
Abstract
Astrocytes are intimately involved in both glutamate and γ-aminobutric acid (GABA) synthesis, and ischemia-induced disruption of normal neuroastrocytic interactions may have important implications for neuronal survival. The effects of middle cerebral artery occlusion (MCAO) on neuronal and astrocytic intermediary metabolism were studied in rats 30, 60, 120, and 240 minutes after MCAO using in vivo injection of [1-13C]glucose and [1,2-13C]acetate combined with ex vivo13C magnetic resonance spectroscopy and high-performance liquid chromatography analysis of the ischemic core (lateral caudoputamen and lower parietal cortex) and penumbra (upper frontoparietal cortex). In the ischemic core, both neuronal and astrocytic metabolism were impaired from 30 minutes MCAO. There was a continuous loss of glutamate from glutamatergic neurons that was not replaced as neuronal glucose metabolism and use of astrocytic precursors gradually declined. In GABAergic neurons astrocytic precursors were not used in GABA synthesis at any time after MCAO, and neuronal glucose metabolism and GABA-shunt activity declined with time. No flux through the tricarboxylic acid cycle was found in GABAergic neurons at 240 minutes MCAO, indicating neuronal death. In the penumbra, the neurotransmitter pool of glutamate coming from astrocytic glutamine was preserved while neuronal metabolism progressively declined, implying that glutamine contributed significantly to glutamate excitotoxicity. In GABAergic neurons, astrocytic precursors were used to a limited extent during the initial 120 minutes, and tricarboxylic acid cycle activity was continued for 240 minutes. The present study showed the paradoxical role that astrocytes play in neuronal survival in ischemia, and changes in the use of astrocytic precursors appeared to contribute significantly to neuronal death, albeit through different mechanisms in glutamatergic and GABAergic neurons.
Astrocytes are intimately involved in both glutamate and γ-aminobutyric acid (GABA) synthesis, and changes in trafficking of metabolites between astrocytes and glutamatergic or GABAergic neurons during ischemia in vivo may have important implications for neuronal survival. In a previous paper on middle cerebral artery occlusion (MCAO), we demonstrated that astrocytic and neuronal intermediary metabolism, and the trafficking of metabolites between these two cellular compartments, can be analyzed in great detail by combining in vivo injection of13C-labeled acetate and glucose with ex vivo13C magnetic resonance (MR) spectroscopy of deproteinized brain extracts (Håberg et al., 1998a). In the present study, this method was used to identify changes in glutamate and GABA synthesis, and in astrocytic metabolism during the first 4 hours MCAO. In cerebral ischemia, glutamate excitotoxicity is considered an important mediator of neuronal death (Choi, 1992; Meldrum, 2000); however, GABA is recognized as an endogenous neuroprotectant in the mature brain (Schwartz-Bloom and Sah, 2001).
The lateral caudoputamen and lower parietal cortex, and the upper frontoparietal cortex ipsilateral to the MCAO, were analyzed separately to gain insight into regionally specific alterations in the metabolic activity of glutamatergic and GABAergic neurons and astrocytes. In MCAO induced with the intraluminal filament technique, the ipsilateral lateral caudoputamen and lower parietal cortex are rendered severely ischemic, whereas the upper frontoparietal cortex represents moderately ischemic tissue. The former is often referred to as the ischemic core, and the latter as the penumbra (Memezawa et al., 1992a; Back, 1998). Inherent to the definition of the penumbra is the possibility of reversing the neuronal failure, but with time the penumbra is incorporated into the ischemic core (Back, 1998). In the present study, neuronal and astrocytic tricarboxylic acid (TCA) cycle activity and the use of astrocytic intermediates for neurotransmitter synthesis in these two regions were followed closely during the initial 240 minutes MCAO to identify changes influencing neuronal survival.
MATERIALS AND METHOD
Animal experiments
All animal procedures adhered to institutionally approved protocol in accordance with the guidelines set by the Norwegian Committee for Animal Research.
Male Wistar rats (Møllegaard Breeding Center, Copenhagen, Denmark) weighing 320 to 340 g were fasted overnight. Anesthesia was induced with 3.5% isoflurane in 70% nitrogen/30% oxygen. During surgery, the animals spontaneously breathed 2% isoflurane in 70% nitrogen/30% oxygen delivered through a face mask. Body temperature was maintained using a feedback-controlled heating blanket connected to a rectal temperature probe. A catheter was introduced into the right femoral vein, externalized at the tail radix, and taped in place. Middle cerebral artery occlusion was induced with the intraluminal filament technique (Longa et al., 1989; Memezawa et al., 1992a). All incisions were sprayed with lidocaine (10 mg/doses) before closure. The rats subjected to MCAO were assigned to four different groups with MCAOs of 30 (n = 7), 60 (n = 7), 120 (n = 7), or 240 minutes (n = 7). Ten rats were sham operated and allowed to recover for either 30 (n = 3) or 240 minutes (n = 7). Isoflurane administration was discontinued at time of MCAO except in rats assigned to MCAO for 120 and 240 minutes, who underwent diffusion-weighted MR imaging immediately after surgery. The imaging was performed 18 minutes after MCAO on a 2.35-T Bruker Biospec (Bruker AG, Fällanden, Switzerland) with a single b value of 1468 mm2/second, echo time 32/repetition time 1500, and number of excitations two-in-eighth transaxial slices with 1.5-mm thick slices that covered the entire hemisphere. Isoflurane was maintained at 2% during MR imaging. To avoid unwanted effects of anesthesia on the cerebral metabolism, rats assigned to either 30 or 60 minutes MCAO did not undergo diffusion-weighted imaging. The rats were returned to individual cages and allowed to recover immediately after surgery or MR imaging. An intravenous injection (1 mL/100 g) 0.3 mmol/L [1-13C]glucose and [1,2-13C]acetate (Cambridge Isotopes Laboratories, Woburn, MA, U.S.A.) in sterile water was given for 2 minutes starting 15 minutes before decapitation. The rats were decapitated into liquid nitrogen 30, 60, 120, or 240 minutes after MCAO or sham operation. Blood was collected from the severed neck vessels of rats subjected to 240 minutes MCAO. A 5-mm coronal slice extending caudally from the chiasma opticum was cut from the frozen brains using a brain matrix (RBM-40000; Activational Systems, Warren, MI, U.S.A.), and the right lateral caudoputamen, lower parietal cortex, and upper frontoparietal cortex were sampled. The brain and plasma samples were extracted as described previously (Håberg et al., 1998a).
13C magnetic resonance spectroscopy
Proton-decoupled 125.5-MHz13C MR spectra were obtained with a Bruker DRX-500 spectrometer (Bruker AG) using a 35 pulse angel, 25-kHz spectral width, and 64-K data points. The acquisition time was 1.3 seconds/scan plus 2.5 seconds of relaxation delay. The number of scans was typically 15,000 for brain samples. To avoid nuclear Overhauser effects, some spectra were broadband decoupled during acquisition only. The respective correction factors were then applied to the spectra obtained.
1H magnetic resonance spectroscopy
The 500-MHz1H MR spectra of the plasma extracts were obtained with a Bruker DRX-500 spectrometer using a 90 pulse angle, 7.5-kHz spectral width, and 16-K data points. The acquisition time was 1.1 second/scan plus 15.0 seconds of relaxation delay. The number of scans was typically 200.
High-performance liquid chromatography analysis
After13C MR spectroscopy, total amino acid concentrations were determined with high-performance liquid chromatography (Specta System Gradient Pump, Freemont, CA, U.S.A.), and fluorescence detection (Shimadzu RF 530, Tokyo, Japan) after derivatization with (o-phthaldialdehyde (Sigma Chemical Co., St. Louis, MO, U.S.A.) using L-aminobutyric acid as an internal standard.
Analysis of13C and1H magnetic resonance spectra and the rationale behind data interpretation
Relevant peaks in the13C and1H MR spectra were identified, and the total amount of13C or1H in the resonance of a particular metabolite was quantified from the integral of the peak area using ethyleneglycol as an internal standard. The total amount of13C includes the naturally abundant13C (1.1% of all carbon atoms). Data analysis is described in detail in Håberg et al. (1998a).
The neuronal and astrocytic compartments can be analyzed simultaneously by concomitant administration of [1-13C]glucose and [1,2-13C]acetate (Badar-Goffer et al., 1990; McLean et al, 1993; Sonnewald et al., 1996; Hålberg et al., 1998a) because of the uneven distribution of enzymes and transporter proteins between neurons and astrocytes.
The astrocytic compartment is recognized by its ability to use acetate as a substrate for the TCA cycle (Van den Berg, 1973) due to the presence of a specific acetate-uptake mechanism (Waniewski and Martin, 1998). In astrocytes, [1,2-13C]acetate is metabolized by acetyl coenzyme A synthetase (EC 6.2.1.1) to acetyl coenzyme A, which enters the TCA cycle and finally gives rise to [4,5-13C]glutamate. [4,5-13C]Glutamate is rapidly converted to [4,5-13C]glutamine by the glia-specific enzyme glutamine synthetase (EC 6.3.1.2) (Norenberg and Martinez-Hernandez, 1979). Glutamate is present only in low concentrations in astrocytes (Ottersen, 1989). [4,5-13C]Glutamine is released from astrocytes and taken up by a high-affinity glutamine transporter that is present on neurons (Varoqui et al., 2000). Glutamate is regenerated from glutamine in the neurons by phosphate-activated glutaminase (PAG) (EC 3.4.1.2) (Kvamme et al., 2000), which converts [4,5-13C]glutamine to [4,5-13C]glutamate. In GABAergic neurons, GABA is synthesized from glutamate by glutamate decarboxylase (GAD) (EC 4.1.1.15), and the direct conversion of [4,5-13C]glutamate results in [1,2-13C]GABA. GABA synthesis only takes place in neurons (Sze, 1979; Sloviter et al., 1996). Label in [1,2-13C]GABA is distributed equally into [3-13C] and [4-13C]GABA in the second turn of the TCA cycle due to the symmetrical succinate step. Likewise, label in [4,5-13C]glutamate/glutamine is distributed into [1,2-13C]- and [3-13C]glutamate/glutamine.
Astrocytes can also metabolize glucose, but to a lesser extent than neurons. However, glucose can enter the TCA cycle of astrocyte cells via the astrocyte-specific anaplerotic enzyme pyruvate carboxylase (PC) (EC 6.4.1.1) (Yu et al., 1983), the brain's principal anaplerotic enzyme (Patel, 1974). In astrocytes, [1-13C]glucose metabolized by PC activity gives rise to [2-13C]glutamine. [2-13C]Glutamine is converted to [2-13C]glutamate in neurons (or to [4-13C]GABA in GABAergic neurons).
Neurons primarily metabolize glucose (Van den Berg, 1973; Künnecke et al., 1993). [1-13C]Glucose enters the neuronal TCA cycle as acetyl coenzyme A solely via pyruvate dehydrogenase (PDH) (EC 1.2.4.1), and finally results in [4-13C]glutamate formation. However, glutamate de novo synthesis occurs in astrocytes and not in neurons (Hertz et al., 1999). The majority of glutamate is found in glutamatergic neurons (Ottersen, 1989). Synaptically released glutamate is predominantly taken up by high-affinity glutamate transporters located on astrocytes (Erecinska, 1987; Tanaka, 2000). In the astrocyte, [4-13C]glutamate either is converted directly to [4-13C]glutamine, or is reintroduced to the TCA cycle (Farinelli and Nicklas, 1992). The exchange of glutamate and glutamine between astrocytes and neurons is termed the glutamate-glutamine cycle (Westergaard et al., 1995; Daikhin and Yudkoff, 2000). After one turn in the astrocytic TCA cycle, the [4-13C]glutamate label will be recovered in [3-13C]- and [2-13C]glutamine. In GABAergic neurons, [4-13C]glutamate is rapidly converted to [2-13C]GABA, and little glutamate is present in these neurons (Ottersen, 1989). After one turn of the TCA cycle, the label that originated in [2-13C]GABA will be distributed equally to [3-13C]- and [4-13C]GABA.
Neurons depend on the transfer of metabolites from astrocytes because of their inability to synthesize glutamate de novo and their lack of anaplerotic activity, combined with the continuous drain of metabolic intermediates from the neuronal TCA cycle due to neurotransmitter production and release (Hertz, 1979; Kaufmann and Driscoll, 1993; Hertz et al., 1999). The contribution from astrocytic precursors to neuronal glutamate and GABA formation can be derived from the PC versus PDH activity ratio, which is an estimate of the anaplerotic (astrocytic) pathway compared with the oxidative (neuronal) pathway in glutamate (Taylor et al., 1996). Furthermore, the acetate versus glucose utilization ratio approximates the relative contribution of the astrocytic TCA cycle precursors compared with the neuronal TCA cycle precursors glutamate, glutamine, and GABA formation (Taylor et al., 1996).
All values are mean ± SD. Statistical comparisons between the groups subjected to MCAO of varying duration and the sham-operated rats who recovered for 240 minutes before decapitation were performed with analysis of variance followed by least-significant difference post-hoc test for multiple comparisons. An unpaired two-way Student's t-test was used to compare the sham-operated rats recovering for 30 and 240 minutes after termination of anesthesia. P < 0.05 was considered significant.
RESULTS
Diffusion-weighted magnetic resonance imaging
The total volume of ischemic tissue 18 minutes after MCAO, as estimated by diffusion-weighted imaging, was 125.5 ± 52.15 mm3(n = 14). The volume of ischemia for each separate slice, from caudal to rostral, was 2.2 ± 4.8, 18.9 ± 12.1, 31.1 ± 16.2, 38.5 ± 10.5, 22.2 ± 10.9, 8.1 ± 10.0, 8.3 ± 2.1, and 1.3 ± 4.2 mm3. The 5-mm slice cut after decapitation to sample brain tissue for further analysis with13C MR spectroscopy was located between slice three and five, the region in which the largest areas of ischemic tissue were found.
Analysis of plasma extracts
In rats subjected to 240 minutes MCAO, the total glucose concentration in plasma as estimated from the1H MR spectra was 16.1 ± 1.1 mmol/L, and only glucose enriched in the C-1 position was detected in significant amounts. The concentrations of [1,2-13C]acetate, lactate, and hydroxybutyrate were 1.7 ± 0.8, 2.1 ± 0.5, and 1.2 ± 0.4 mmol/L, respectively. There was no significant13C enrichment in lactate or hydroxybutyrate; therefore, the observed amino acid labeling originated from cerebral [1-13C]glucose and [1,2-13C]acetate metabolism. Furthermore, neither the duration of fasting nor the stress of cerebral ischemia influenced the plasma composition, because similar amounts of13C-labeled substrate were present in plasma at 90 minutes MCAO (Håberg et al., 1998a).
Effects of middle cerebral artery occlusion on total amino acid content
Lateral caudoputamen and lower parietal cortex.
In this severely ischemic region, the total glutamate content was significantly decreased 30 minutes after MCAO, and continued to decline for the next 240 minutes (Fig. 1A). The glutamine content remained at control levels until 240 minutes when it was significantly decreased compared with 60 minutes MCAO (Fig. 1A). The total GABA content was more than doubled at 240 minutes MCAO (Fig. 1C). The alanine levels started to increase markedly from 60 minutes MCAO, and had doubled by 240 minutes (Fig. 1E). The total amount of aspartate decreased from 60 minutes MCAO, and was approximately half of that found in sham-operated rats after 240 minutes MCAO (Fig. 1E).
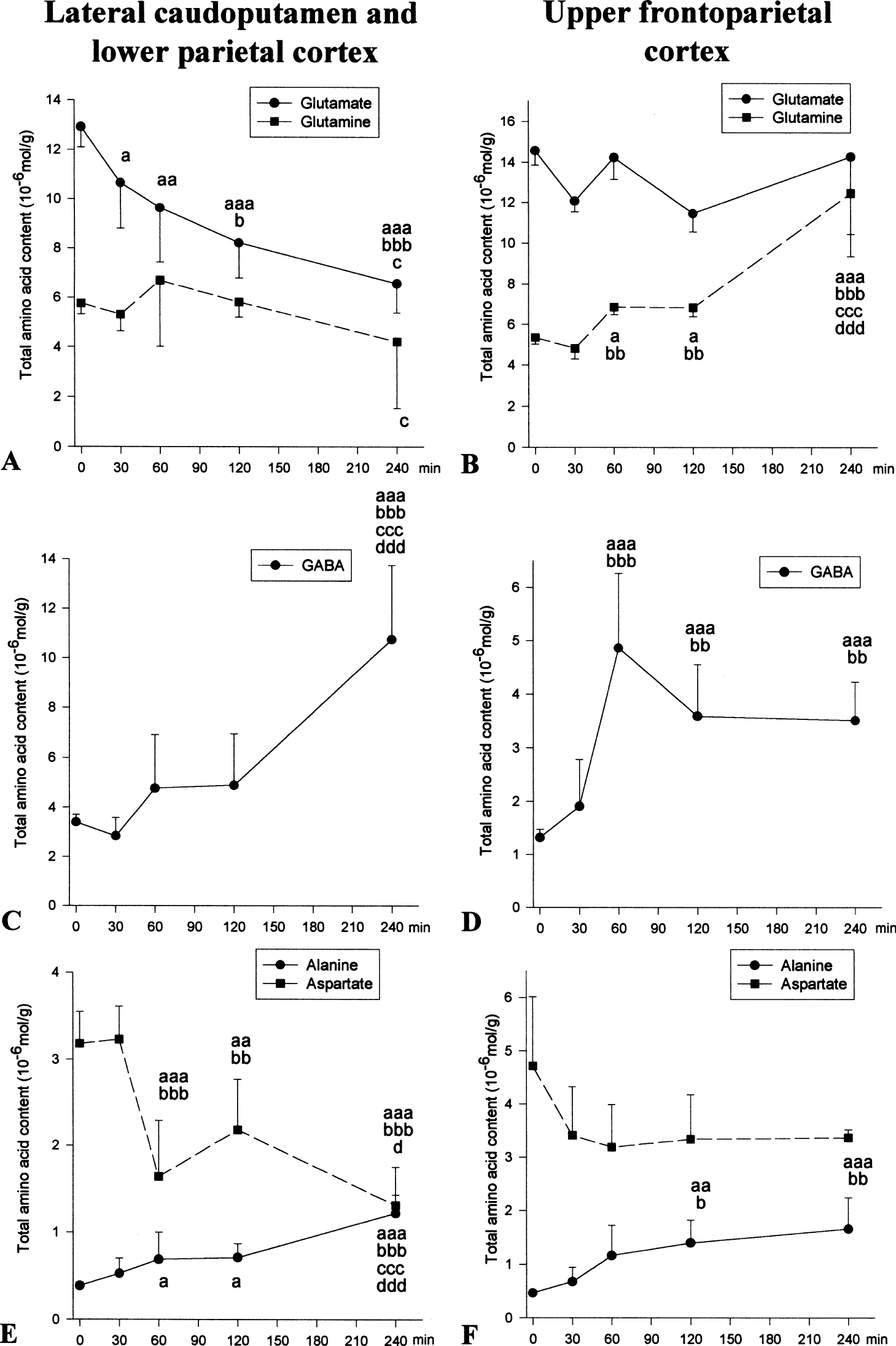
Effect of MCAO on total amino acid content. Time zero represents sham-operated rats allowed to recover for 240 minutes before time of death. The amino acid content was analyzed with high-performance liquid chromatography (see Materials and Methods). The lateral caudoputamen and lower parietal cortex (
Upper frontoparietal cortex.
In this moderately ischemic region, glutamate remained at control levels during the 240 minutes MCAO (Fig. 1B). The glutamine content was significantly increased at 60 minutes MCAO and nearly doubled at 240 minutes (Fig. 1B). The GABA content was also significantly increased at and beyond 60 minutes MCAO (Fig. 1D). Alanine levels were not markedly increased before 120 minutes (Fig. 1F), and aspartate concentration was at control levels during 240 minutes MCAO (Fig. 1F).
13C magnetic resonance spectroscopy of brain extracts: effects of middle cerebral artery occlusion on cerebral metabolism
[1-13C]Glucose and [1,2-13C]acetate.
In the lateral caudoputamen and lower parietal cortex, [1-13C]glucose content was significantly increased to a similar degree at all time points after MCAO (Fig. 2A). Unmetabolized [1,2-13C]acetate was present at all times after MCAO (Fig. 2A).
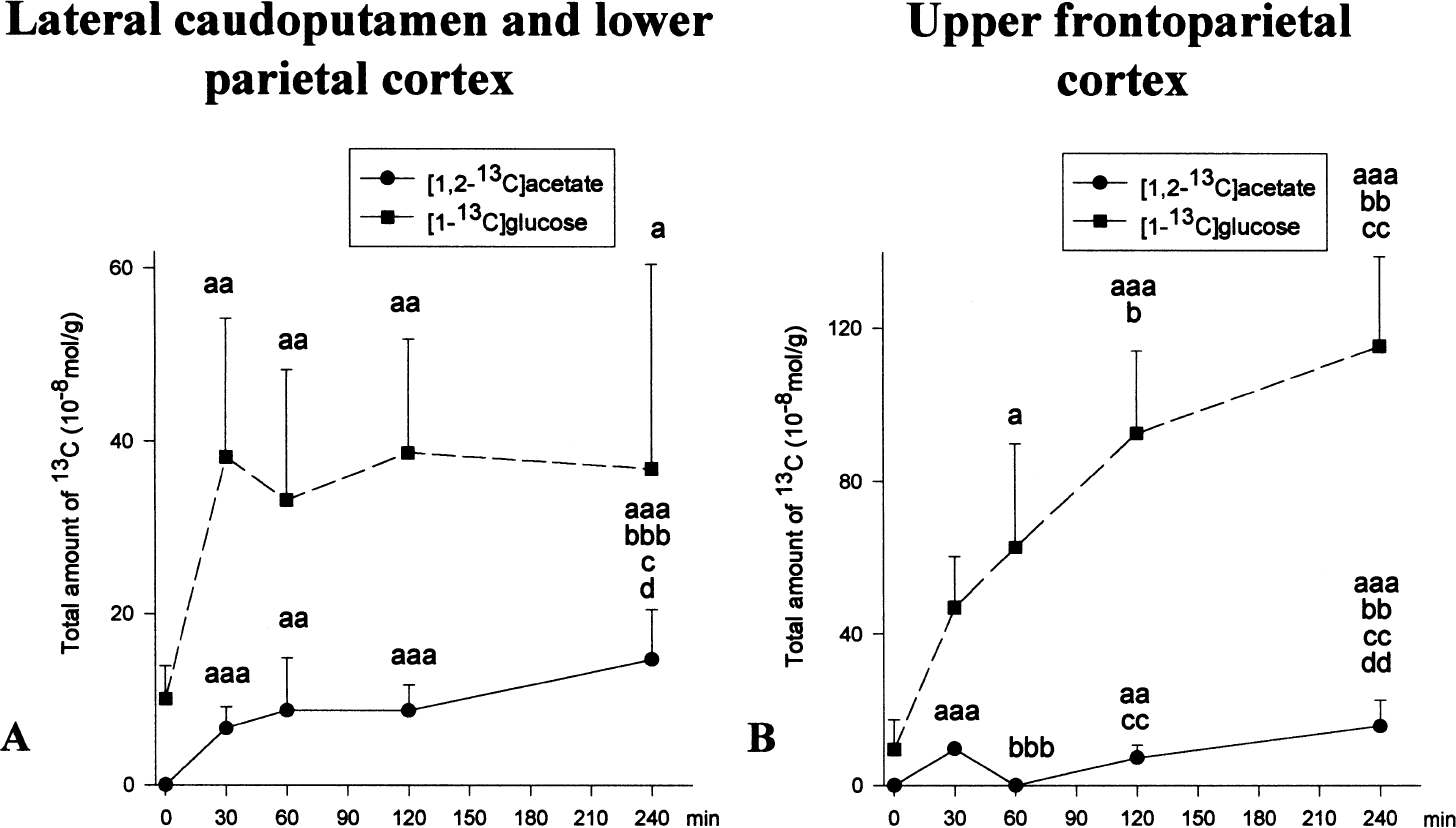
The total amount of [1-13C]glucose and [1,2-13C]acetate in ischemic tissue after MCAO. Time zero represents sham-operated rats allowed to recover for 240 minutes before time of death. The total amount of13C (10−8mol/g tissue) was analyzed with13C MR spectroscopy (see Materials and Methods), and was not corrected for naturally abundant13C (1.1%). The lateral caudoputamen and lower parietal cortex (
In the frontoparietal cortex, [1-13C]glucose content increased steadily and significantly from 60 to 240 minutes MCAO (Fig. 2B). [1,2-13C]Acetate was present at 30 and 120 minutes MCAO, but not at 60 minutes. At 240 minutes, the [1,2-13C]acetate content was significantly increased compared with all previous time points (Fig. 2B).
Glutamate and glutamine.
In the lateral caudoputamen and lower parietal cortex, the total amount of [4-13C]glutamate and [4-13C]glutamine decreased rapidly during the first 60 minutes MCAO, then leveled out before decreasing 240 minutes after MCAO (Fig. 3A). This decrease was most notable in [4-13C]glutamate. The total amount of label in [4,5-13C]glutamate was also significantly reduced from 30 minutes MCAO (Fig. 3C), but the use of acetate compared with glucose as precursor for glutamate was markedly increased throughout the 240 minutes MCAO (Table 1). In glutamine, the total amount of label in [4,5-13C]glutamine was slightly reduced during the initial 120 minutes, then declined steeply at 240 minutes MCAO (Fig. 3C). The acetate-glucose utilization ratio was significantly higher at 120 minutes MCAO, but returned to control levels at 240 minutes (Table 1). In [3-13C]glutamate, the total amount of13C decreased markedly to the same range as the total amount of label in [3-13C]glutamine from 60 minutes MCAO (Fig. 3E). Pyruvate carboxylase activity in glutamate was present at all times after MCAO, but was significantly reduced from 120 minutes MCAO (Table 1). In glutamine, the PC/PDH activity ratio was markedly increased at 60 minutes MCAO, but at 240 minutes MCAO no PC activity was detectable (Table 1).
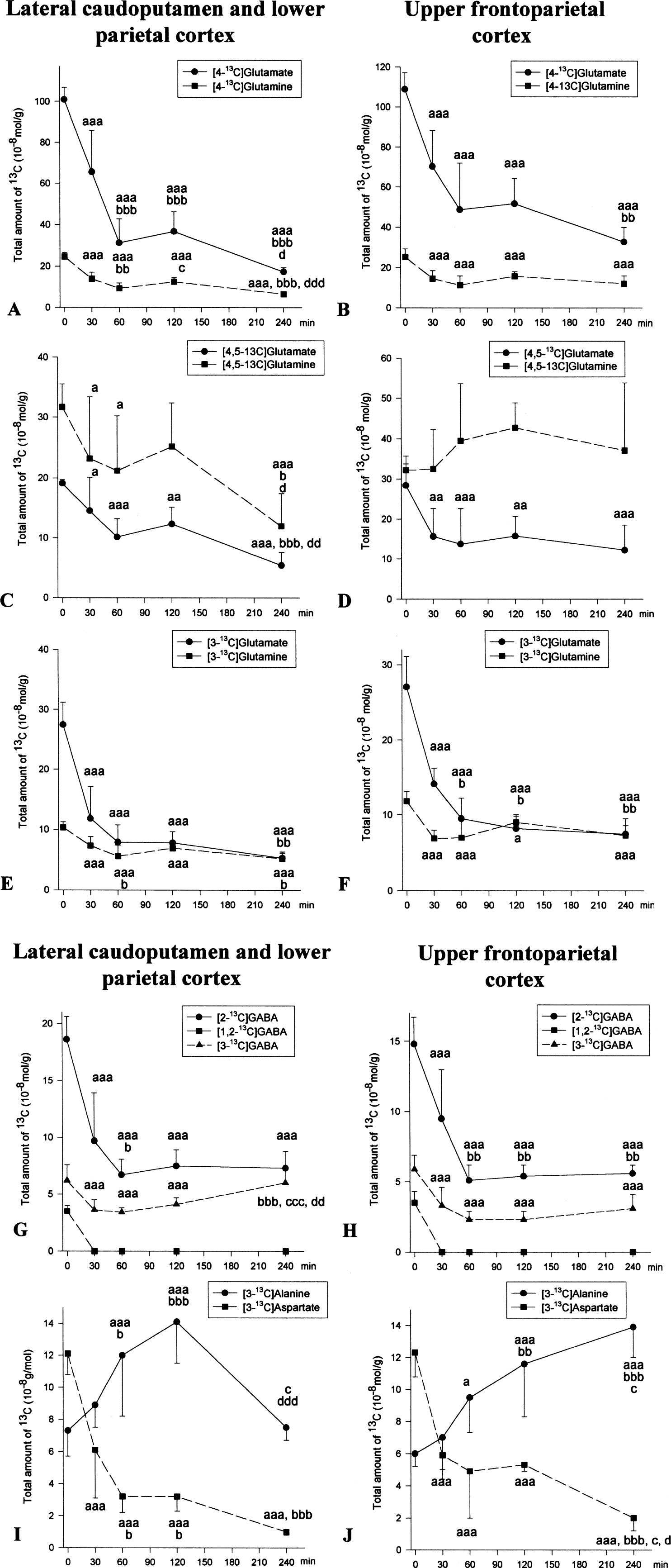
Effect of MCAO on incorporation of label from [1-13C]glucose and [1,2-13C]acetate in amino acids. Time zero represents sham-operated rats allowed to recover for 240 minutes before time of death. The total amount of13C (10−8mol/g tissue) was analyzed with13C MR spectroscopy (see Materials and Methods), and was not corrected for naturally abundant13C (1.1%). All values are mean ± SD; n = 7 in each group. The lateral caudoputamen and lower parietal cortex (
Effects of MCAO on the use of precursors derived from astrocytic metabolism compared with neuronal metabolism in glutamate, glutamine, and GABA formation
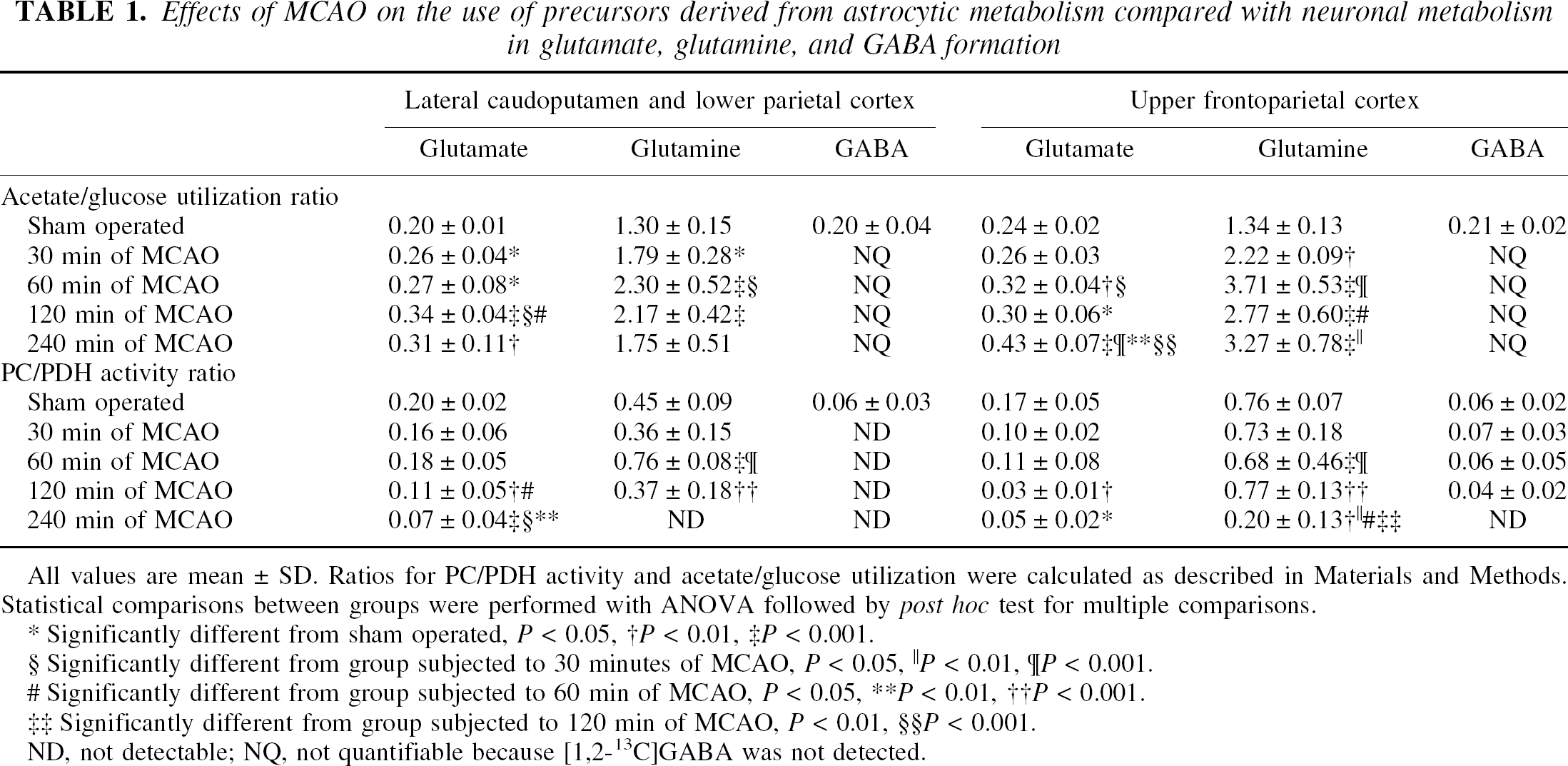
All values are mean ± SD. Ratios for PC/PDH activity and acetate/glucose utilization were calculated as described in Materials and Methods. Statistical comparisons between groups were performed with ANOVA followed by post hoc test for multiple comparisons.
Significantly different from sham operated, P < 0.05
P < 0.01,
P < 0.001.
Significantly different from group subjected to 30 minutes of MCAO, P < 0.05
P < 0.01
P < 0.001
Significantly different from group subjected to 60 min of MCAO, P < 0.05
P < 0.01,
P < 0.001.
Significantly different from group subjected to 120 min of MCAO,P < 0.01
P < 0.001.
ND, not detectable; NQ, not quantifiable because [1,2-13C]GABA was not detected.
The total amount of label in [4-13C]glutamate in the upper frontoparietal cortex was significantly reduced to about the same level the initial 120 minutes MCAO, and then declined further at 240 minutes MCAO (Fig. 3B). In [4-13C]glutamine the total amount of13C was markedly reduced to the same extent at all times after MCAO (Fig. 3B). In [4,5-13C]glutamate the total amount of label was reduced to the same level from 30 to 240 minutes MCAO (Fig. 3D). However, from 60 minutes MCAO acetate became increasingly important as precursor in glutamate formation indicated by the significantly elevated acetate/glucose utilization ratio in glutamate which was almost doubled at 240 minutes (Tbl. 1). There was no decline in the total amount of13C in [4,5-13C]glutamine for 240 minutes MCAO (Fig. 3D), and the acetate/glucose utilization ratio was elevated from 30 minutes MCAO and continued to increase with time (Table 1). The total amount of13C in [3-13C]glutamate declined rapidly and was in the same range as the total amount of13C in [3-13C]glutamine from 60 minutes MCAO (Fig. 3F). In glutamate the PC/PDH activity ratio was significantly lower from 120 minutes MCAO, whereas in glutamine the PC/PDH activity was markedly lowered first at 60 minutes and then at 240 minutes MCAO (Table 1).
γ-Aminobutyric acid.
In the lateral caudoputamen and lower parietal cortex, the total amount of label in [2-13C]GABA was significantly reduced at all times after MCAO (Fig. 3G). In [3-13C]GABA, the total amount of13C decreased significantly after MCAO, but between 120 and 240 minutes MCAO the amount of label in [3-13C]- and [4-13C]GABA (data not shown) increased markedly (Fig. 3G). At 240 minutes MCAO, the total amount of13C was similar in [2-13C]-, [3-13C]- and [4-13C]GABA (Fig. 3G). This finding implied a cessation of GABAergic TCA cycle activity, because the amount of label in [2-13C]GABA must exceed the amount of label found in the subsequent turns using the present infusion scheme. The stop in GABAergic TCA-cycle activity was verified by the lack of incorporation of13C above the natural abundance level of 1.1% in the GABA isotopomers at 240 minutes MCAO. [1,2-13C]GABA was not detected at any time after MCAO, indicating that there was no direct use of [4,5-13C]glutamine in GABA synthesis after MCAO (Fig. 3G). Furthermore, there was no detectable PC activity in GABA (Table 1).
In the upper frontoparietal cortex, the total amount of label in [2-13C]GABA decreased rapidly during the initial 60 minutes MCAO and then leveled out during the remainder of the experimental period (Fig. 3H). The total amount of label in [3-13C]- and [4-13C]GABA (data nor shown) was significantly decreased in this region at all times after MCAO, but not as markedly as for [2-13C]GABA (Fig. 3H). [1,2-13C]GABA was not detected at any time after MCAO, but PC activity was present from 30 to 120 minutes MCAO (Table 1).
Aspartate and alanine.
In the lateral caudoputamen and lower parietal cortex, the total amount of13C in [3-13C]aspartate was approximately halved at 30 minutes MCAO and continued to decline for 240 minutes (Fig. 3I). However, the total amount of13C in [3-13C]alanine was significantly increased at 60 and 120 minutes MCAO, but at 240 minutes had decreased to a level similar to that in sham-operated rats (Fig. 3I).
The total amount of amount of13C in [3-13C]aspartate in the upper frontoparietal cortex was significantly reduced to the same level at 30 to 120 minutes MCAO, followed by a further marked reduction at 240 minutes MCAO (Fig. 3J). In [3-13C]alanine, the total amount of label increased steadily and significantly throughout the 240 minutes MCAO (Fig. 3J).
Effects of isoflurane on cerebral metabolism
Lateral caudoputamen and lower parietal cortex.
In sham-operated rats recovering for 30 minutes after termination of anesthesia, the total amount of label in [3-13C]aspartate, [2-13C]GABA, [4-13C]glutamate, and [4-13C]glutamine was significantly lower compared with sham-operated rats recovering for 240 minutes (Table 2). Moreover, [1,2-13C]acetate (4.1 ± 1.7 10−8mol/g) was detected in all samples from the 30 minutes sham group, but was not found in the 240 minutes group.
Delayed effects of isoflurane on cerebral metabolism in sham-operated rats
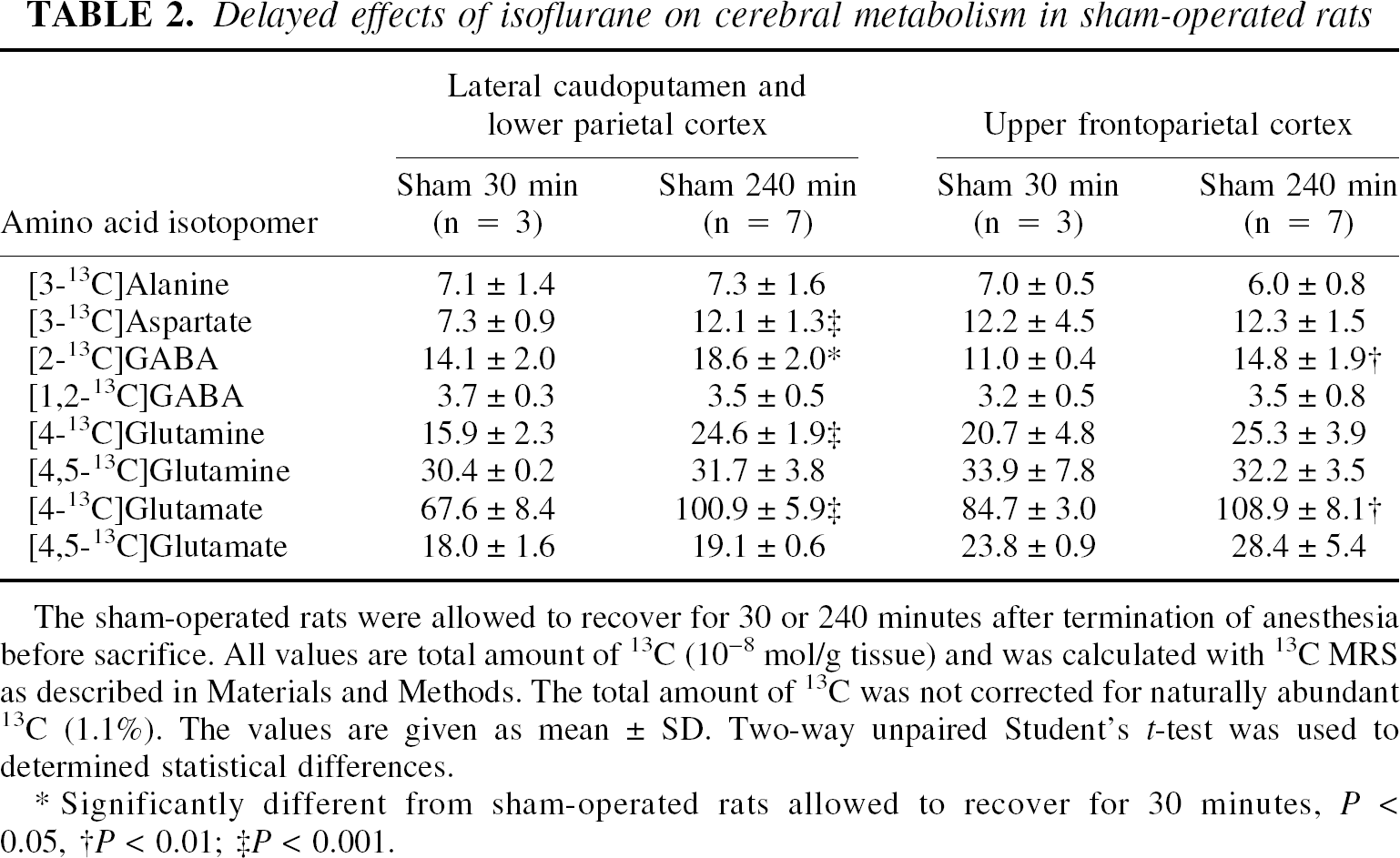
The sham-operated rats were allowed to recover for 30 or 240 minutes after termination of anesthesia before sacrifice. All values are total amount of 13C (10−8 mol/g tissue) and was calculated with 13C MRS as described in Materials and Methods. The total amount of 13C was not corrected for naturally abundant 13C (1.1%). The values are given as mean ± SD. Two-way unpaired Student's t-test was used to determined statistical differences.
Significantly different from sham-operated rats allowed to recover for 30 minutes, P < 0.05
P < 0.01
P < 0.001
Upper frontoparietal cortex.
In the sham-operated group recovering for 30 minutes after anesthesia, the total amount of label was significantly decreased in [2-13C]GABA and [4-13C]glutamate compared with 240 minutes sham group (Table 2). [1,2-13C]Acetate (4.5 ± 1.7 10−8mol/g) was present in the former group but not in the latter sham group.
These results clearly show that isoflurane exerted prolonged effects on neuronal and, to some extent, astrocytic metabolism. Thirty minutes after termination of isoflurane administration, the anesthetic agent still affected incorporation of the label from [1-13C]glucose into some amino acids via PDH. Previously reduced incorporation of the label into amino acids from glucose but not from acetate has been shown during pentobarbital anesthesia (Cremer and Lucas, 1971; Shank et al., 1993). The reduction in glucose metabolism was more prominent in the lateral caudoputamen and lower parietal cortex than in the upper frontoparietal cortex; however, isoflurane affects subcortical structures more than cortical structures (Hansen et al., 1988; Hoffman et al., 1991). Furthermore, the marked reduction of label in [4-13C]glutamine in this region, which is mostly derived from [4-13C]glutamate, implied that isoflurane specifically reduced neuronal glutamate release there. These results show that isoflurane predominantly affects neuronal metabolism and activity. Agreeing with that, astrocytic metabolism was also reduced, as shown by the presence of unmetabolized [1,2-13C]acetate in the 30-minute sham group but not in the 240-minute group. Therefore, the presence of unmetabolized [1,2-13C]acetate in the upper frontoparietal cortex 30 minutes after MCAO was most likely an isoflurane artifact because [1,2-13C]acetate was not detected at 60 minutes MCAO in the same region. Although isoflurane reduced cerebral metabolism 30 minutes after it had been turned off, it was evident only 30 minutes after induction that MCAO exerted its own distinct effects on the cerebral metabolism, as illustrated by the significant reduction in label incorporation from [1,2-13C]acetate into amino acids.
DISCUSSION
The present study clearly shows that MCAO rapidly induces profound changes in neuronal and astrocytic metabolism, and in the trafficking of metabolites between these two cellular compartments. The changes depended both on the duration of ischemia and on the degree of ischemia. There were both similarities and distinct differences in the metabolic response to MCAO in severely ischemic tissue in the lateral caudoputamen and lower parietal cortex, and in moderately ischemic tissue in the upper frontoparietal cortex.
Label incorporation into amino acids from [1-13C]glucose via PDH and the TCA cycle was depressed almost to the same extent in the two ischemic regions, showing the high sensitivity of neuronal metabolism to reduction of CBF. However, decreased neuronal glucose metabolism in MCAO does not indicate irreversible neuronal damage, because reperfusion within 30 minutes salvages substantial tissue volumes from infarction in the lateral caudoputamen, and the upper frontoparietal cortex can be saved with reperfusion within 90 to 120 minutes (Memezawa et al., 1992b; Müller et al., 1995; Garcia et al., 1995). Thus, the development of metabolic insufficiency in neurons appeared to arise from a combination of metabolic disturbances, including alterations in the use of astrocytic precursor.
The most obvious difference between the severely and moderately ischemic tissue was found in the acetate metabolism. Reduced astrocytic metabolism, as indicated by reduced formation of [4,5-13C]glutamine and/or the presence of unmetabolized [1,2-13C]acetate, was detected at 30 minutes in severely ischemic tissue and at 120 minutes MCAO in moderately ischemic tissue. These time points coincide with the time-windows for successful reperfusion of the lateral caudoputamen and lower parietal cortex and of the upper frontoparietal cortex, respectively, suggesting that reduced metabolism in astrocytes may be an indicator of irreversible tissue injury.
MCAO induced changes in the glutamate-glutamine cycle
Lateral caudoputamen and lower parietal cortex.
Profound changes in the glutamate metabolism were revealed in this severely ischemic region. The normal equilibrium between glutamate and glutamine content and, thus, the glutamate-glutamine cycle were disturbed from 30 minutes MCAO. There was a rapid decline in glutamate synthesis from glucose-derived acetyl coenzyme A, representing neuronal-derived glutamate synthesis. At the same time, a reduction was seen in use of glutamine in glutamate formation. This reduction may result from reduced activity in astrocytic or neuronal glutamine transporters in ischemia and/or from reduced PAG activity, because both ammonia accumulation and acidosis, which are present in ischemia, inhibit PAG (Benjamin, 1981; Kvamme et al., 1982; Hogstad et al., 1988). However, astrocytic glutamine was a more important precursor for glutamate than precursors derived from neuronal TCA cycle activity at all times after MCAO, suggesting that astrocytic glutamine contributed significantly to excitotoxicity in vivo. This observation concurs with previous studies using cell cultures (Goldberg et al., 1988; Rosenberg, 1991; Huang and Hertz, 1994). The reduced glutamate synthesis present from 30 minutes MCAO cannot fully explain the reduced glutamate content detected in the same region. The decreased glutamate levels were most likely connected to increased glutamate use, including oxidation and/or loss of glutamate into systemic circulation and cerebrospinal fluid during MCAO. Astrocytic glutamate uptake and subsequent conversion to glutamine was shown by the better preservation of label in [4-13C]glutamine than in [4-13C]glutamate throughout the 240 minutes MCAO, which is in agreement with earlier studies (Aas et al., 1993; Takagi et al., 1993; Swanson, 1992; Torp et al., 1993; Ottersen et al., 1996). Increased entry of glutamate into the TCA cycle in astrocytes was suggested by the better preservation of the total amount of label in [3-13C]glutamine than in [3-13C]glutamate at 60 minutes MCAO and beyond. Previous in vitro and in vivo data have shown increased glutamate metabolism via the astrocytic TCA cycle when extracellular glutamate concentrations were increased and during ischemia (McKenna et al., 1996; Håberg et al., 1998a; Pascual et al., 1998). Glutamate may also be consumed in GABA and glutamine synthesis, and increased alanine synthesis may reduce glutamate content by transamination. In addition, glutamate may be lost into the systemic circulation and cerebrospinal fluid. Elevated glutamate levels have been demonstrated in the cerebrospinal fluid of rats subjected to focal ischemia (Cataltepe et al., 1996), and in both plasma and cerebrospinal fluid of patients with acute ischemic stroke (Castillo et al., 1996). Loss of glutamate from the brain parenchyma gives an indication of the mismatch between glutamate synthesis and consumption and of glutamate release and uptake during MCAO, illustrating the serious metabolic insufficiency in this region. Despite this continuous loss of metabolites, glutamatergic neurons maintained a low level of glutamate synthesis throughout the 240 minutes MCAO.
Upper frontoparietal cortex.
Glutamate metabolism was also affected in moderately ischemic tissue, but the changes in the glutamate-glutamine cycle were somewhat distinctive in this region. As in the severely ischemic tissue, glutamate synthesis was reduced from both neuronal and astrocytic precursors at 30 minutes MCAO and beyond, but in moderately ischemic tissue the contribution from astrocytic precursors compared with neuronal precursors did not increase significantly before 60 minutes MCAO. That neuronal glutamine use in glutamate formation was reduced despite the presence of normal [4,5-13C]glutamine levels, and that the total amount of [4,5-13C]glutamate was at the same level at all times after MCAO, indicates some functional inhibition of either glutamine transporters and/or PAG activity independent of the duration of ischemia. For example, PAG activity may be reduced by the presence of normal glutamate concentrations (Hogstad et al., 1988). From 60 minutes MCAO, glutamine became increasingly important in glutamate synthesis while the neuronal TCA cycle activity gradually declined. Glutamine principally replenishes the neurotransmitter pool of glutamate (Laake et al., 1995), the pool from which glutamate release is considered to originate in moderately ischemic tissue (Takagi et al., 1993; Obrenovitch, 1996). The constellation of gradually declining neuronal metabolic activity and continued replenishment of the neurotransmitter pool of glutamate renders neurons extremely susceptible to glutamate excitotoxicity. There is increasing evidence for a delayed component in glutamate-mediated neuronal damage in cerebral ischemia, and based on the present data it appears to be connected to the continued astrocytic glutamine production and the subsequent neuronal use of glutamine in glutamate neurotransmitter synthesis. The current finding of persistent glutamate synthesis from glutamine and, to some extent, from the neuronal TCA cycle precursors for 240 minutes MCAO implies that administration of glutamate receptor antagonists has to be continued for a prolonged period. In line with this observation is the finding that AMPA (α-amino-3-hydroxy-5-methyl-4-isoxasole propionate) receptor antagonist administration lasting 4 hours after MCAO does not reduce final infarct volume, whereas administration lasting 24 hours salvages the upper frontoparietal cortex (Håberg et al., 1998b). Furthermore, inhibiting glutamine synthetase activity and thereby reducing glutamine synthesis has been shown to reduce infarct size in the cortex of rats subjected to MCAO (Swanson et al., 1990). Finally, because increased extracellular glutamine concentrations stimulate neuronal glutamate release (Szerb and O'Regan, 1985), the increased glutamine concentrations detected from 60 minutes MCAO may have deleterious effects.
Results from the sham-operated rats showed that astrocytic glutamine was a more important precursor for glutamate in the upper frontoparietal cortex than in the lateral caudoputamen and lower parietal cortex. This finding is in agreement with the theory that PAG density and activity is higher in cortex than in caudate-putamen/striatum (Aoki et al., 1991; Wallace and Dawson, 1993).
MCAO-induced changes in GABA metabolism
Middle cerebral artery occlusion completely changed GABA metabolism in severely ischemic tissue in the lateral caudoputamen and lower parietal cortex, and also in moderately ischemic tissue in the upper frontoparietal cortex. [1,2-13C]GABA was not detectable in either region showing a dramatic reduction in the conversion of astrocytic glutamine to GABA after MCAO. Furthermore, the PC activity was undetectable at all times after MCAO in severely ischemic tissue, suggesting the complete cessation of astrocytic precursor use for GABA formation. In moderately ischemic tissue, PC activity was present in GABA from 30 to 120 minutes MCAO, indicating that astrocytic precursors participated to some extent in GABA synthesis. This reduction in glutamine use in GABA formation was at great variance from the increased use of glutamine in glutamate synthesis during MCAO. However, as in glutamatergic neurons, GABA synthesis from neuronal precursors was markedly lowered after MCAO.
The build up of GABA, which can proceed in the absence of functioning mitochondria, was present from 60 minutes MCAO in moderately ischemic tissue and at 240 minutes in severely ischemic tissue. Cerebral ischemia stimulates GABA synthesis via GAD (Sze, 1979; Erecinska et al., 1996), and at the same time inhibits GABA breakdown by GABA transaminase (E.C.2.6.1.19) (Schousboe et al., 1973) consequently increasing the GABA content (Erecinska et al., 1984). Decreased GABA transaminase activity will impede the re-entry of carbon-atoms from GABA into the TCA cycle, thereby decreasing the activity of the GABA-shunt (Balazs, 1970), resulting in loss of TCA cycle intermediates and subsequently also of aspartate (see below). However, in severely ischemic tissue additional mechanisms causing GABA build up may be operating. The substantial increase in unlabeled GABA from 120 to 240 minutes MCAO, which was accompanied by a corresponding decline in glutamate levels, may arise from direct conversion of unlabeled glutamate to GABA taking place either intracellularly or extracellularly. A substantial increase in GABA levels have been demonstrated both in the cells (Torp et al., 1993) and in the extracellular compartment (Matsumoto et al., 1993) during cerebral ischemia. The increased extracellular GABA level is considered to be due to reversal of GABA transporters; however, based on the present results, the possibility that GAD leaks into the extracellular space and leads to extracellular production of GABA from extracellular glutamate cannot be excluded.
In the GABAergic neurons, MCAO compromised the flux through the TCA cycle by a (1) significant decrease or complete stop in the use of astrocytic glutamine, (2) rapid and marked reduction in use of neuronal precursors, and (3) progressive decrease in GABA-shunt activity. At 240 minutes MCAO, no TCA cycle flux was detected in the region rendered severely ischemic in the lateral caudoputamen and lower parietal cortex, suggesting the death of GABAergic neurons. Selective death of GABAergic neurons in the striatum has previously been shown in rats after transient forebrain ischemia (Francis and Pulsinelli, 1982). However, GABAergic neurons in the hippocampus and cortex are considered to be particularly resistant to ischemia and N-methyl- d -aspartate excitotoxicity (Tecoma and Choi, 1989; Gonzales et al., 1992; Johansen and Diemer, 1991). These differences in ischemia susceptibility between different populations of GABAergic neurons may be because GABAergic neurons are diverse in size and function. Based on the present data, GABAergic neurons in the lateral caudoputamen and lower parietal cortex were more sensitive to ischemia than glutamatergic neurons in the same region. However, it is not certain what properties lead to the increased sensitivity of GABAergic neurons in the lateral caudoputamen and lower parietal cortex to focal ischemia. The current findings imply that the complete interruption in the use of glutamine in GABA synthesis in this region may contribute substantially to the death of GABAergic neurons, because the reduction in glucose use and GABA-shunt activity was at least as noticeable in the region with moderate ischemia during the initial 120 minutes MCAO.
Because aspartate is found mainly in GABAergic neurons (Ottersen and Storm-Mathisen, 1985), the MCAO-induced changes in aspartate labeling and content were most likely connected to the changes in GABA metabolism. The significant reduction in aspartate synthesis in both ischemic regions from 30 minutes MCAO and beyond could be connected to the reduced glutamine use in GABAergic neurons, because in vitro data suggest that aspartate is predominantly synthesized from glutamine in GABAergic neurons (McKenna et al., 2000). The rapid decline in aspartate labeling found between 120 and 240 minutes MCAO in the upper frontoparietal cortex coincided with the complete stop in the use of astrocytic precursors, lending further support to the notion that glutamine is important in aspartate synthesis. In addition, aspartate consumption is increased by reduced GABA transaminase activity (Hassel et al., 1998), which was also found in the current study. In summary, aspartate consumption was probably increased because of decreased GABA shunt activity, whereas aspartate synthesis decreased because of reduced glutamine use.
MCAO-induced changes in astrocyte metabolism
Lateral caudoputamen and lower parietal cortex.
In this region of severe ischemia, astrocytic metabolism was reduced at 30 minutes, but was not severely impaired before 240 minutes MCAO. The first sign of declining astrocytic metabolism was reduced acetate metabolism, both oxidative and for use in amino acid synthesis. Still, acetyl coenzyme A synthetase, PC, and glutamine synthetase, all of which are adenosine triphosphate-dependent enzymes, operated throughout the experimental period. All enzymes appeared to maintain a slightly lower level of activity during the initial 120 minutes MCAO, but we detected dramatically reduced activity in the label incorporation via these enzymes at 240 minutes MCAO. No PC activity was detected in glutamine, and only a low level of PC activity was present in glutamate; furthermore, the acetate-glucose use ratio was at control levels in glutamine but increased in glutamate at 240 minutes MCAO. These findings suggest that glutamate metabolism in astrocytes is compartmentalized and that the compartment synthesizing glutamate for glutamine production is more susceptible to ischemia. Alternatively, ischemia reduces the transport of glutamate produced in the astrocytic mitochondria over the mitochondrial membrane to the cytosol, where glutamine synthetase is located. Previously, mitochondrial glutamate accumulation has been demonstrated in astrocytes in global ischemia (Torp et al., 1993). Alanine synthesis may also be considered a marker for astrocytic metabolism because it is preferentially synthesized in astrocytes in vitro (Westergaard et al., 1993). Furthermore, alanine synthesis is considered to increase specifically in astrocytes in cerebral ischemia (Bachelard, 1998). In severely ischemic tissue, alanine synthesis was significantly increased between 60 and 120 minutes MCAO but declined from 120 to 240 minutes MCAO, giving further evidence of rapidly declining astrocyte metabolism at the end of the experimental period.
Ischemic frontoparietal cortex.
In moderately ischemic tissue, astrocytic metabolism was relatively unaffected by MCAO until 120 minutes MCAO, at which time unmetabolized [1,2-13C]acetate appeared. [1,2-13C]Acetate has previously been detected in this region after 90 minutes MCAO (Håberg et al., 1998a). There was reduced PC activity at 60 minutes MCAO, but activity was notably reduced at 240 minutes MCAO. Both acetyl coenzyme A synthetase and glutamine synthetase activity were unaffected by MCAO. In addition, the increasing alanine synthesis present throughout the 240 minutes MCAO gave yet another indication of better preservation of metabolism in astrocytes of moderately ischemic tissue.
CONCLUSION
The present study showed the paradoxical role that astrocytes play in neuronal survival in ischemia. Although the presence of normal astrocytic metabolism appeared to be critical for neuronal survival, it was evident that alterations in the use of astrocytic precursors contributed significantly to neuronal death, albeit through different mechanisms in glutamatergic and GABAergic neurons. In glutamatergic neurons astrocytic precursors sustained the neurotransmitter pool of glutamate despite clear indications of failing neuronal metabolism. This finding was especially notable in the upper frontoparietal cortex (often referred to as the penumbra), thus showing the importance of glutamine in glutamate excitotoxicity. In GABAergic neurons, the complete stop in the use of glutamine in GABA formation in the ischemic core in the lateral caudoputamen and lower parietal cortex may have contributed significantly to the death of these neurons at 240 minutes MCAO. Therefore, neuronal survival during focal cerebral ischemia depends on astrocytic metabolism and neuronal use of astrocytic precursors.
Footnotes
Acknowledgment:
The authors thank Kristin Antonsen and Torild Krogstad for performing the high-performance liquid chromatography analysis at the Institute of Biotechnology, Norwegian University of Science and Technology.