
Abstract
Cerebrovascular events have emerged as a central feature of the clinical syndrome associated with Sars-CoV-2 infection. This increase in infection-related strokes is marked by atypical presentations including stroke in younger patients and a high rate of hemorrhagic transformation after ischemia. A variety of pathogenic mechanisms may underlie this connection. Efforts to identify synergism in the pathophysiology underlying stroke and Sars-CoV-2 infection can inform the understanding of both conditions in novel ways. In this review, the molecular cascades connected to Sars-CoV-2 infection are placed in the context of the cerebral vasculature and in relationship to pathways known to be associated with stroke. Cytokine-mediated promotion of systemic hypercoagulability is suggested while direct Sars-CoV-2 infection of cerebral endothelial cells may also contribute. Endotheliopathy resulting from direct Sars-CoV-2 infection of the cerebral vasculature can modulate ACE2/AT1R/MasR signaling pathways, trigger direct viral activation of the complement cascade, and activate feed-forward cytokine cascades that impact the blood-brain barrier. All of these pathways are already implicated as independent mechanisms driving stroke and cerebrovascular injury irrespective of Sars-CoV-2. Recognizing the overlap of molecular pathways triggered by Sars-CoV-2 infection with those implicated in the pathogenesis of stroke provides an opportunity to identify future therapeutics targeting both Sars-CoV-2 and stroke thereby reducing the impact of the global pandemic.
Introduction
In late 2019, the emergence of a severe acute respiratory syndrome caused by the novel coronavirus Sars-CoV-2 in China rapidly progressed to a worldwide pandemic, infecting millions of individuals and causing numerous deaths. 1 While neurologic complications were previously documented in patients with the 2003 outbreak seen in the clinical syndrome associated with Sars-CoV-1, cerebrovascular complications were uncommon. 2 In contrast, early on in the characterization of the clinical syndrome associated with Sars-CoV-2 infection, cerebrovascular events, predominantly ischemic stroke, appeared as a prominent feature of the COVID-19 clinical syndrome. A number of studies suggest a significantly increased risk of ischemic stroke in those with COVID-19 infections. 3 In a subset of hospitalized patients from the origin of the Sars-CoV-2 outbreak, nearly 5% of these patients had an acute cerebrovascular event, especially those with severe infection. 4 In the United States, anecdotal reports suggest an increase in patients positive for COVID-19 presenting with large-vessel strokes, with many of the patients under the age of 50 and lacking the typical risk factors for stroke 5 (Figure 1). In a New York City hospital network, the incidence of ischemic stroke admission increased seven times compared to a normal flu season, suggesting a fundamental association of Sars-CoV-2, COVID-19, and cerebrovascular events. 6 Another study in pandemic-stricken New York City showed that rates of stroke as a whole were not necessarily higher amongst COVID-19 patients, but the reported causes of stroke were disproportionately cryptogenic and patients had a higher mortality rate than those who had a stroke but were not positive for COVID-19. 7 In the largest collection of neurologic complications of COVID-19 patients, the CoroNerve Study Group in the UK reported a staggering 62% rate of cerebrovascular events in hospitalized patients with COVID-19. 8 This suggests that there may be a link between strokes that occur specifically in the context of COVID-19 and certain individuals may be more susceptible than others.
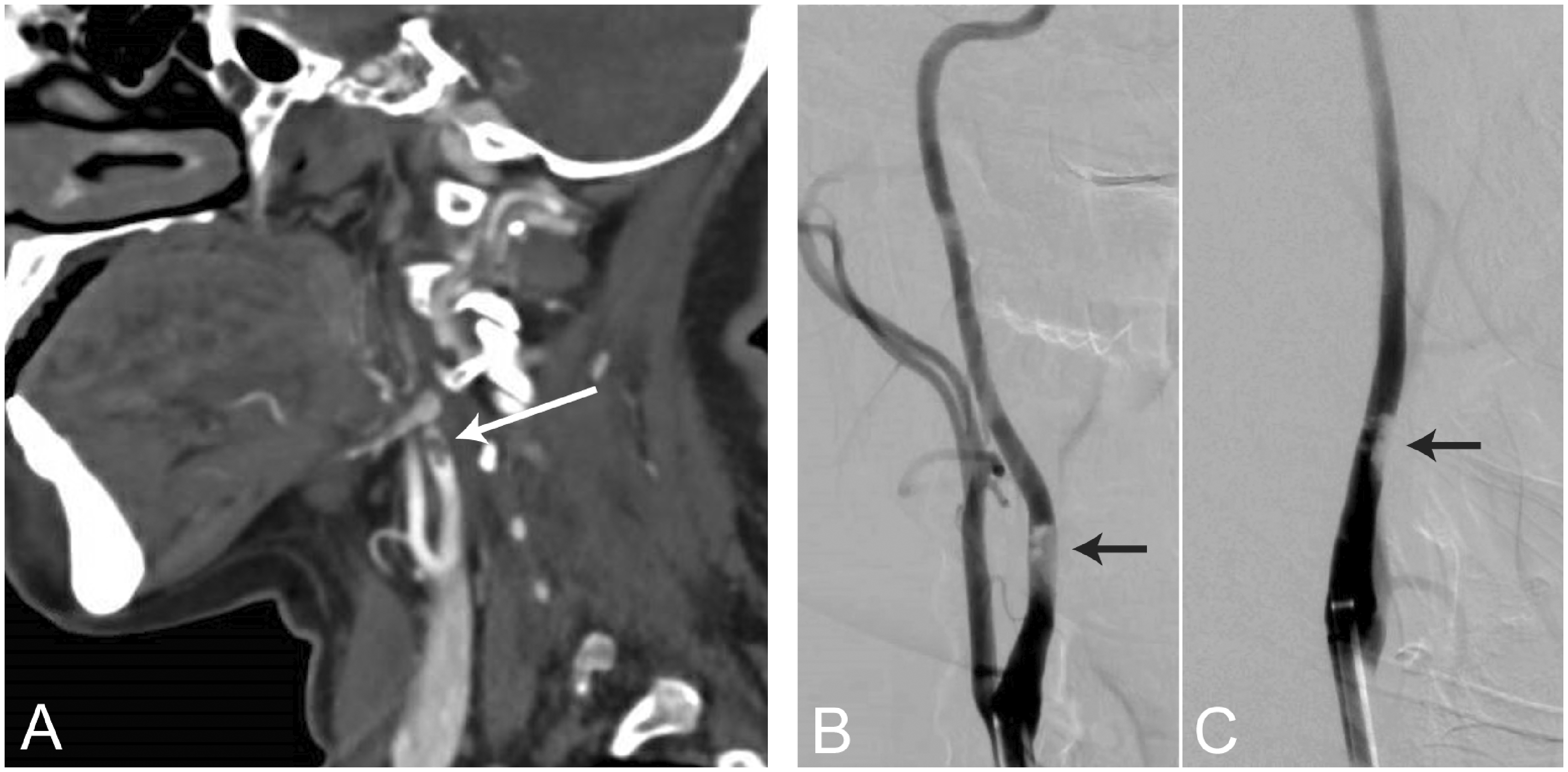
Atypical stroke syndromes associated with Sars-CoV-2 infection. A 43-year-old female with no past medical history presented with left-sided weakness and spatial neglect, but without any respiratory symptoms. CT angiogram (a) and digital subtraction angiograms in the lateral (b) and anterior-posterior (c) views showed partially occlusive thrombus formation on the posterior wall in the cervical segment of the right internal carotid artery, several centimeters distal to the carotid bulb. On admission, polymerase chain reaction (PCR) from a nasopharyngeal swab sample was positive for Sars-CoV-2. Her blood test revealed lymphocytopenia (0.6 x 10 9 cells/L), elevated D-dimer (4,765 µg/L) and IL-6 (3.8 pg/µL) levels. Ultrasound imaging was negative for thrombosis in the heart or deep veins.
Worldwide efforts to develop new treatments and vaccines against Sars-CoV-2 infection are underway and likely to reduce the impact of COVID-19-related strokes. However, a number of critical questions arise when considering this association. What is the most critical factor leading to thrombosis? If Sars-CoV-2 infection triggers systemic coagulation, are there specific pathways in the brain vasculature that may increase the likelihood of localized thrombosis and stroke? Can Sars-CoV-2 directly infect the cerebral endothelium? Are the molecular pathways similar to the primary site of infection in the lung? What therapeutic options may be considered to specifically target COVID-19 stroke? A deeper understanding of the potential mechanisms underlying Sars-CoV-2 infection of the cerebral endothelium can inform stroke pathogenesis both in COVID-19 infections and perhaps in other forms of stroke. With age as a primary driver of stroke incidence, does the presentation of younger stroke patients infected with Sars-CoV-2 provide an opportunity to inform our understanding of the precise role of aging in stroke mechanisms? In this review, we present the existing evidence supporting the susceptibility of the cerebral endothelium to Sars-CoV-2 infection, what is known about the molecular mechanisms unique to the brain’s vasculature that can promote cerebrovascular thrombosis due to viral infection, and potential therapeutic pathways to target COVID-19-associated strokes.
Sars-CoV-2 mediated systemic hypercoagulability as a driver of stroke
One potential explanation for the increased risk of stroke in patients with Sars-CoV-2 infection is the systemic susceptibility for thrombotic events and hypercoagulability. In a study conducted in Wuhan, China, 19% of all patients and 50% of the non-survivor patients that tested positive for COVID-19 were reported to have coagulopathy as an outcome. 9 In addition, non-survivors from this study had significantly elevated D-dimer levels, further reinforcing a systemic coagulopathy as a potential consequence of severe Sars-CoV-2 infection. Another study reporting data across 3 Dutch hospitals showed that nearly 31% of ICU patients positive for Sars-CoV-2 infection had some type of thrombotic event (with around 27% having a venous thromboembolic event, and around 3% having an arterial thrombotic event). 10 Other thrombotic events reported for these patients include acute pulmonary embolism, deep-vein thrombosis, ischemic stroke, myocardial infarction, or systemic arterial embolism, all of which are heavily associated with increased risk of ischemic stroke and other cerebrovascular complications. The association of Sars-CoV-2 infection with systemic prothrombotic is further supported by an extensive post-mortem study of 67 subjects that found evidence of microthrombi in multiple organ systems (including the brain). 11 Some reports have suggested that mechanical thrombectomy and revascularization procedures in patients with virus-associated strokes are more difficult due to various coagulation abnormalities and large amounts of clots in various areas of the brain. 12
Elevated D-dimer and fibrinogen levels have been seen in patients with Sars-CoV-2 infection. 13 Specifically, younger patients under the age of 50 presenting with COVID-19-related strokes and no stroke risk factors have notably elevated D-dimer levels. 14 Because elevation of these thrombotic markers can be associated with stroke and its various subtypes, this is a potential indicator of a systemic shift towards prothrombotic cascades in the body, thus leading to an increased risk of stroke through either local thrombosis or systemic embolism. Systemic treatment with anticoagulants, specifically low molecular weight heparin (LMWH), reduced mortality rates of Sars-CoV-2 patients, further suggesting that a state of hypercoagulability could be contributing to the increased risk of stroke. 15
A number of viruses, including HSV, Dengue, and Ebola, are capable of hijacking the coagulation cascade through direct molecular interaction. 16 Inadvertent incorporation of membrane-bound tissue factor into the viral capsid during viral shedding is known to occur in HSV infection. 17 Some viral coat proteins such as HSV glycoprotein C can directly activate the coagulation cascade and downstream cellular signaling through protease-activated receptors (PARs) making cells more susceptible to viral infection. 18 Similarly, adenovirus serotype 5 can bind factor X to increase cellular attachment and avoid complement-mediated degradation. 19 This type of feed-forward prothrombotic signaling cascade helps to coat infected cells with fibrin, potentially shielding them from immune cell targeting. Importantly, individuals with higher levels of circulating tissue factor harbor a 2-fold increased risk of stroke 20 that may place them in a baseline prothrombotic state and therefore more susceptible to virally-mediated activation of coagulation. Whether similar direct molecular interactions between viral proteins and proteins in the coagulation cascade occurs in Sars-CoV-2 infection is largely unknown but given the high rate of prothrombotic complications including stroke seen in severe COVID-19 infections, this molecular hijacking deserves further study.
Viral-mediated complement activation in Sars-CoV-2 also seems to be a driver of thrombosis. Activation of the complement system can drive coagulation and promote thrombosis, as these two systems work closely together. 21 The N protein of Sars-CoV-2 binds MASP-2, a critical serine protease in the complement pathway, leading to the overactivation of the complement system. 22 In addition, knocking out MASP-2 decreased incidence of thrombosis, indicating that Sars-CoV-2 N protein activation of the complement system could lead to increased risk of thrombosis. In a report of 5 cases of COVID-19, including younger patients without significant medical history or chronic risk factors, microvascular injury and thrombosis was observed and associated with the deposition of terminal complement components. 23 This study also demonstrated that Sars-CoV-2 S protein co-localized with these complement components, suggesting a relationship between Sars-CoV-2 complement activation and increased coagulation, leading to thrombosis. Complement activation is known to play a significant role in human and experimental models of stroke 24 as well as cerebral small vessel disease. 25 Ischemia-induced exposure of neo-epitopes on cerebral endothelium can also trigger complement activation. 26 MASP-2 knockout mice exposed to transient middle cerebral artery occlusions have reduced stroke volumes, improved neurologic outcomes, and reduced C3 deposition in the cortex. 27 This effect can also be preserved by exogenous MASP-2 blockade, suggesting a potential therapeutic pathway to block Sars-CoV-2-mediated cerebrovascular pathology. The synergy between Sars-CoV-2 and localized activation of the complement cascade in the cerebral vasculature may reinvigorate the therapeutic targeting of complement as an adjunctive stroke treatment.
It is clear that Sars-CoV-2 is contributing to a state of systemic hypercoagulation through various mechanisms, including activation of the complement system, which can ultimately lead to stroke. While these may explain a subset of strokes that are occurring or may explain general systemic susceptibility to thrombotic events, it is also possible that COVID-19-related strokes are due to in situ thrombosis resulting from direct Sars-CoV-2 infection of the cerebral endothelium. In the small vessels of the brain, Sars-CoV-2 also may be causing endothelitis with thrombotic microangiopathy and microbleeds in pattern consistent with leukoencephalopathy. 28 This raises key questions: are there brain-specific molecular pathways that may facilitate Sars-CoV-2 infection of the cerebral endothelium and are they perhaps primed by chronic stroke risk factors?
Potential mechanisms of cerebral endothelial susceptibility
In addition to its primary infectious route into the body through the pulmonary alveoli, systemic vascular endothelial infection seems to be a hallmark of Sars-CoV-2 infection. 29 In the lung, ACE2 is the primary cellular receptor for the spike S protein of Sars-CoV-2. Endothelial membrane disruption and microvascular occlusion in the lungs, paired with subsequent angiogenesis, suggest that infected tissues may be experiencing vascular damage and increasing the risk for thrombotic events. 30 Direct viral invasion of the cerebral endothelium could be a significant driver of Sars-CoV-2-mediated stroke. A number of molecular pathways are implicated in endothelial complications following Sars-CoV-2 infection including vascular leakage through ACE2-mediated kallikrein-bradykinin pathway, sphingosine 1 phosphate receptor 1-mediated amplification of cytokine release, and VEGF-mediated angiogenesis. 31 Yet, given the unique biology of the brain endothelium, which of these pathways are relevant to the brain and stroke and what molecular pathways unique to brain vasculature are involved in Sars-CoV-2 infection remain unknown.
Viral entry via furin-mediated and plasmin-mediated S protein cleavage
Furin, a serine protease that has been previously implicated in highly pathogenic forms of influenza virus, has recently been found to be important in the binding of Sars-CoV-2 S protein to ACE2. 32 Furin can pre-activate the S protein of the virus through furin-dependent cleavage, thereby increasing the ability of Sars-CoV-2 in binding certain ACE2-expressing cells. 32 This furin-dependent cleavage of Sars-CoV-2 S protein reduces the dependency on host cell interactions for viral entry and may facilitate infection in cell types that have low expression of suggested viral entry co-factors.
Furin protein expression levels have been shown to be high in the brain, suggesting an intrinsic susceptibility of the cerebral endothelium to Sars-CoV-2 penetration. 33 Furin expression within the brain has specifically been seen in the pericytes and capillary endothelial cells, with robust detection on the luminal surface of continuous capillary endothelial cells. 34 Furin inhibition can prevent inflammation-induced hypoxic conditions that can break down the blood-brain barrier. 35 Furin is also thought to be important in the worsening of atherosclerotic lesions by inhibiting the activation of matrix metalloproteinases. 36 Inhibition of furin was shown to reduce vascular endothelial inflammation and limit the expression of various cytokine genes in vitro. 36 Furin has also been implicated in various pathways downstream of the endothelium relating to stroke, as it has been found to promote pathways that lead to NMDA calcium excitotoxicity. 37
Another suggestion that furin may play an important role in the cerebrovascular susceptibility to Sars-CoV-2 infection is the lack of TMPRSS2 expression in the brain. TMPRSS2 is a serine protease that cleaves the S protein of the Sars-CoV-2 virus, priming it for its entry into the cell. 38 TMPRSS2 shows nominal expression in the brain. 39 Detailed analysis using both ACE2 and TMPRSS2 gene expression levels suggests that oligodendrocyte progenitor cells and astrocytes are low level expressors of both genes. However, the degree to which these genes are robustly regulated at the gene expression level is not known. Surface protein expression of ACE2 and TMPRSS2 may be more dynamic and variable. In the absence of robust TMPRSS2 in the brain, furin may be the primary activator of the S protein of Sars-CoV-2 and a key co-factor for cerebrovascular susceptibility of infection. This evidence of furin expression, furin-dependent processes, and the effects of furin inhibition in the brain during stroke, along with the unique observation that furin can cleave the Sars-CoV-2 S protein (a quality not associated with Sars-CoV-1), suggests that unique pathways of viral entry in the brain and cerebrovasculature may exist.
In addition to furin, another important protein in the cleavage of the Sars-CoV-2 S protein is plasmin. Plasmin is an important down-regulator of stroke pathophysiology: it is responsible in promoting fibrinolysis, subsequently breaking down clots. tPA-mediated activation of plasmin is the primary treatment for ischemic stroke. Sars-CoV-1 S protein was previously shown to be cleaved by plasmin, enhancing ACE2-mediated viral entry into various cell types. 40 Though there are critical structural differences between S protein of Sars-CoV-2 compared to Sars-CoV-1, 41 plasmin is predicted to bind the S protein of Sars-CoV-2. 42 Plasmin activation in response to systemic hypercoagulation could potentially increase virulence, as shown for influenza, 43 and promote cerebrovascular infection. While this data suggests that plasmin may increase virulence of Sars-CoV-2, this is still something that needs to be studied further. With various fibrinolytic therapies being utilized for the treatment of pulmonary complications of Sars-CoV-2, understanding the relevant molecules that mediate viral entry of Sars-CoV-2 into cerebral endothelia and potentially promote in situ thrombosis in the cerebral vasculature is critical.
Viral entry via ACE2 in endothelial cells and pericytes causes a weakened blood-brain barrier
One of the major risk factors for stroke, hypertension, has been found to be heavily associated with poor prognosis in Sars-CoV-2 infection. 44 When Sars-CoV-2 binds to ACE2, it disrupts the normal functioning of the renin-angiotensin system (RAS) in regulating blood pressure throughout the body. 45 Within the nervous system, neurogenic hypertension has been shown to be induced by ACE2 shedding induced by ADAM-17. 46 ACE2 catalyzes the cleavage of Angiotensin-II (AngII) into Angiotensin-1,7 (Ang-1,7), shifting renin-angiotensin signaling away from AngII-mediated, vasoconstrictive and pro-inflammatory Angiotensin-1 receptor (AT1R) activation towards Ang-1,7-mediated AT2R signaling that is thought to be vasodilatory and anti-inflammatory. 47 Previous work has shown that the non-AT1R arm of the renin-angiotensin system acting through Angiotensin-1,7, which binds to the Mas receptor (MasR), has downstream neuroprotective effects against ischemic injury and stroke.48,49 Concurrent AT2R and MasR activation improves cerebral blood flow after stroke.50,51 AT2R knockout mice have enlarged infarct size, worsened recovery, and a blunted response to AT1R inhibition, suggesting a critical role for AT2R in regulating cerebral blood flow during ischemia. 52 This brain-specific alteration in ACE2/AT2R/MasR signaling can indirectly promote inflammatory signaling in the cerebral endothelium. 53 These inflammatory changes triggered by a shift in AT2R/MasR signaling may weaken blood-brain barrier endothelial integrity and promote localized thrombosis.
Prior work indicates that ACE2 mRNA and protein are expressed in the endothelial cells of the brain. 54 The avid binding of Sars-CoV-2 to ACE2 likely accelerates ADAM-17-mediated cleavage of ACE2 as was noted with Sars-CoV-1. 55 Furthermore, coronavirus S protein binding of ACE2 downregulates ACE2 protein expression. 56 This may further attenuate the neuroprotective effects of Angiotensin-1,7 signaling. With ACE2 expression known to play a role in stabilizing and potentially reducing atherosclerosis, 57 changes in ACE2 expression on the surface of the cerebral endothelium may destabilize intracranial atherosclerotic lesions. This highlights a critical need to better understand the cerebrovascular distribution of ACE2 expression and how stroke risk factors may potentiate its local vascular expression.
Another potential route for Sars-CoV-2-mediated infection of the cerebral vasculature is through infection of mural support cells including pericytes. Pericytes are a critical cell-type for the integrity of the blood-brain barrier (BBB) and play increasingly recognized roles in stroke injury and chronic BBB maintenance. 58 They are also implicated in the pathogenesis of Sars-CoV-2-associated myocarditis. 59 Single-cell sequencing databases of the mouse brain vasculature suggest that ACE2 is a predominant marker gene for pericytes and vascular smooth muscle cells. 60 Recent work from a collection of single-cell sequencing databases from brain also indicates that ACE2 is expressed in both human brain endothelial cells and pericytes. 61 Comparatively little is understood about ACE2 biology in pericytes. When pericytes are partially lost by genetic deletion of PDGFRβ, fibrin and platelet markers are upregulated, promoting increased thrombotic activity and significant BBB disruption.62,63 A number of pericyte-endothelial signaling cascades including TGF-β and Tie-2, both critical to BBB integrity and vascular integrity, 64 are potentially disrupted by viral infection of pericytes. Other viruses including Japanese encephalitis virus (JEV) and human cytomegalovirus (CMV) can target brain pericytes, 65 degrade the BBB 66 and drive inflammatory cascades including up-regulation of IL-6. 67 Chronic compromise of pericyte health and BBB integrity by stroke risk factors including aging, may increase the susceptibility of pericytes to direct viral invasion by Sars-CoV-2 and pericyte-endothelial interactions could prove critical to understanding COVID-19 stroke risk.
A catastrophic consequence of a weakened BBB is the potential for ischemic stroke lesions to progress to include a hemorrhagic component that increases stroke severity and worsens outcome. This appears to be common in Sars-CoV-2-related strokes with a 20.8% hemorrhagic transformation rate in pandemic-stricken New York City. 68 Hemorrhagic stroke may also be prominent due to the increasing trend of treating Sars-CoV-2 infections with therapeutic anticoagulation. 69 Down-regulation of ACE2 by Sars-CoV-2 binding may trigger relative down-regulation of the protective effects of ACE2/AT2R/MasR signaling and weaken the BBB.70,71 Alternatively, endothelial invasion of the virus may activate a variety of inflammatory cascades that are known to impact BBB integrity. As in the case of identifying molecular pathways promoting viral susceptibility, recognition of the influence of these downstream signaling mechanisms on BBB integrity may inform our broad understanding of hemorrhagic stroke.
Downstream inflammatory pathways initiating and perpetuating ischemic injury
Inflammatory processes, including the systemic or localized onset of a cytokine storm syndrome, could also be important in the increased risk of stroke amongst patients with Sars-CoV-2 infection.72,73 Inflammatory contributions to thrombosis through the local release of cytokines can accelerate vascular endothelial damage, and reciprocally, thrombosis can also worsen inflammation, leading to a vicious feed-forward cycle of cellular damage. Severe Sars-CoV-2 has similar clinical features to macrophage activating syndrome (MAS) and secondary hemophagocytic lymphohistiocytosis (HLH), 9 both of which are syndromes characterized by runaway inflammation and hypercoagulability in end-organ tissues and are associated with stroke.74,75 Evidence from the lung suggests that a similar immunothrombosis cycle is dramatically provoked by Sars-CoV-2 infection. Whether Sars-CoV-2 infection of the cerebral vessels triggers the same response and through what mechanisms remains unclear. The existing literature about the hyperinflammatory response of Sars-CoV-2 juxtaposed with the known action of these cytokines on the brain endothelium suggests this virus can trigger cerebrovascular immunothrombosis.
Early on in the COVID-19 outbreak, elevated levels of interleukin-6 (IL-6) amongst non-survivor patients compared to patients who survived were observed. 9 IL-6 is a pleotropic cytokine and a known inflammatory marker driving JAK/STAT signaling cascades that are generally thought to worsen ischemic stroke. 76 However, mechanistic studies suggest a more nuanced role for IL-6 with IL-6-null mice demonstrating improved long-term outcomes after stroke associated with pro-angiogenic effects of IL-6 produced locally by resident brain cells. 77 As a proximate cause of stroke, increases in circulating IL-6 provoked by Sars-CoV-2 infection may be promoting increased cerebrovascular AT1R signaling and associated endothelial dysfunction. 78 Establishment of a feed-forward cycle of endothelial production of IL-6 is also possible and may be associated with threshold levels of IL-6 that can indicate the severity of Sars-CoV-2 infection. 79
The IL-1 family of pro-inflammatory cytokines are also heavily implicated in Sars-CoV-2 and dramatically upregulated in severe Sars-CoV-2 infection. 80 IL-1 has also been associated with inflammation in stroke at various stages, including prior to and after the occurrence of a stroke. 81 IL-1β-driven systemic inflammation is associated with comorbid risk factors, such as atherosclerosis, hypertension, and diabetes, and can increase the risk of stroke and chronic cerebrovascular disease. Targeting of IL-1β using canakinumab may reduce stroke. 82 IL-1 signaling cascades can perpetuate detrimental inflammatory processes following the onset of a stroke, including poorly regulated angiogenesis and the release of additional pro-inflammatory cytokines.
These inflammatory cascades are implicated in fulminant acute respiratory distress syndrome and have been observed in Sars-CoV-2 infected patients with severe infections accelerated by the so-called “cytokine storm” triggered by the novel coronavirus. This robust feed-forward activation of cytokine signaling is in part regulated by NF-κB, a transcription factor whose canonical signaling pathway is provoked by innate inflammatory signals such as IL-1 and various viral infections 83 and has also been tied to stroke pathology. NF-κB has been associated with the development of atherosclerotic plaques and the subsequent plaque rupture pathways that can lead to stroke. 84 Additionally, within the cerebral vasculature, activation of NF-κB can promote inflammation and neuronal loss in the context of stroke, while pharmacologic inhibition of the NF-κB pathway is associated with reduced stroke size and severity in animal models. 83
STAT3 is a transcription factor that is activated by IL-6 and necessary for activation of NF-κB inflammatory pathways. 85 The IL-6-STAT3 axis is being studied as a potential promoter of runaway inflammatory responses in Sars-CoV-2 86 and cerebral endothelial expression of IL-6R triggering is required for the brain’s pyrogenic response. 87 In normal endothelial cells, STAT3 regulates angiogenesis through VEGFR signaling and may play a protective role after ischemic injury. 88 However, sustained activation of STAT3 may be detrimental in stroke with evidence suggesting that inhibition of an inflammatory pathway involving JAK2 and STAT3 conferred neuroprotection following ischemic injury in rat brains. 89 The extent to which these transcription factor pathways are activated directly by Sars-CoV-2 infection or indirectly by inflammatory signaling cascades remains unknown but controlling runaway inflammation within the cerebral vasculature will likely prove critical to the pathogenesis of virally-mediated strokes.
The cell surface C-C chemokine receptor type 5 (CCR5), known for its co-factor role in mediating HIV infection, also appears to participate in the feed-forward cytokine acceleration predominant in Sars-CoV-2 infection. 90 CCR5 functions to regulate T-cell migration towards sites of inflammation where they become active. CCR5 is also expressed by cerebral endothelial cells and can mediate other neuroinfectious syndromes that affect the cerebral vessels. 91 Cerebral ischemia promotes CCR5 expression in T-cells and is necessary for T-cell homing to cerebral endothelial cells that up-regulate CCR5 ligands in a model of stroke. 92 Interestingly, CCR5 may also promote brain repair after stroke by regulating microglial pruning of synapses in peri-infarct brain tissue. 93
Therapeutic strategies to target Sars-CoV-2 action in the cerebral vasculature
A number of therapeutic strategies for Sars-CoV-2 infection are currently in clinical trial with a focus on slowing severe lung infection and reducing the complications associated with severe infection. In this section, we focus on therapeutic approaches to protect the cerebral vasculature from Sars-CoV-2 infection and the downstream molecular pathways triggered by infection in their context as stroke therapeutics (Table 1).
Therapeutic strategies targeting mechanisms implicated in both Sars-CoV-2 infection and stroke.

Therapeutic modalities under study for effectiveness in Sars-CoV-2 infection are listed along with mechanism of action. Evidence for benefit in stroke was classified into five categories: 1) Not studied in stroke; 2) Pre-clinical evidence of benefit in stroke; 3) Clinical trial evidence of benefit in stroke; 4) No clinical evidence of benefit in stroke; and 5) Evidence of potential harm.
Pathways to block viral binding
Interfering with Sars-CoV-2 viral entry is an attractive therapeutic approach that has evidence of efficacy from HIV. Because the mechanisms of viral entry for Sars-CoV-2 appear similar to those for Sars-CoV-1, involving viral S protein binding to human ACE2, the development of therapeutic approaches in this area has occurred at lightning speed. Monoclonal antibodies against viral proteins including the Sars-CoV-2 S protein are in development and beginning to enter clinical trials. If effective, these drugs should reduce the ability of the virus to enter the cerebral vessels and therefore reduce the risk of stroke associated with direct viral invasion.
Another approach to prevent viral binding and cellular entry is the use of the soluble form of the Sars-CoV-2 receptor, ACE2. 94 Soluble hACE2 was already being developed as a therapeutic for ARDS 94 and it can block Sars-CoV-2 infection of blood vessels in vitro in tissue organoids. 95 Because of the earlier development of a recombinant soluble hACE2 molecule as a clinical grade therapeutic, this viral blocking approach has also already entered clinical trial. By targeting the primary interaction of Sars-CoV-2 with target cells, this approach may also reduce viral infection in brain endothelia. Close monitoring of the reduction in Sars-CoV-2-mediated strokes in this clinical trial will be important to establish this specific mechanism.
Targeting blockade of the necessary co-factors that mediate viral entry is also being explored. Furin inhibitors can be used to prevent viral binding by blocking furin from cleaving the S protein. Furin inhibition has previously been shown to prevent endothelial dysfunction and blood-brain barrier inflammation in hypoxic conditions, indicating that it can be used safely in the brain. 35 In their short-term use, furin inhibitors have been shown to be safe 96 and may be effective against Sars-CoV-2 virus. However, furin is also responsible for several normal functions in embryonic development, such as ventral closure, indicating that its long-term use would need to be carefully monitored and studied further. 97
Non-selective inhibitors of TMPRSS2, including camostat, already had therapeutic indications and thus quickly entered clinical trials to prevent viral entry of Sars-CoV-2 infection. 98 With nominal expression of TMPRSS2 in the brain, blocking this viral entry co-factor is unlikely to prevent Sars-CoV-2 infection in the cerebral vessels. New pre-clinical studies are testing the efficacy of a broader spectrum serine protease inhibitor that would inhibit both TMPRSS2 and furin. 99
Anticoagulation therapies, such as low molecular weight heparin (LMWH), may be promising in preventing stroke risk by limiting coagulopathy and inflammation. 15 While tissue plasminogen activator (tPA), a commonly used acute ischemic stroke treatment, has shown some promise in temporarily decreasing the severity of Sars-CoV-2 infection by improving blood flow in the lung, LMWH may be a safer fibrinolytic alternative for patients noting that plasmin may increase the virulence of Sars-CoV-2. 100
Modulating downstream renin-angiotensin signaling
By binding to ACE2 as a cellular receptor, Sars-CoV-2 can also shift the balance of AT1R/AT2R signaling towards the pro-inflammatory, pro-thrombotic, and vasoconstrictive pathways downstream of AT1R. Agonism of the AT2R signaling, including Ang1,7-mediated activation of MasR, may block the feed-forward inflammatory signaling that Sars-CoV-2 appears to be adept at. Direct agonism of MasR using AVE0991 protected neurons from ischemic injury in vitro but failed to show benefit in functional or histologic outcomes in a mouse MCAO model of stroke. 49 Nonetheless, the synergism of the evidence around AT2R/MasR signaling in both Sars-CoV-2 infection and cerebrovascular disease suggests that modulating renin-angiotensin signaling may prove particularly effective in preventing the consequences of Sars-CoV-2 infection in the brain.
Blocking downstream inflammatory pathways
Various immunosuppressive drugs could be beneficial in not only alleviating symptoms of severe infection, but also decelerating the cytokine surge associated with a novel viral infection. Through receptor inhibition, these therapies aim to specifically target receptors throughout the body but can also be expected to reduce the significance of inflammatory signaling on the cerebral vessels and the blood-brain barrier.
Tocilizumab, a recombinant anti-human IL-6 receptor monoclonal antibody, has been shown to safely reduce severe Sars-CoV-2 infection and decrease mortality. 101 Methotrexate, an anti-inflammatory drug, also helps to decrease IL-6 levels in T cells and could be used to suppress cerebrovascular susceptibility. 102 Low-dose methotrexate was not effective in reducing stroke risk prospectively, 103 though retrospective data indicate chronic use in rheumatoid arthritis is associated with a reduced rate of ischemic stroke. 104 With IL-6 positioned as a key regulator of virus-associated cerebrovascular events, these therapies could prove beneficial in preventing virally-mediated stroke. The anti-gout medication, colchicine can also reduce IL-6 levels and may have benefit in treating cardiovascular disease including reducing ischemic stroke rates. 105 Because of this IL-6 lowering effect, colchicine has also entered clinical testing in severe Sars-CoV-2 infection. 106 If these IL-6 focused therapeutics prove effective in reducing Sars-CoV-2 mediated strokes, re-examination of the role of IL-6 in ischemic stroke seems warranted.
High-dose Anakinra, an IL-1 receptor antagonist, has also been shown to be safe and is associated with improvement of Sars-CoV-2 infection. 107 Anakinra has been studied in acute ischemic stroke and is shown to reduce inflammatory markers including IL-6. Despite a robust effect on plasma levels of inflammation, Anakinra was not associated with a favorable outcome after stroke. 108 Additional analysis suggested a potential benefit when correcting for baseline IL-6 levels, but in routine ischemic stroke, IL-1 receptor antagonism may prove to upstream in the inflammatory cascade resulting in off-target effects. However, in the setting of more widespread inflammation triggered by Sars-CoV-2 infection, there may still be benefit of anti-IL-1 targeting to prevent cerebrovascular complications.
Targeted antagonism of CCR5 with leronlimab was initially developed as an HIV therapeutic. Its use in Sars-CoV-2 infection is designed to prevent the cytokine storm seen in severe infection and may also reduce viral load. 90 Leronlimab has not been tested in stroke but CCR5 antagonism may have a role in stroke treatment. Based on its ability to promote new axon formation and motor recovery after stroke, the oral CCR5 antagonist, maraviroc, is currently being evaluated to promote recovery after stroke.
JAK inhibitors can help prevent inflammation related to Sars-CoV-2. 109 Ruxolitinib, an inhibitor of the JAK1/JAK2 pathway, has been suggested as a systemic therapy for Sars-CoV-2 infection as an attenuator of hyperinflammation. Because of the involvement of JAK2 and its downstream activation IL-6 and STAT-3 in the pathophysiology of stroke, JAK inhibitor therapy could also decrease the risk of stroke and reduce severe infection.110,111 Thromboembolism has been seen in some patients using the JAK inhibitor Barcitinib for rheumatoid arthritis, but other JAK inhibitors, such as Tofacitinib, do not seem to pose this risk. 112 Because of this potential complication, JAK inhibitors are least likely to show benefit in reducing stroke risk associated with Sars-CoV-2 infection.
Conclusion
The COVID-19 pandemic has spurred a number of novel research avenues harnessing insights from existing datasets, including massive single-cell sequencing resources. With cerebrovascular diseases as a prevalent cause of disability and mortality, understanding the association of stroke with this novel virus can provide insights into targeted treatments. Even though virus-associated strokes and cerebrovascular complications are still considered to be uncommon, it is increasingly clear that Sars-CoV-2 is a disease with a predominant component affecting the vasculature through direct infection, hijacking of inflammatory cascades, and a feed-forward cycle that can promote systemic and localized thrombosis (Figure 2). This is evidenced by increased incidence of COVID-19-related strokes in younger patients who lack typical risk factors for stroke, suggesting a unique cerebrovascular susceptibility that needs to be further understood.
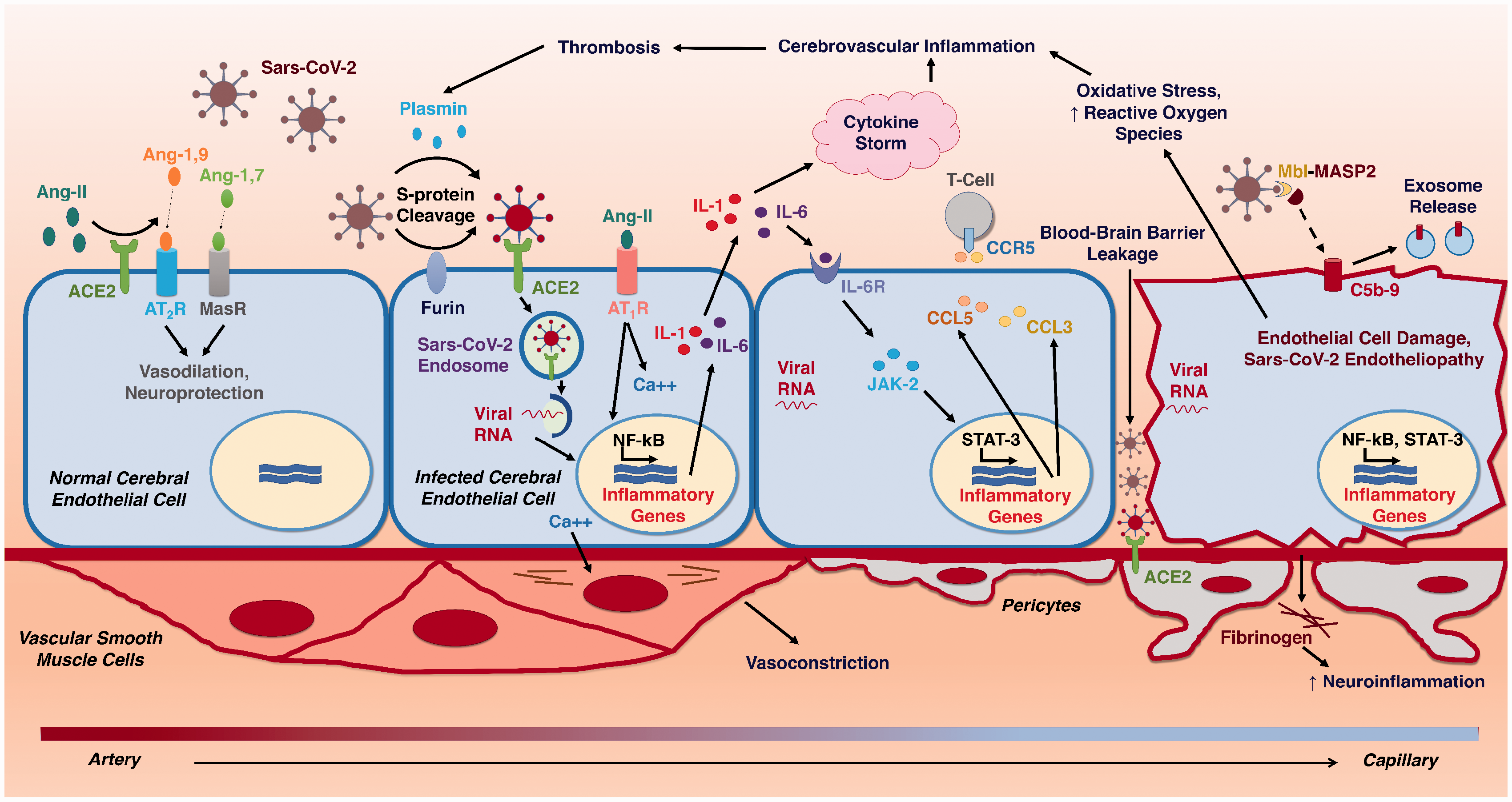
Pathophysiologic Mechanisms of Cerebral Endotheliopathy and Stroke due to Sars-CoV-2. Schematic diagram illustrating the progressive molecular cascades triggered in the cerebral endothelium by Sars-CoV-2 infection. ACE2 – Angiotensin-converting enzyme 2; Ang-1,7 – Angiotensin-1,7; Ang-1,9 – Angiotensin-1,9; Ang-II – Angiotensin II; AT1R – Angiotensin-II Receptor, Type 1; AT2R – Angiotensin-II Receptor, Type 2; C5b-9 – Complement C5b-9; Ca++ - Calcium2+; CCL3 – C-C Motif Chemokine ligand 3; CCL5 – C-C Motif Chemokine ligand 5; CCR5 – C-C chemokine receptor type 5; IL-1 – Interleukin-1; IL-6 – Interleukin-6; IL-6R – Interleukin-6 Receptor; JAK-2 – Janus kinase 2; MASP-2 – Mannose-binding protein-associated serine protease 2; MasR – Mas receptor; MBL – Mannose-binding lectin; NF-kB – Nuclear factor kappa B; STAT-3 – Signal transducer and activator of transcription 3.
The developing body of literature around Sars-CoV-2 infection suggests that there are certain brain-specific mechanisms and inflammatory pathways that may specifically be increasing the risk of acute cerebrovascular events in cases of severe infection. Viral entry into the brain endothelial cells can be mediated by proteins present in the cerebral vasculature but also increased by conditions that inherently increase the risk for stroke such as hypertension-induced changes in ACE2 expression and atherosclerotic lesions with elevated levels of furin, plasmin, and inflammatory signaling cascades. The pro-inflammatory triggered by Sars-CoV-2 include IL-1, IL-6, and STAT-3 as well as NF-κB; all of which have had previous associations with stroke. These processes suggest a strong susceptibility of the cerebral endothelia to Sars-CoV-2-mediated molecular pathways that can increase the risk of stroke. One potential way of studying this better is through the creation of dynamic models of the brain vasculature using endothelialized 3 D printed models. These ex vivo models of the cerebral vasculature can play an important role in helping to elucidate the brain-specific pathways targeted by Sars-CoV-2. 113 A greater focus on Sars-CoV-2-mediated effects on the brain will be crucial in preventing and treating virus-related strokes.
With research in this emerging field evolving rapidly, developing a strong basis for the pathogenesis of stroke in the context of Sars-CoV-2 infection could prove beneficial in determining which patients are at risk for virus-associated cerebrovascular complications. An improved understanding of inflammatory signaling in the cerebral vasculature can not only identify therapeutic targets to ameliorate COVID-19-related strokes but also clarify the emerging role of inflammatory signaling triggered by more traditional risk factors for stroke including hypertension, diabetes, obesity, and age. Spurred on by the urgency required of the COVID-19 pandemic, stroke research may be poised to usher in a new era of precision diagnostics and anti-inflammatory therapies into the toolbox of stroke care.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by a Rapid Response COVID-19 Award from the American Heart Association No. 20203858 (JDH), the UCLA W. M. Keck Foundation COVID 19 Research Award Program (NK, JDH), and the National Institute for Neurologic Disorders and Stroke (NS112799) (NK, DSL, JDH).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.