabolic rates (CMR) of glucose oxidation in glutamatergic and GABAergic neurons have been shown to be reduced in transgenic13–15 and chemical 16 models of AD. In contrast, the rate of acetate oxidation, a marker of astroglial metabolic activity, has been reported to be increased in AβPP-PS1 mice at the stage of high plaque load. 13
Although cerebral glucose hypometabolism is proposed to be the initial event in the entire cascade of AD pathology, there is a limited quantitative measurement of neuronal and astroglial metabolic activity in STZ-treated mice. In this study, we have used 1H-[13C]-NMR spectroscopy in conjunction with infusion of [1,6-13C2]glucose and [2-13C]acetate to quantitatively evaluate the neuronal and astroglial metabolic activity, respectively, in ICV-STZ-treated mice. Our findings revealed a significant reduction in the rates of neuronal glucose oxidation and neurotransmitter cycling in the cerebral cortex and hippocampus of STZ-treated mice.
Materials and methods
Animal preparation
All experimental procedures with animals were approved by the Institutional Animal Ethics Committee of CSIR-CCMB, and conducted in accordance with the guidelines established by the Committee for the Purpose of Control and Supervision of Experiments on Animals, Ministry of Environment and Forests, Government of India. ARRIVE guidelines were followed for the preparation of the manuscript. C57BL6/J mice were procured from the Jackson Laboratory (New Harbor, Maine, USA), bred and maintained in the CCMB Animal House Facility. Mice were housed in individually ventilated cages, and maintained at standard room temperature (25 ± 2°C) in a 12/12 h light and dark cycle with ad libitum access to food and water. Six-month-old male mice weighing 28 to 30 g were used for the study. Two sets of experiments were performed to understand the dose dependence of STZ (3 and 5 mg/kg). In a pilot study involving lower dose (3 mg/kg), a total of 16 mice were randomly assigned into two groups: ICV-STZ (n = 8) and Control (vehicle-injected) (n = 8). For the final experiment that involved STZ dose 5 mg/kg, mice were allocated equally to the ICV-STZ (n = 18) and control (n = 18) group. Investigators were familiar with the animals allocated to ICV-STZ and control groups, and there was no blinding used during the study.
Intracerebroventricular injection of streptozotocin
Mice were anesthetized using a combination of ketamine:xylazine (100:7.5 mg/kg, intraperitoneal), and positioned on a stereotaxic apparatus (Stoelting Co., USA). Two holes were drilled in the cranium (1.7 mm lateral, 0.3 mm posterior and 2.6 mm ventral from the bregma with an angle of 16° lateral from the dorso-ventral axis) for bilateral administration of STZ. 17 A fresh solution of STZ was prepared in artificial cerebrospinal fluids (aCSF, 20 mmol/L citrate buffer, pH 4.2) for the individual mouse to provide the required dose in 2 µl. STZ (1 µL) was administered into each lateral ventricle using a 5 µL micro-syringe mounted with a 32-gauge needle (Hamilton, USA) in 150 s. Control mice received the same volume of vehicle (aCSF in citrate buffer). One animal from each group died in the experiment with the lower dose of STZ (3 mg/kg), whereas three animals from each group died at the higher dose (5 mg/kg) during the entire study period (Figure 1(a)).
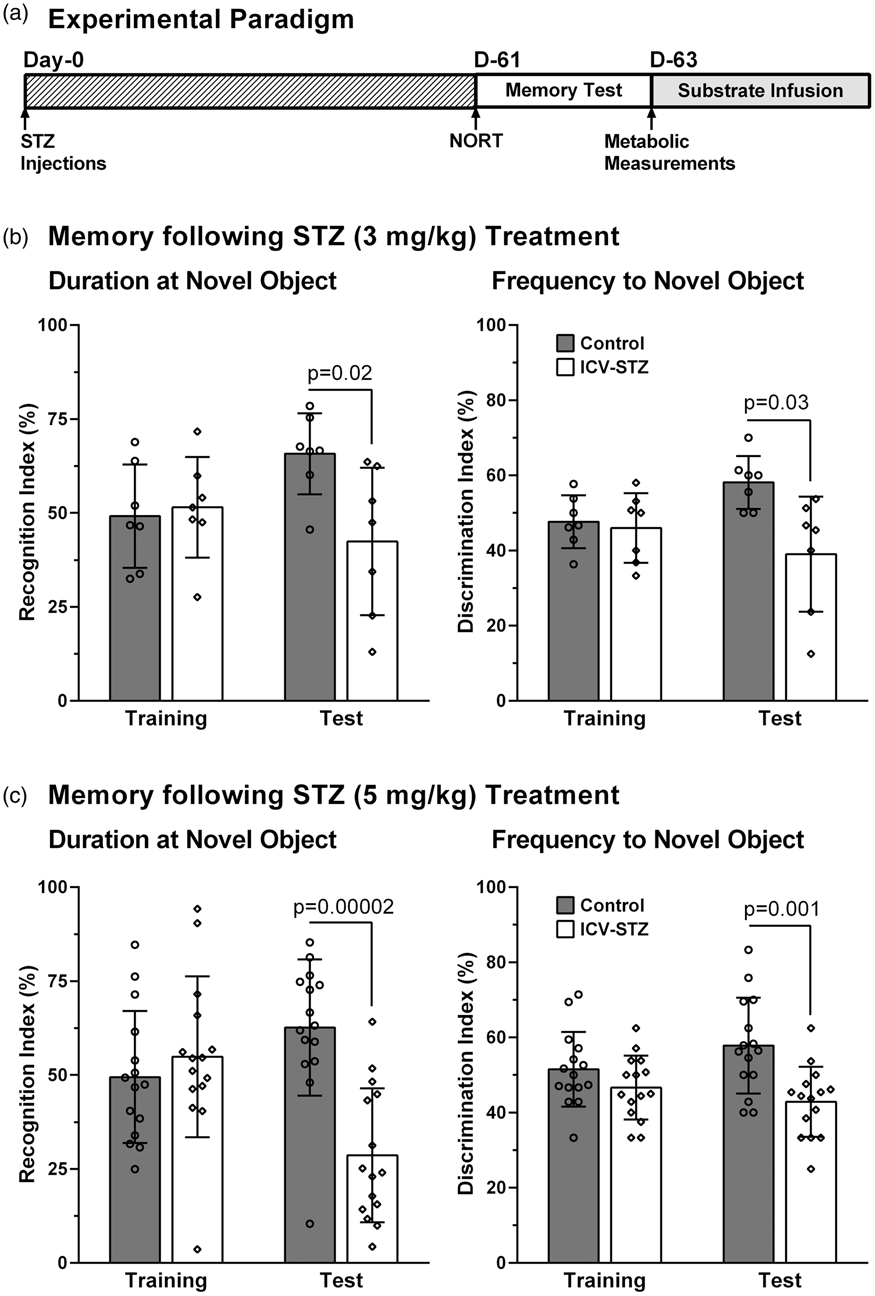
(a) Schematic representation of experimental paradigm. STZ was administered on the 0th day. The memory assessment was carried out on the 61st day, and the neurometabolic analysis on the 63rd day following infusion of [1,6-13C2]glucose/[2-13C]-acetate. Memory of the mice after (b) ICV-STZ (3 mg/kg), (c) ICV-STZ (5 mg/kg) treatment. The left and right panels depict the recognition and discrimination indices, respectively. Mice were first trained with two identical objects for 5 min. The working memory of mice was evaluated 1 h after the training by replacing the first object with a novel one. The recognition and discrimination indices were calculated using equations (1) and (2), respectively. The bar represent the mean±SD of the groups, while symbols denote individual values.
Memory assessment
The memory of mice was assessed two months post-STZ treatment using the Novel Object Recognition Test (NORT).
18
In brief, mice were initially trained to explore two identical objects placed at two corners in a rectangular wooden box (35×45×30 cm3) for a period of 5 min. One-hour post-training, each mouse was further reintroduced into the box, wherein one of the familiar objects was replaced with a novel one, and the mouse was allowed to explore the objects again for 5 min. The recognition index (RI) of mice for the novel object was obtained as follows:

The ability of the animal to discriminate between objects was assessed using the frequency of visits to the novel object. The discrimination index (DI) for novel object was calculated as follows:

Infusion of [1,6-13C2]glucose and [2-13C]acetate
Mice were fasted for 4 h before metabolic measurement. The lateral tail vein was catheterized for the infusion of 13C-labeled substrates. 19 In brief, [1,6-13C2]glucose (Cambridge Isotope Laboratories, Andover, MA, USA) dissolved in water (0.225 mol/L) was administered with an initial bolus rate of 1575 µmol/kg/min for the first 20 s. The infusion rate was stepped down exponentially at every 20 s to attain the final rate of 247 µmol/kg/min at 100 s, and was continued till 120 s. For assessment of astroglial metabolic activity, a solution of sodium [2-13C]acetate (1 mol/L, Cambridge Isotope Laboratories, Andover, MA, USA) and D-glucose (0.225 mol/L, Sigma-Aldrich Inc., St. Louis, MO, USA) was prepared, and pH adjusted to 7.0. The sodium [2-13C]acetate solution was delivered in mice using an infusion protocol. 20 A bolus of [2-13C]acetate was administered with an initial rate of 10 mmol/kg/min during the first 15 s, and the rate was stepped down exponentially in four steps to 0.50 mmol/kg/min by 75 s, which continued till 120 s. Mice were released to home cage immediately after completion of the substrate infusion. The brain metabolism in mice infused with [1,6-13C2]glucose and [2-13C]acetate was arrested using Focused Beam Microwave Irradiation system (4 kW, 1 s), (MMW-05, Muromachi Kikai Co., Ltd. Japan) 21 at 7 and 10 min, respectively.
Extraction of metabolites
The cerebral cortex and hippocampus were dissected from Microwave fixed brain. Tissue was weighed and homogenized with 0.1 N HCl in methanol (2:1 vol/wt) using a battery-operated tissue homogenizer (Argos Technologies, France) in a wet-ice bath. 22 [2-13C]Glycine (100 µl, 2 mmol/L, Cambridge Isotope Laboratories, Andover, MA, USA) was added as an internal concentration reference. The mixture was homogenized in 60% ethanol (6:1 vol/wt). The homogenate was centrifuged at 15000 g for 45 min, and the supernatant was lyophilized. The freeze-dried powder was dissolved in 550 µl D2O containing 0.25 mmol/L sodium 3-(trimethylsilyl)-2,2′,3,3′-D4-propionate (TSP) for NMR analysis.
Acetate, being a volatile molecule, is lost during lyophilization in the routine extraction procedure. Hence, 13C labeling of brain [2-13C]acetate was measured in the brain stem extract. In brief, the brain stem was homogenized in 550 µl D2O containing 1 mmol/L sodium formate. The homogenate was centrifuged, and the supernatant was used for NMR analysis.
1H-[13C]-NMR spectroscopy of brain tissue extracts
The NMR spectra of brain tissue extracts were recorded using a 600 MHz NMR spectrometer (Bruker Biospin, Ettlingen, Germany). The 1H NMR spectra of brain stem extracts were obtained using pulse acquired sequence. The concentrations of metabolites in the cerebral cortex and hippocampus were measured in 1H-[13C]-NMR spectra of brain tissue extracts.23,24 The levels of cerebral metabolites were calculated from the area of the respective peaks in the unedited spectrum relative to [2-13C]glycine (added during metabolites extraction) measured in the 13C edited spectrum. The percent 13C enrichment of metabolites at various carbon positions was determined from the ratio of the area in the difference spectrum to the non-edited. The 13C enrichments were reported after subtracting the natural abundance (1.1%) of carbon-13.
Estimation of astroglial and neuronal flux
The astroglial metabolic activity was investigated by monitoring the labeling of brain amino acids from [2-13C]acetate, which is selectively transported into astroglia.
25
Acetate metabolism through the astroglial TCA cycle transfers 13C label into the astroglial (small compartment) glutamate-C4 (GluC4) pool,
26
which is subsequently converted to glutamine-C4 (GlnC4)20,
27
by glutamine synthetase (glutamate ammonia ligase), an enzyme exclusively localized in the astrocytes.
28
The labeling of GluC4 and GABA-C2 (GABAC2) occurs through glutamate-glutamine and GABA-glutamine cycling, respectively. Aspartate-C2/C3 (AspC2/C3), GABAC3/C4 and GluC2/C3 are labeled due to further metabolism of GABAC2 and GluC4 in the respective TCA cycle. The kinetics of 13C labeling of amino acids from [2-13C]acetate is shown to be linear till 30 min in the rat cerebral cortex.20,29 The cerebral metabolic rate of acetate oxidation (CMRAce(Ox)) was calculated based on the 13C label trapped into different amino acids from [2-13C]acetate in 10 min using the following expression:
13
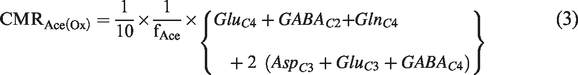
The oxidation of [1,6-13C2]glucose in the glutamatergic and GABAergic neurons through the TCA cycle labels α-ketoglutarate-C4, which undergoes transamination to label GluC4.13,19 In GABAergic neurons, GluC4 is decarboxylated to GABAC2 by glutamate decarboxylase. Astrocytic GlnC4 is labeled via cycling of GluC4 and GABAC2 followed by the action of glutamine synthetase. Further metabolism of glutamate and GABA in the TCA cycles labels AspC2/C3, GluC2/C3, GABAC3/C4 and GlnC2/C3. The labeling of amino acids has been shown to be linear till 20 min of glucose infusion in rats30,31, mice
19
and human brain
32
. The rate of glucose oxidation in glutamatergic neurons (CMRGlc(Glu)) was estimated from label trapped into amino acids from [1,6-13C2]glucose as described previously.31,33
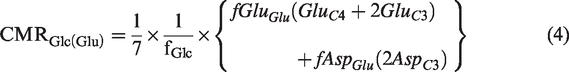
The rate of glucose oxidation in GABAergic neurons (CMRGlc(GABA)) was determined as follows:
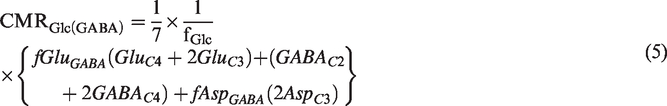
where GABACi represents the concentration of labeled GABA at 'ith' carbon from [1,6-13C2]glucose; fAsp(GABA) and fGlu(GABA) represent the fraction of aspartate and glutamate present in GABAergic neurons as described earlier. 19
The total rate of glucose oxidation including neurons and astrocytes was estimated as follows:
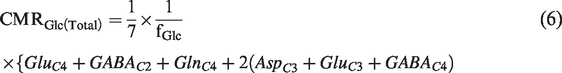
Estimation of rate of ATP synthesis
The rate of ATP synthesis was estimated by accounting for all the energy-rich molecules such as NADH, FADH2 including ATP/GTP that are produced during the metabolism of glucose via glycolysis and TCA cycle. The rates of glucose oxidation in glutamatergic and GABAergic neurons derived using equations (4) and (5) were used as primary flux together with a stoichiometry of 32 ATP for each glucose molecule.
Statistics
Unless stated, the data presented in the results correspond to the 5 mg/kg dose of STZ. All the statistical analyses were carried out using GraphPad Prism software (Ver 6.01, San Diego, USA). The Kolmogorov-Smirnov test was performed to assess the distribution of each data set. The significance of the difference of various measurements in ICV-STZ-treated and control mice was determined from the 2-tailed Student's T-test with Welch’s correction. For a few data sets that did not pass the normality, the significance of difference was obtained using the Mann-Whitney U test. The data resulting in a p-value less than 0.05 were considered different from each other.
Results
Memory of ICV-STZ-treated mice
The memory of mice was assessed 60 days post STZ treatment using NORT. There was no significant difference (p ≥ 0.46) in recognition index (RI) and discrimination index (DI) of STZ-treated mice (3 and 5 mg/kg) during training when compared to controls (Figure 1(b) and (c)). However, during the test phase, when one of the objects was replaced with a novel object, RI and DI of STZ-treated mice decreased significantly (p ≤ 0.03) when compared with controls (Figure 1(b) and (c)).
Neurometabolites homeostasis in STZ-treated mice
The levels of neurometabolites were measured in the non-edited 1H-[13C]-NMR spectra from cortical and hippocampal tissue extracts (Figures 2 and 1S). There was a minor though significant (p ≤ 0.02) reduction in NAA, taurine and creatine levels in the cerebral cortex and hippocampus of mice treated with the lower dose of STZ (3 mg/kg) (Table 1S). Additionally, there was a reduction (p = 0.04) in the cortical glutamate with STZ-treatment. The changes in brain metabolite homeostasis were more pronounced at the higher dose of STZ (5 mg/kg). This includes a reduction in glutamate (STZ 11.6 ± 0.8 µmol/g, n = 15; Control 12.4 ± 0.6 µmol/g, n = 15, p = 0.005), GABA (2.2 ± 0.1 vs. 2.3 ± 0.2 µmol/g, p = 0.01), NAA (6.0 ± 0.4 vs. 6.5 ± 0.4 µmol/g, p = 0.001) and creatine (12.1 ± 0.9 vs. 13.1 ± 1.2 µmol/g, p = 0.02) levels in the cerebral cortex (Table 1). Likewise, levels of glutamate (STZ 10.8 ± 0.6 µmol/g, n = 15; Control 11.6 ± 0.7 µmol/g, n = 15, p = 0.002), GABA (2.7 ± 0.1 vs. 3.0 ± 0.4 µmol/g, p = 0.005), NAA (6.0 ± 0.3 vs. 6.6 ± 0.5 µmol/g, p = 0.002), taurine (7.8 ± 0.6 vs. 8.6 ± 1.2 µmol/g, p = 0.03) and creatine (12.7 ± 0.5 vs. 13.5 ± 0.9 µmol/g, p = 0.002) were decreased in the hippocampus of mice treated with STZ. In contrast, the level of myo-Inositol was increased in the cerebral cortex (p = 0.04) and hippocampus (p = 0.02) of STZ-treated mice (Table 1).
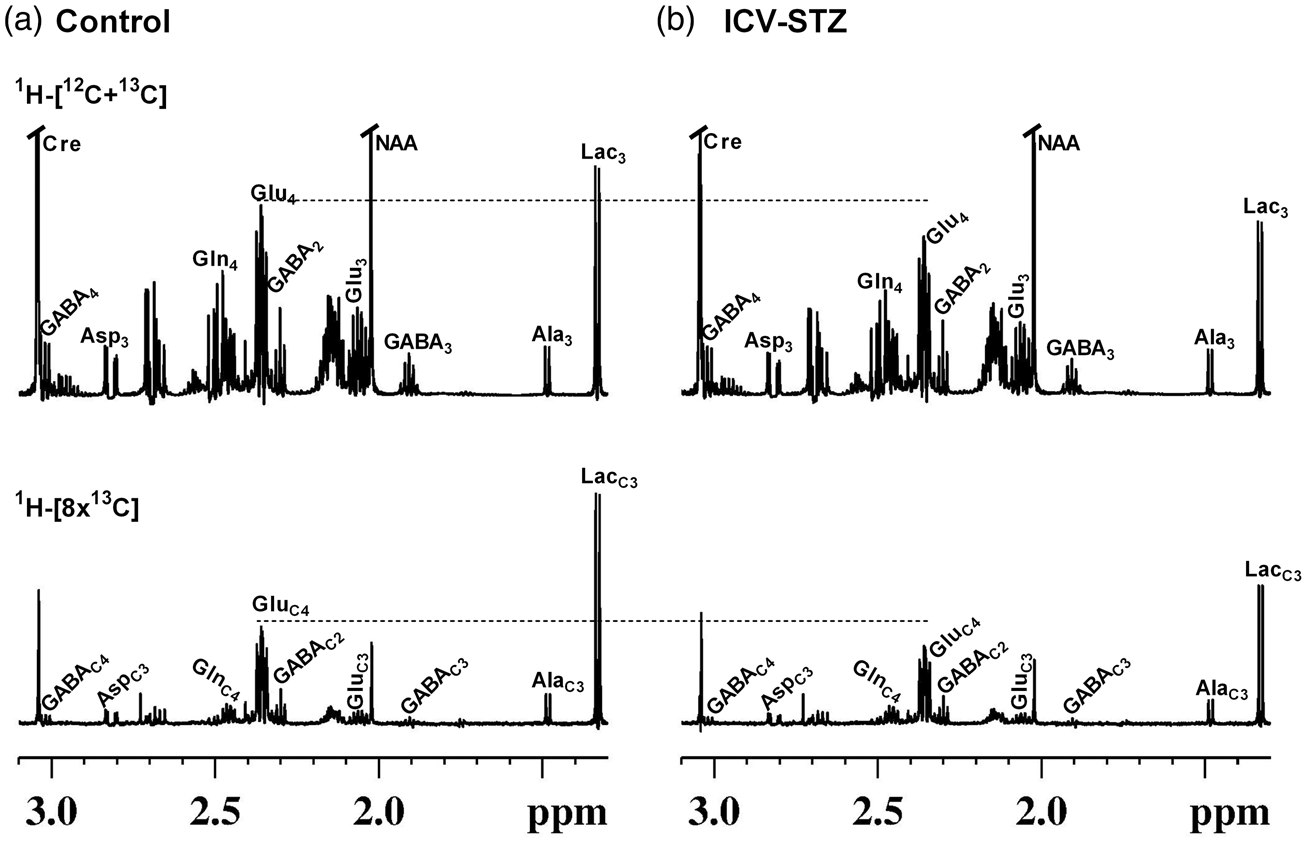
Representative 1H-[13C]-NMR spectra of cortical extracts from (a) Control, and (b) ICV-STZ (5 mg/kg) treated mice. Mice were infused with [1,6-13C2]glucose, and the brain metabolism was arrested using a focused beam microwave irradiation system immediately at 7 min of the infusion. Cortical metabolites were extracted using ethanol extraction protocol, and the NMR spectra were recorded using 600 MHz NMR spectrometer. Abbreviation used are: AlaC3, alanine-C3; AspC3, aspartate-C3; Cre, creatine; GABAC2, γ-aminobutyric acid-C2; GABAC3, γ-aminobutyric acid-C3; GABAC4, γ-aminobutyric acid-C4; GluC3, glutamate-C3; GluC4, glutamate-C4; GlnC4, glutamine-C4; LacC3, Lactate-C3; NAA, N-acetyl aspartate.
Concentration (µmol/g) of brain metabolites in STZ-treated (5 mg/kg) mice.
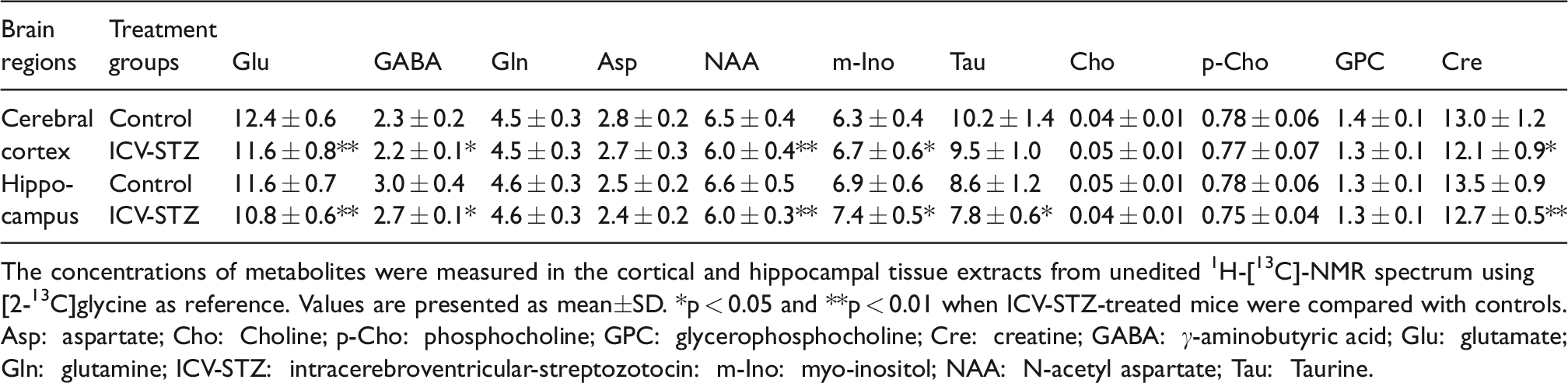
The concentrations of metabolites were measured in the cortical and hippocampal tissue extracts from unedited 1H-[13C]-NMR spectrum using [2-13C]glycine as reference. Values are presented as mean±SD. *p < 0.05 and **p < 0.01 when ICV-STZ-treated mice were compared with controls.
Asp: aspartate; Cho: Choline; p-Cho: phosphocholine; GPC: glycerophosphocholine; Cre: creatine; GABA: γ-aminobutyric acid; Glu: glutamate; Gln: glutamine; ICV-STZ: intracerebroventricular-streptozotocin: m-Ino: myo-inositol; NAA: N-acetyl aspartate; Tau: Taurine.
Astroglial metabolic activity in STZ-treated mice
Representative 1H-[13C]-NMR spectra of cortical tissue extract of mice infused with [2-13C]acetate are presented in Figure 1S. Prominent signals of GlnC4 (2.46 ppm) and GluC4 (2.34 ppm) could be seen in the 13C edited spectrum (lower panel) in the control and STZ-treated mice (5 mg/kg) (Figure 1S). Additionally, labeling of AspC3, GABAC2 and GluC3 could be seen. There was no significant difference in the 13C labeling of cortical (p ≥ 0.14) and hippocampal (p ≥ 0.45) amino acids in STZ-treated mice when compared with respective controls (Table 2S). Consequently, the cerebral metabolic rate of acetate oxidation (CMRAce(Ox)) was unperturbed in the cerebral cortex (STZ 0.139 ± 0.010 µmol/g/min, n = 8; Control 0.142 ± 0.008 µmol/g/min, n = 8, p = 0.55) and hippocampus (0.152 ± 0.010 vs. 0.151 ± 0.012 µmol/g/min, p = 0.89) of STZ-treated mice. (Table 2S).
Metabolism of [1,6-13C2]glucose in STZ-treated mice
1H-[13C]-NMR spectra showing the labeling of cortical amino acids from [1,6-13C2]glucose in ICV-STZ-treated (5 mg/kg) mice and controls are presented in Figure 2. Labeling of different metabolites could be seen in the 13C edited spectrum (lower panel) from control and ICV-STZ-treated mice (Figure 2(a) and (b)). Mice treated with the lower dose of STZ (3 mg/kg) exhibited a small but significant reduction in the labeling of GluC4 (P < 0.05) and GABAC2 (P < 0.01) from [1,6-13C2]glucose in the cerebral cortex and hippocampus (Table 3S). Moreover, the concentration of GluC3 and GlnC4 was decreased in the cerebral cortex. Treatment with higher dose of STZ (5 mg/kg) leads to a reduction in the labeling of AspC3 (STZ 0.27 ± 0.03 µmol/g, n = 7; Control 0.32 ± 0.03 µmol/g, n = 7, p = 0.007) together with GABAC2 (0.29 ± 0.03 vs. 0.34 ± 0.02 µmol/g, p = 0.001), GlnC4 (0.37 ± 0.02 vs. 0.43 ± 0.04 µmol/g, p = 0.004) and GluC3 (0.49 ± 0.06 vs. 0.60 ± 0.04 µmol/g, p = 0.005) and GluC4 (2.16 ± 0.24 vs. 2.59 ± 0.22 µmol/g, p = 0.005) in the cerebral cortex (Figure 2, Table 2). The hippocampus exhibited profound reduction (P ≤ 0.009) in the labeling of these amino acids in the STZ-treated (5 mg/kg) mice (Table 2). The reduction in 13C labeled AspC3, GluC4/C3, and GABAC2 is suggestive of decreased cerebral glucose oxidation in the STZ-treated mice. Additionally, the reduced 13C labeling of GlnC4 from [1,6-13C2]glucose suggests compromised neurotransmitter cycling in the cerebral cortex and hippocampus of STZ-treated mice.
Concentration (µmol/g) of 13C labeled amino acids from [1,6-13C2]glucose in STZ-treated (5 mg/kg) mice.
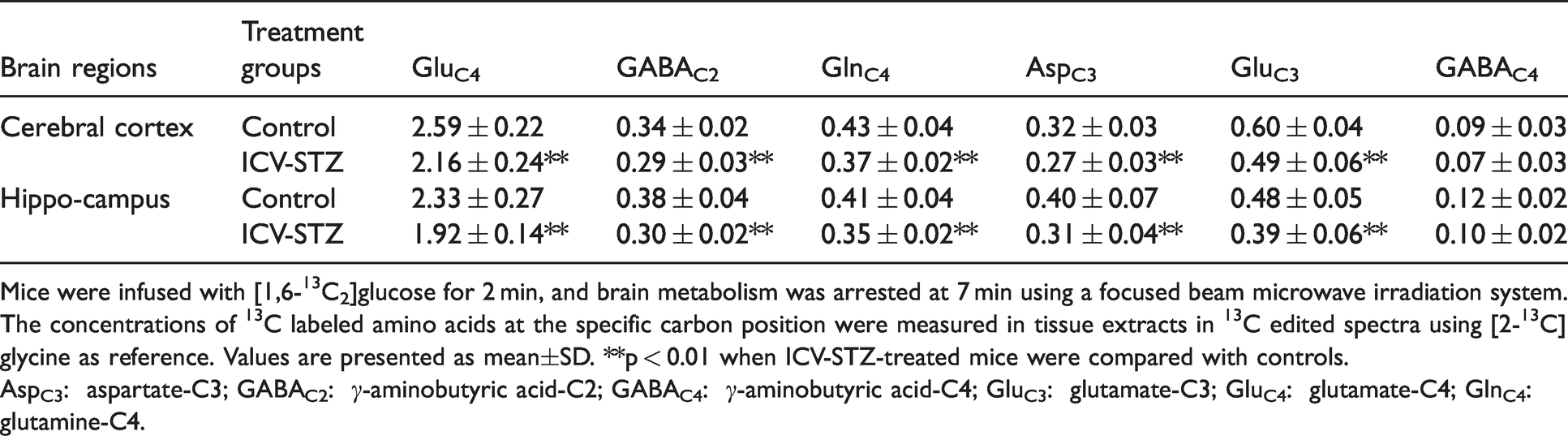
Mice were infused with [1,6-13C2]glucose for 2 min, and brain metabolism was arrested at 7 min using a focused beam microwave irradiation system. The concentrations of 13C labeled amino acids at the specific carbon position were measured in tissue extracts in 13C edited spectra using [2-13C]glycine as reference. Values are presented as mean±SD. **p < 0.01 when ICV-STZ-treated mice were compared with controls.
AspC3: aspartate-C3; GABAC2: γ-aminobutyric acid-C2; GABAC4: γ-aminobutyric acid-C4; GluC3: glutamate-C3; GluC4: glutamate-C4; GlnC4: glutamine-C4.
Neurometabolic activity in STZ-treated mice
The rates of glucose oxidation in glutamatergic and GABAergic neurons were decreased (p ≤ 0.04) slightly (∼9%) following the lower dose of STZ-treatment (3 mg/kg) (Figure 3). The higher dose of STZ (5 mg/kg) resulted in further reduction in glucose oxidation in the cerebral cortex (STZ: 0.641 ± 0.050 µmol/g/min, n = 7; Control: 0.767 ± 0.053 µmol/g/min, n = 7, p = 0.0008) (Figure 4(a)). The decrease in the rate of glucose oxidation in the STZ-treated mice was contributed by a similar degree of reduction (∼17%) in glutamatergic (0.401 ± 0.043 vs. 0.482 ± 0.033 µmol/g/min, p = 0.003) and GABAergic (0.103 ± 0.008 vs. 0.123 ± 0.013 µmol/g/min, p = 0.01) neuronal metabolic activity. Similarly, hippocampus exhibited a reduction (−18%) in glucose oxidation (0.599 ± 0.041 vs. 0.733 ± 0.068 µmol/g/min, p = 0.001) that was contributed by a decline in the rate of glucose oxidation in glutamatergic (0.370 ± 0.029 vs. 0.452 ± 0.042, p = 0.002) and GABAergic neurons (0.119 ± 0.010 vs. 0.147 ± 0.015, p = 0.003) (Figure 4(c)).
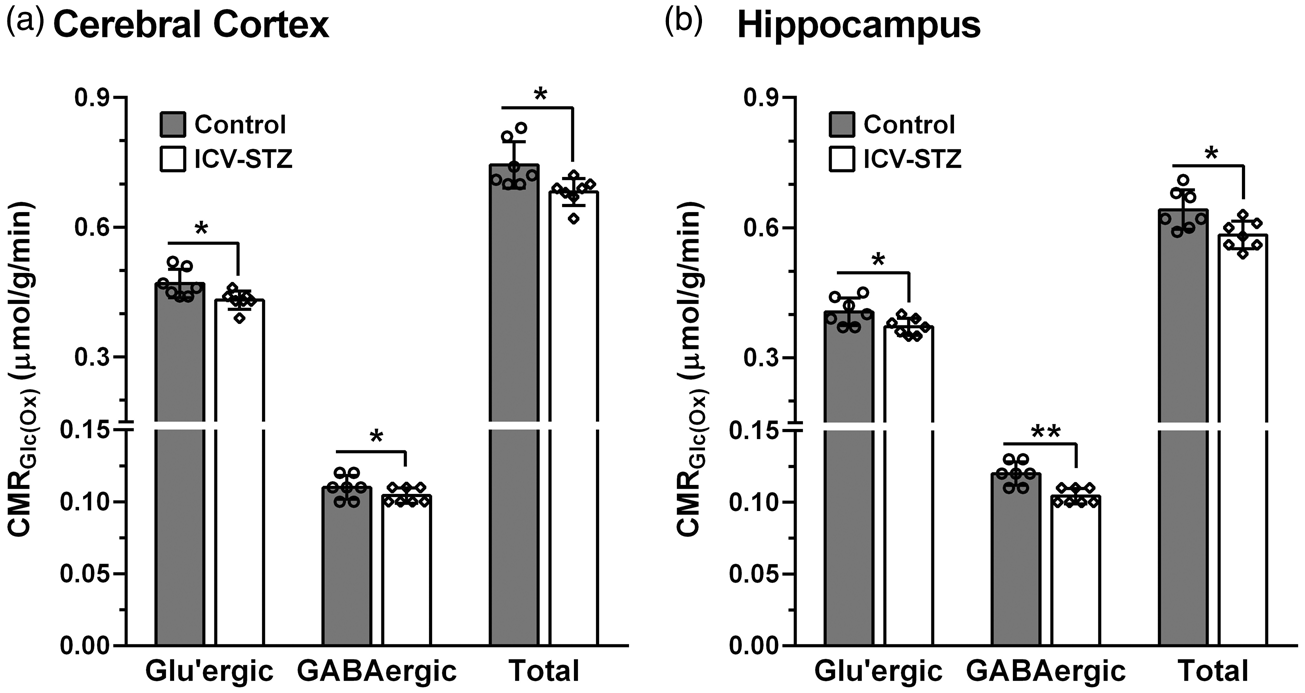
Cerebral Metabolic rates of glucose oxidation (CMRGlc(Ox)) in (a) Cerebral cortex and (b) Hippocampus of ICV-STZ (3 mg/kg) treated mice. The 13C labeling of amino acids from [1,6-13C2]glucose was measured in the tissue extracts using 1H-[13C]-NMR spectroscopy. The CMRGlc(Ox) was calculated using equations (4) to (6). The vertical bar represents the mean±SD of the group, while the symbols depict individual values. *p < 0.05 and **p < 0.01 when ICV-STZ-treated mice were compared with controls.
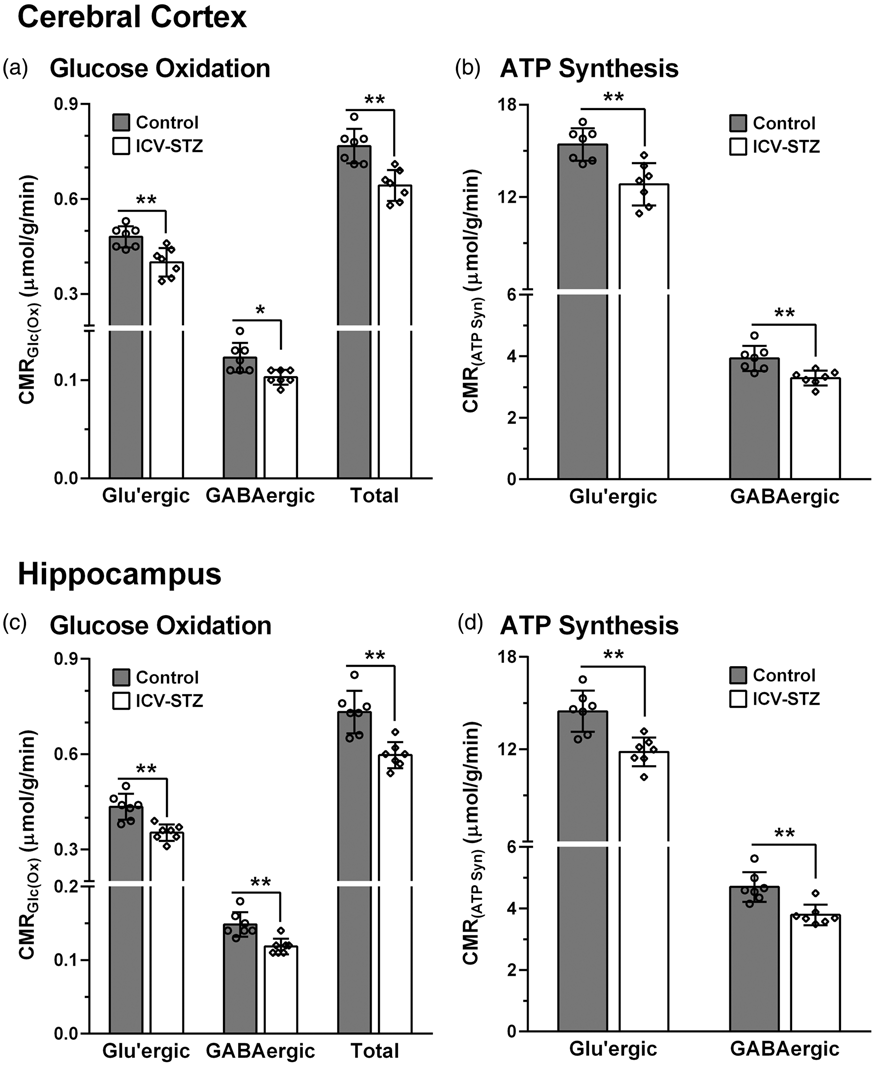
Cerebral Metabolic rates of: (a) Glucose oxidation (CMRGlc(Ox)) and (b) ATP synthesis (CMRATP(Syn)) in the cerebral cortex of ICV-STZ-treated (5 mg/kg) mice; (c) CMRGlc(Ox), and (d) CMRATP(Syn) in the hippocampus of STZ-treated mice. The 13C labeling of amino acids from [1,6-13C2]glucose was measured in the tissue extracts using 1H-[13C]-NMR spectroscopy. The CMRGlc(Ox) was calculated using Eqns. (4) to (6). The ATP synthesis rate was determined by multiplying the CMRGlc by a factor of 32. The vertical bar represents the mean±SD of the group, while the symbols depict individual values. *p < 0.05 and **p < 0.01 when ICV-STZ-treated mice were compared with controls.
ATP synthesis in STZ-treated mice
The rate of ATP synthesis was reduced significantly (p = 0.0006) in the cerebral cortex of ICV-STZ-treated (5 mg/kg) mice (20.5 ± 1.6 µmol/g/min) when compared with sham controls (24.6 ± 1.7 µmol/g/min). This reduction was due to a similar decline in ATP synthesis in glutamatergic (12.8 ± 1.4 vs. 15.4 ± 1.1 µmol/g/min, p = 0.002) and GABAergic neurons (3.3 ± 0.2 vs. 3.9 ± 0.4 µmol/g/min, p = 0.006) (Figure 4(b)). The hippocampus of STZ-treated mice also exhibited a similar reduction in the rate of ATP synthesis (19.2 ± 1.3 vs. 23.5 ± 2.2 µmol/g/min, p = 0.001) that was contributed by a decrease in the ATP production in glutamatergic (11.8 ± 0.9 vs. 14.5 ± 1.3 µmol/g/min, p = 0.001) and GABAergic (3.8 ± 0.3 vs. 4.7 ± 0.5 µmol/g/min, p = 0.002) neurons (Figure 4(d)).
Correlation between memory and neurometabolic activity
In order to understand the relation between memory and neurometabolic activity, a correlation analysis was carried out between the discrimination index (DI) and cerebral metabolic rate of glucose oxidation. A significant correlation was observed for cortical CMRGlc (R2 = 0.42; p = 0.01) and CMRGlc(Glu) (R2 = 0.44; p = 0.01) with DI, whereas the correlation of CMRGlc(GABA) with DI was not very significant (R2 = 0.09; p = 0.20) (Figure 5(a)). Likewise, the correlation of CMRGlc (R2 = 0.46; p = 0.007) and CMRGlc(Glu) (R2 = 0.48; p = 0.007) with DI was significant in the hippocampus (Figure 5(b)). Additionally, CMRGlc(GABA) exhibited a fair relationship (R2 = 0.35; p = 0.02) with DI. It is noteworthy that the correlation between neurometabolic and memory measures in the hippocampus (R2 = 0.35 to 0.48) was reasonably better than that observed in the cerebral cortex (R2 = 0.09 to 0.44).
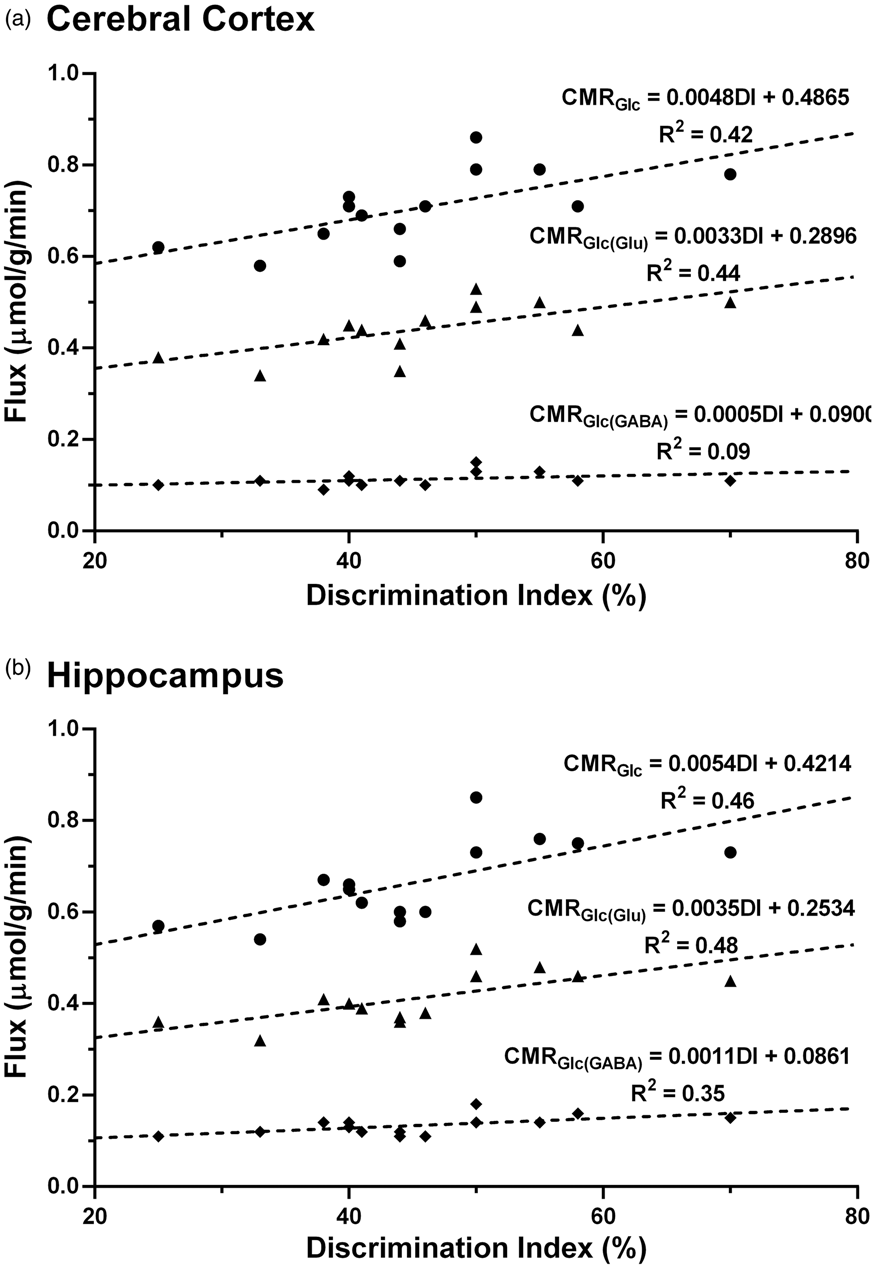
The correlation between memory and neurometabolic measures (● CMRGlc,
Discussion
The very first case of AD was reported more than a century ago but the actual mechanism involved in its onset and progression is still not clear. Amyloid-β protein from AD brain was purified and characterized for the first time by Glenner and Wong in the early eighties. 34 This led to the identification of several mutations in APP, PS1 and PS2 proteins responsible for the formation of Aβ plaques. These findings supported the genetic basis of the disease in a subset of AD patients, and facilitated the generation of transgenic rodent models. However, these models could not provide the detailed mechanism of the disease. Hence, there is a pressing need for the use of alternate models for AD studies. ICV-STZ-administered mice and rats have been routinely used to explore the pathophysiology of sporadic AD. The current study was aimed at establishing the energetics involved in neuronal (glutamatergic and GABAergic) and astroglial metabolism in ICV-STZ-treated mice. Similar to previous reports, our results show impaired memory in STZ-treated animals. 35
1H NMR spectroscopy has been used extensively to assess the levels of several key brain metabolites in patients and animal models of AD. These studies suggested a reduction in the levels of cortical NAA, 36 GABA and glutamate, 15 while an increase in that of myo-inositol 37 in AD. Our findings of a reduction in levels of glutamate, GABA, creatine and NAA together with an increased myo-inositol in STZ-treated mice is in consistence with previous reports in transgenic AβPP-PS1 mouse,13,14 AlCl3-treated C57BL6/J mouse 16 and ICV-STZ-treated rat 38 models of AD. The concurrent reduction of glutamate and NAA levels in STZ-treated mice suggests reduced viability or population of glutamatergic neurons. Glutamate is known to be involved in the control of different cognitive functions including learning and memory through long-term potentiation. The reduction in glutamate level probably explains the poor memory of ICV-STZ-treated mice. The increased level of myo-inositol in STZ-treated mice suggests an increase in the population or activity of astroglial cells, possibly due to increased inflammatory response that exists in the AD brain. 39 Additionally, hypertonic extracellular condition increases inflow of myo-inositol into cells leading to an increase in its cellular content. 40
Although the brain contributes only ∼2% of total body weight, it utilizes ∼20% of the total oxygen and glucose indicating the overwhelming energy demand of the brain. Such high energy is required to support the neuronal functions associated with action potentials, resting potentials, neurotransmitter release and recycling. 41 Decrease in the rate of glucose metabolism leads to reduced ATP production, and increased risk of oxidative damage, which have profound consequences on brain functioning. 42 The failure to fulfill the energy demand of the brain is known to increase the risk of Aβ formation and aggregation in APPSWE mice. 43 In fact, glucose hypometabolism is one of the prominent and key characteristics of the AD brain. PET studies have indicated a widespread reduction in brain glucose consumption in the posterior cingulate, lateral temporoparietal and occipital cortices of AD patients.44,45 Additionally, glucose hypo-metabolism has been observed in the temporoparietal and posterior cingulate brain regions of subjects with mild cognitive impairment. 46 Moreover, we have reported a reduction in brain glucose oxidation in 6-month-old AβPP-PS1 mice, which bears resemblance to the preclinical stage of AD. 14 There is very limited information about the status of cerebral glucose metabolism in ICV-STZ animals. A reduction in brain glucose utilization in the parietal cerebral cortex (−19%), frontal cerebral cortex (−13%) and different hippocampal regions (−12%) have been reported in the STZ-treated Wistar rats. 47 Consistent with this report, we observed a decrease in the rate of glucose oxidation in the cerebral cortex (−16%) and hippocampus (−18%).
Acetate metabolism has been used as a marker of astroglial metabolic activity and neuroinflammation. Analysis of neurometabolism in 12-month-old AβPP-PS1 mice revealed an increase in cerebral metabolic rate of acetate oxidation, indicating astrocyte-mediated neuroinflammation in AD. 13 Our attempts to understand the functional status of astroglia in STZ-treated mice revealed no change in the 13C labeling of cortical and hippocampal amino acids from [2-13C]acetate, suggesting that astroglial metabolic activity is unperturbed in these mice (Table 2S). Hence, the finding of increased myo-inositol level in STZ-treated mice suggests hypertonicity of extracellular fluids in cortical and hippocampal volume. 40
This study sheds light on the functional status of excitatory and inhibitory neurons in ICV-STZ-treated mice. Our data indicated a reduction in the label incorporation in GluC4/C3 and GABAC2/C4 from [1,6-13C2]glucose in the cerebral cortex and hippocampus of STZ-treated mice (Table 2). These findings suggest a reduction in the excitatory and inhibitory activity related to glutamatergic and GABAergic neurons, respectively (Figure 4(a) and (c)). The reduction of glucose oxidation in the cerebral cortex and hippocampus of STZ-treated mice was contributed by almost an identical reduction in the rates of glucose oxidation in excitatory (−17%) and inhibitory neurons (−18%). The neuronal glucose oxidation flux is shown to be stoichiometrically coupled with the rate of neurotransmitter cycling.48,49 Hence, a reduction in the neuronal glucose oxidation in glutamatergic and GABAergic neurons in the ICV-STZ-treated mice suggests reduced synaptic transmission in these mice. In fact, the level of 13C label trapped into GlnC4, which is labeled via glutamate-glutamine and GABA-glutamine cycling, was reduced in the cerebral cortex (−14%) and hippocampus (−16%) of STZ-treated mice (Table 2) further corroborate the interpretation.
Our analysis of the association between memory and neurometabolic measures showed a fairly good correlation for different fluxes in both brain regions (Figure 5). The total as well as glutamatergic neurometabolic rates were in a fairly good association with the memory in the cerebral cortex and hippocampus. However, a significant correlation (R2) of GABAergic flux with episodic memory could be observed only in the hippocampus. This further validates our findings, as glutamate is known to play a direct role in the process of learning and memory through long term potentiation. Additionally, the correlation of GABAergic neurometabolic activity with the memory in the hippocampus emphasizes its role as the primary brain region responsible for memory.
Glucose metabolism serves as the primary source of energy through the process of oxidative phosphorylation via electron transport chain (ETC) in mitochondria. The derived energy is utilized to power neurotransmission and other vital neuronal functions in the brain. The disruption of ETC not only reduces the yield of ATP, but also increases the burden of oxidative stress, which is known to be a contributing factor in the majority of neurodegenerative disorders including AD. 42 Our analysis indicated that the efficiency of ATP synthesis was reduced to ∼82% in the cerebrum of STZ-treated mice. The reduced availability of ATP may be driving the brains of STZ-treated mice to an AD-like condition. 43
There are a few limitations with regard to the current study. Firstly, the estimation of the cerebral metabolic rates in the current study assumes complete trapping of labels into amino acids. Any loss of label due to glutamine efflux from the brain will violate the assumption, and will underestimate the metabolic rates to the extent of the efflux. However, glutamine efflux (PC flux) is reported to be very small relative to pyruvate dehydrogenase. 50 Secondly, the expression used for the estimation of glucose oxidation in glutamatergic and GABAergic neurons does not account for the flow of label into glutamine. However, the labeling of glutamine in the control and STZ-treated mice is very small. Lastly, a small amount of 13C label may be lost as CO2 by oxidation in subsequent turns of the TCA cycle. Hence, the metabolic flux associated with glutamatergic and GABAergic neurons is likely to be underestimated. However, it could be argued that such losses would occur in control and ICV-STZ-mice. Therefore, although the absolute rates may be slightly lower in both groups, the directional changes in flux should remain valid.
The current study examines the impact of ICV-STZ administration on behavioral and cerebral metabolic measures in the mouse. Most importantly, it deciphers the energetics of astroglia, excitatory glutamatergic and inhibitory GABAergic neurons under the sporadic AD-like condition. The findings of the study revealed a compromised state of excitatory as well as inhibitory neurons, and reduced synaptic transmission in the ICV-STZ-administered mice. These results provide experimental evidence for impairment of neurotransmitter metabolism in STZ-treated mice, a model of sporadic AD.
Supplemental Material
sj-pdf-1-jcb-10.1177_0271678X21996176 - Supplemental material for Brain energy metabolism in intracerebroventricularly administered streptozotocin mouse model of Alzheimer’s disease: A 1H-[13C]-NMR study
Supplemental material, sj-pdf-1-jcb-10.1177_0271678X21996176 for Brain energy metabolism in intracerebroventricularly administered streptozotocin mouse model of Alzheimer’s disease: A 1H-[13C]-NMR study by Narayan D Soni, Akila Ramesh, Dipak Roy and Anant B Patel in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study was supported by funding from CSIR-CCMB (NCP/MLP0139).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
All authors have read and approved the final manuscript.
Supplemental material
Supplemental material for this article is available online.
Acknowledgements
The authors wish to thank Dr. Robin de Graff at Yale University for providing the POCE pulse sequence, and Dr. T Ramakrishna (ex CCMB Scientist) for critical editing of the manuscript. We acknowledge the efforts of Ms. B. Jyothi Lakshmi and Mr. N. Sai Ram of the Animal House for the maintenance of quality animals. We also thank the CCMB Animal House and Behavioral Facility for providing the facility for surgical procedures and behavioral experiments. All NMR experiments were carried out in the NMR Microimaging and Spectroscopy Facility, CSIR-CCMB, Hyderabad, India.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.