
Abstract
Stroke has enormous clinical, social, and economic implications, and demands a significant effort from both basic and clinical science in the search for successful therapies. Atherosclerosis, the pathologic process underlying most coronary artery disease and the majority of ischemic stroke in humans, is an inflammatory process. Complex interactions occur between the classic risk factors for atherosclerosis and its clinical consequences. These interactions appear to involve inflammatory mechanisms both in the periphery and in the CNS. Central nervous system inflammation is important in the pathophysiologic processes occurring after the onset of cerebral ischemia in ischemic stroke, subarachnoid hemorrhage, and head injury. In addition, inflammation in the CNS or in the periphery may be a risk factor for the initial development of cerebral ischemia. Peripheral infection and inflammatory processes are likely to be important in this respect. Thus, it appears that inflammation may be important both before, in predisposing to a stroke, and afterwards, where it is important in the mechanisms of cerebral injury and repair. Inflammation is mediated by both molecular components, including cytokines, and cellular components, such as leukocytes and microglia, many of which possess pro- and/or antiinflammatory properties, with harmful or beneficial effects. Classic acute-phase reactants and body temperature are also modified in stroke, and may be useful in the prediction of events, outcome, and as therapeutic targets. New imaging techniques are important clinically because they facilitate in vivo dynamic evaluation of tissue damage in relation to outcome. Inflammatory conditions such as giant cell arteritis and systemic lupus erythematosus predispose to stroke, as do a range of acute and chronic infections, principally respiratory. Diverse mechanisms have been proposed to account for inflammation and infection-associated stroke, ranging from classic risk factors to disturbances of the immune and coagulation systems. Considerable opportunities therefore exist for the development of novel therapies. It seems likely that drugs currently used in the treatment of stroke, such as aspirin, statins, and modulators of the renin–angiotensin–aldosterone system, act at least partly via antiinflammatory mechanisms. Newer approaches have included antimicrobial and antileukocyte strategies. One of the most promising avenues may be the use of cytokine antagonism, for example, interleukin-1 receptor antagonist.
Atherosclerosis is the common pathologic entity underlying the majority of vascular disease, including cerebral and cardiac ischemia. The interplay between classic risk factors for atherosclerotic disease such as hypertension, smoking, dyslipidemia, and diabetes mellitus is poorly understood. There is increasing evidence that inflammatory mechanisms are involved in both the development and progression of atherosclerosis and its clinical manifestations (Ross, 1999). Inflammation is certainly important in the pathophysiology of cerebral ischemia in the setting of stroke (Barone and Feuerstein, 1999). It also appears that in cerebral ischemia occurring after subarachnoid hemorrhage, head injury, or cardiac arrest, inflammatory mechanisms play an important role in pathophysiology (Fassbender et al., 2001; Mussack et al., 2001).
There is now evidence that low-grade inflammation, identified by an elevated C-reactive protein (CRP) level, may be an additional risk factor for the development of
ischemic stroke or transient ischemic attack (TIA) (Ridker et al., 1997; Rost et al., 2001). Even in the absence of significant atherosclerosis, for example, in pediatric stroke, inflammatory mechanisms are important. Preceding systemic infections are also linked to stroke risk (Syrjänen et al., 1988; Grau et al., 1995a), perhaps by eliciting a systemic inflammatory response. Resolving the debate over whether inflammation confers harm, benefit, or perhaps a combination of both, presents a significant challenge (del Zoppo et al., 2001; Feuerstein and Wang, 2001). Clinical investigations in stroke are usually limited to blood or cerebrospinal fluid (CSF) sampling after the event. New approaches such as novel imaging techniques are becoming increasingly important because they facilitate in vivo observations of inflammation as a dynamic process in relation to brain tissue damage and neurologic outcome.
Stroke is the third biggest killer and the leading cause of adult disability, and it accounts for a significant proportion of health service budgets, yet stroke research remains disproportionately underfunded, particularly in the United Kingdom (Rothwell, 2001). Various problems, usually attributable to either unsuitability of the preclinical model or suboptimal clinical trial design, have hampered the translation of experimentally promising treatments into clinical practice. Considerable attention is now being directed toward overcoming these difficulties (Stroke Therapy Academic Industry Roundtable II [STAIR-II], 2001). Closer collaboration between basic and clinical science appears to be the key to achieving the ultimate goal of delivering safe and effective clinical therapies for a condition that is at last beginning to receive the attention it warrants.
This review has several objectives: (1) to summarize the evidence showing that inflammatory mechanisms are involved in the pathophysiology of clinical stroke, and that they influence outcome; (2) to outline the inflammatory conditions and acute and chronic infections associated with clinical stroke; (3) to summarize the proposed mechanisms linking inflammatory and infective processes to stroke; and (4) finally, and most importantly in the clinical setting, to discuss the emerging opportunities for novel therapeutic strategies.
COMPONENTS OF INFLAMMATION IN ACUTE STROKE
The pathophysiologic mechanism ultimately responsible for the majority of ischemic strokes is occlusion of carotid or cerebral vessels (Fig. 1), as a result of atherothrombosis, thromboembolism, or cardioembolism. Other mechanisms contribute to small vessel occlusive disease and hemorrhagic stroke. In the central ischemic core, where there is virtually no blood flow, tissue is lost rapidly as a consequence of cell death from hypoxia and lack of energy substrates. As ischemia progresses and then reperfusion of the affected tissue occurs, various processes ensue including inflammation, excitotoxicity, nitric oxide production, free radical damage, and apoptosis. These mechanisms, collectively known as the ischemic cascade, play a role in cell death in the ischemic core. More important from the clinical perspective, these events also lead to delayed cell death in the area of intermediate blood flow surrounding the core, known as the ischemic penumbra (Astrup et al., 1981). The various components of the ischemic cascade offer potential therapeutic targets to reduce the ultimate tissue loss and neurologic deficit by lessening the proportion of penumbral tissue recruited into the infarcted core.
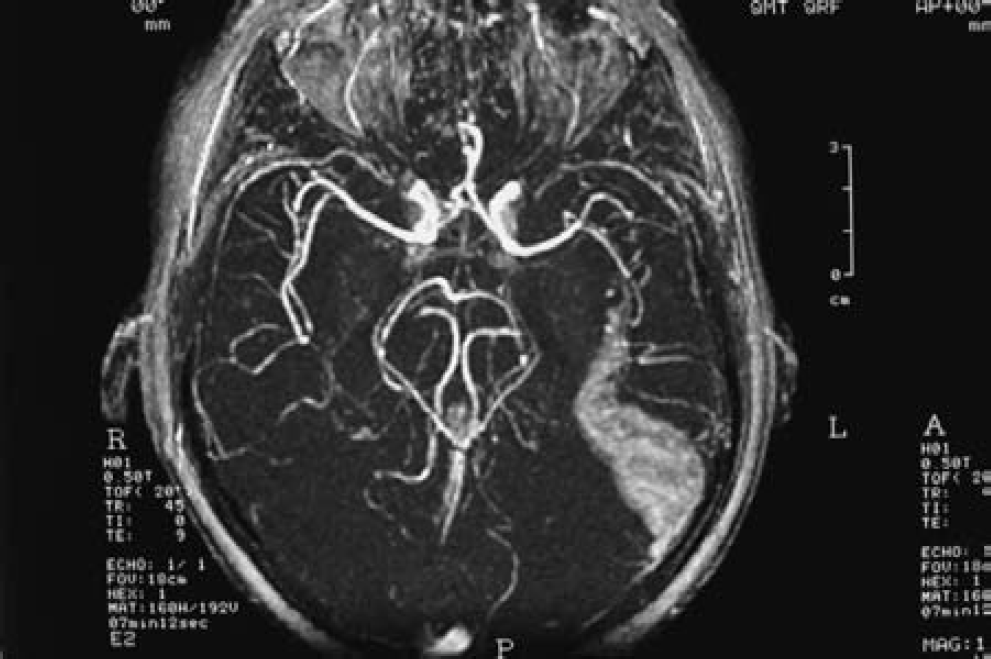
Transverse section of a magnetic resonance angiogram showing circle of Willis. A left middle cerebral artery branch occlusion has a cerebral infarct extending from it (courtesy of I. W. Turnbull).
Inflammation comprises both molecular and cellular components. Experimental studies of ischemia have shown that a key cellular inflammatory event occurs at the blood–microvascular endothelial cell interface (Hallenbeck, 1996). Locally produced cytokines such as interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α) released by microglia, astrocytes, endothelial cells, and neurons influence this process (del Zoppo et al., 2000). Within minutes to hours of reduction of cerebral blood flow, leukocyte recruitment, activation, and adhesion to the endothelium of the cerebral microvasculature occur. Activated leukocytes obstruct the affected cerebral microvasculature (del Zoppo et al., 1991), and transmigration of neutrophils and monocytes/macrophages occurs into the cerebral infarct (Garcia et al., 1994). Reperfusion may also influence the extent of inflammatory injury; furthermore, it is possible that inflammation associated with reperfusion may limit the efficacy of thrombolytic therapy in acute stroke (Jean et al., 1998). In addition to the central inflammatory response, systemic inflammatory processes also occur, both before and after acute stroke. Where appropriate clinical (or pathologic) evidence of these processes is available, the various molecular and cellular components of inflammation associated with clinical stroke are outlined below.
Molecular components of inflammation
Interleukin-1. Experimental stroke models have advanced our understanding of IL-1 in neurodegeneration (Rothwell, 1999; Allan and Rothwell, 2001), and suggest a direct functional role for IL-1 in cerebral ischemic injury. Central or peripheral administration of recombinant IL-1ra to rodents markedly reduces brain damage caused by cerebral ischemia (Relton and Rothwell, 1992; Relton et al., 1996), whereas overexpression of IL-1ra in the brain also inhibits ischemic brain damage (Betz et al., 1995). Administration of a neutralizing antibody to IL-1ra increases ischemic brain damage in the rat (Loddick et al., 1997), whereas injection of an anti–IL-1β antibody is neuroprotective (Yamasaki et al., 1995).
To date, there have been only a few clinical reports on IL-1 and IL-1ra in patients who have had stroke. Elevated serum concentrations of IL-1β protein have not been detected (Fassbender et al., 1994; Tarkowski et al., 1995), although intrathecal production of IL-1β may occur (Tarkowski et al., 1995). Increased IL-1β messenger RNA (mRNA) expression in peripheral mononuclear cells 1 to 3 days after onset of symptoms, returning to normal by 20 to 31 days, showed moderate correlation with degree of neurologic impairment (Kostulas et al., 1999). Plasma concentrations of IL-1ra are elevated in patients within 4 ± 2 days (mean ± SD) of acute ischemic stroke, with and without infection, in comparison with healthy controls (Beamer et al., 1995).
Interleukin-6. A role for IL-6 in the response to focal ischemic brain damage is supported by induction of its mRNA (Wang et al., 1995), and a dramatic increase in IL-6 bioactivity in the ischemic hemisphere (Loddick et al., 1998) after middle cerebral artery occlusion (MCAO) in the rat. Intracerebroventricular injection of recombinant IL-6 significantly reduces ischemic brain damage after MCAO, suggesting that this cytokine is an important endogenous inhibitor of neuronal death during cerebral ischemia (Loddick et al., 1998).
In a small study of patients with acute cerebral ischemia, circulating IL-6 concentrations increase significantly, reaching a plateau between 10 hours and 3 days, before returning to baseline by 7 days (Fassbender et al., 1994). Elevated IL-6 concentrations showed significant positive correlation with volume of computed tomography brain lesion as well as poor functional and neurologic outcome. There have been various other reports of elevated plasma IL-6 concentrations in patients who have had acute strokes (Beamer et al., 1995; Tarkowski et al., 1995; Kim et al., 1996; Fassbender et al., 1997; Carlstedt et al., 1997; Ferrarese et al., 1999; Vila et al., 2000a; Vila et al., 2000b). Stimulated blood cells from patients who have had a stroke show a significant increase in IL-6 release from days 1 to 2 until 1 month (Ferrarese et al., 1999). Cerebrospinal fluid IL-6 concentrations are significantly higher than serum concentrations, peaking on days 2 and 3, with initial CSF IL-6 concentrations significantly correlating with magnetic resonance imaging infarct volume at 2 to 3 months (Tarkowski et al., 1995). These data suggest intrathecal production of IL-6 in patients with stroke, but it remains unclear whether these findings reflect overproduction of IL-6 associated with greater tissue injury or a directly damaging effect of IL-6 itself. By contrast, IL-6 may have antiinflammatory (Tilg et al., 1994) and neuroprotective (Loddick et al., 1998) effects as well as proinflammatory effects. Raised circulating IL-6 levels have also been proposed as part of the inflammatory signature of advanced atherosclerosis (Erren et al., 1999).
Tumor necrosis factor-α. Evidence is accumulating for a role for TNF-α in ischemic brain injury. Induction of TNF-α mRNA occurs in ischemic cortex after both permanent MCAO (Liu et al., 1994; Buttini et al., 1996) and transient MCAO (Wang et al., 1994) in rats, in addition to the presence of neuronal TNF-α protein after permanent MCAO (Liu et al., 1994; Buttini et al., 1996). Focal cerebral ischemic injury is reduced by inhibition of TNF-α activity using soluble TNF-receptor I (sTNF-RI) (Dawson et al., 1996; Barone et al., 1997) or anti–TNF-α monoclonal antibody (Barone et al., 1997), whereas it is exacerbated by administration of TNF-α (Barone et al., 1997). Tumor necrosis factor-α may also have a neuroprotective role because mice genetically deficient in TNF receptors show enhanced ischemic injury (Bruce et al., 1996).
Tumor necrosis factor-α is upregulated in the postmortem brain tissue of patients with acute cerebral infarction (Tomimoto et al., 1996), and appears sequentially in infarction core and periinfarct areas before expression in contralateral hemisphere and brain areas remote from the infarction (Sairanen et al., 2001). Cerebrospinal fluid TNF-α concentrations are elevated in patients with acute ischemic stroke (Vila et al., 2000a; Zaremba et al., 2001), including those with pronounced white matter lesions (Tarkowski et al., 1997). Serum concentrations of TNF-α have been elevated in most studies of acute ischemic stroke patients (Intiso et al., 1997; Carlstedt et al., 1997; Vila et al., 2000a; Zaremba et al., 2001), and raised plasma TNF-α concentrations in patients with lacunar infarction are associated with early neurologic deterioration and poor functional outcome (Castellanos et al., 2002). In one study, however, no such rise in serum TNF-α concentrations was seen (Fassbender et al., 1994). A significant increase in TNF-α release from stimulated blood cells persists for at least 3 months after stroke (Ferrarese et al., 1999). Elevated serum sTNF-RI and sTNF-RII concentrations are associated with carotid atherosclerosis (Elkind et al., 2002), and sTNF-RI concentrations are increased in patients with acute ischemic stroke and infection (Fassbender et al., 1997).
Transforming growth factor-β. Experimental evidence exists for a role for members of the TGF-β family in acute neurodegeneration, including cerebral ischemia (Allan and Rothwell, 2001). Increased expression of TGF-β1 mRNA and protein occurs in brain tissue after ischemic stroke in humans, particularly in infarct border zones (Krupinski et al., 1996). This observation may be in keeping with a neuroprotective action of TGF-β1 within the ischemic penumbra. Serum concentrations of TGF-β have been reported to decrease in patients with acute stroke, regardless of stroke subtype (Kim et al., 1996), although a subsequent study demonstrated no difference in serum TGF-β1 concentrations between acute ischemic stroke patients and control subjects (Slevin et al., 2000). Because TGF-β expression is prominent in the recovery phase of some models of CNS disease, it has been proposed that this cytokine contributes to disease recovery (Benveniste, 1998).
Interleukin-10. The antiinflammatory cytokine IL-10 is neuroprotective in experimental focal cerebral ischemia (Spera et al., 1998), and elevated numbers of peripheral blood mononuclear cells secreting IL-10 are seen in patients with acute ischemic and hemorrhagic stroke (Pelidou et al., 1999). Cerebrospinal fluid concentrations of IL-10 are also elevated in acute ischemic stroke (Tarkowski et al., 1997). Recent clinical data suggest that subjects with low IL-10 production levels have an increased risk of stroke, supporting a protective role for this molecule (van Exel et al., 2002).
Other cytokines. Various other cytokines may have important roles in the regulation of inflammation after cerebral ischemia. Limited experimental and/or clinical evidence exists for several such molecules, including interferon-γ (Tomimoto et al., 1996; Li et al., 2001), IL-2 (Kim et al., 2000), IL-4 (Kim et al., 2000), IL-16 (Schwab et al., 2001), IL-17 (Li et al., 2001), and granulocyte-macrophage colony-stimulating factor (Tarkowski et al., 1999).
Interleukin-8. A systemic increase of IL-8 mRNA expressing mononuclear cells in blood and IL-8 plasma concentrations occurs in patients with acute ischemic stroke, and it has been suggested that this chemokine may have a role in recruiting peripheral neutrophils to sites of cerebral ischemia (Kostulas et al., 1998). Furthermore, IL-8 concentrations are higher in CSF compared with plasma in patients with ischemic stroke, indicating that the CNS may be the predominant site of production (Kostulas et al., 1999). Cerebrospinal fluid IL-8 concentrations differ between patients with large infarcts or gray matter infarcts, and patients with small lesions mainly located in the white matter. In the former group, IL-8 concentrations are initially elevated and then fall rapidly, suggesting a role for IL-8 in acute inflammation. In the latter group, IL-8 concentrations remain elevated up to 3 months, which may be suggestive of a neuroprotective role (Tarkowski et al., 1997).
Monocyte chemoattractant protein-1. This is a potent chemokine specific for monocytes, and MCP-1 is thought to have a significant role in monocyte/macrophage infiltration in experimental cerebral ischemia. A significant increase of CSF MCP-1 concentration occurs in patients with acute ischemic stroke (Losy and Zaremba, 2001).
Cyclooxygenase-2 mRNA and protein are present at the border of the ischemic territory in experimental cerebral ischemia, and COX-2 reaction products, including toxic prostanoids and reactive oxygen species, are thought to exacerbate ischemic damage (del Zoppo et al., 2000). In postmortem specimens from acute ischemic stroke patients, COX-2 is upregulated both locally, in regions of ischemic damage (Iadecola et al., 1999), and more globally, including regions remote from the infarct (Sairanen et al., 1998). Microglia are the major source of COX-2 expression in postmortem brain tissue of patients with chronic cerebral ischemia (Tomimoto et al., 2000). Immunologic NOS and COX-2 upregulation in the human brain suggest an involvement of these two mediators in the mechanisms of cerebral ischemia in clinical stroke.
Localized ICAM-1 expression occurs in histologically normal human carotid bifurcation (Endres et al., 1997), a high-risk region for the development of atherosclerotic plaque, whereas endothelial ICAM-1 expression is increased in symptomatic versus asymptomatic carotid plaque (DeGraba et al., 1998). Patients with carotid atherosclerosis who may be described as “prestroke” have raised concentrations of soluble (s)ICAM-1 (Hwang et al., 1997; Blann et al., 1999) and E-selectin (Hwang et al., 1997). Chronic alteration of adhesion molecule expression also occurs in the presence of risk factors for atherosclerosis, with an increase in sICAM-1 serum concentration and a decrease in sL-selectin (Fassbender et al., 1995). Adhesion molecules are also expressed by human brain microvascular endothelium. The expression of ICAM-1, VCAM-1, and E-selectin by cerebral microvascular endothelial cells is increased by IL-1β, TNF-α, lipopolysaccharide (Hess et al., 1994; Stanimirovic et al., 1997), and in vitro ischemia-like insults (Stanimirovic et al., 1997). Intercellular adhesion molecule-1 expression by brain microvessels is significantly increased in the cerebral infarcts of patients who die after ischemic stroke (Lindsberg et al., 1996).
Several studies have also demonstrated a rise in serum concentrations of soluble adhesion molecules after cerebral ischemia. Soluble intercellular adhesion molecule-1 concentrations are elevated (Shyu et al., 1997) and may peak (Bitsch et al., 1998) in patients within 24 hours of acute ischemic stroke. Soluble vascular adhesion molecule-1 concentrations are elevated between days 1 and 5 (Fassbender et al., 1995; Blann et al., 1999) with a peak at 5 days (Bitsch et al., 1998). A transient increase in circulating concentration of soluble endothelial leukocyte adhesion molecule-1 also occurs (Fassbender et al., 1995). Furthermore, elevated serum levels of sICAM-1 and sE-selectin occur in patients with both large-vessel and small-vessel cerebrovascular disease (Fassbender et al., 1999). One study reported decreased concentration of sICAM-1 in acute stroke in parallel with increased in vitro neutrophil adhesion (Clark et al., 1993).
Cellular components of inflammation
The major inflammatory cells to be activated and that accumulate within the brain after cerebral ischemia are blood-derived leukocytes and resident microglia. Leukocytes clearly perform vital roles in normal host defense, and there is increasing evidence that neutrophils in particular may be mediators of secondary brain damage in cerebral ischemia and reperfusion. The experimental work addressing neutrophils and monocytes/macrophages in this context has been reviewed in detail (Kochanek and Hallenbeck, 1992). Microglia are the first nonneuronal cells to respond to CNS injury, are the major CNS source of cytokines and other immunomolecules, and become phagocytic when fully activated by neuronal death.
In patients with acute ischemic stroke, peripheral leukocyte counts are increased (Pozzilli et al., 1985a; D'Erasmo et al., 1991). Labeled leukocyte uptake by infarcted areas (Pozzilli et al., 1985b) and infiltration in areas of perfusion defect (Wang et al., 1993) have also been described. Selectively labeled neutrophil accumulation in the brain occurs 6 to 12 hours after stroke onset, progresses for up to 24 hours, and then declines (Akopov et al., 1996). Histopathologic studies in human postmortem specimens have shown that neutrophil accumulation around the infarct core is maximal at 48 to 72 hours, and is replaced by mononuclear cells by 4 to 6 days (Adams and Sidman, 1968). Furthermore, in serial CSF samples of patients with acute stroke, neutrophil outflow is maximal on day 4 and modest increases in macrophages and monocytoids reach maxima toward the end of the first week (Sörnäs et al., 1972).
Classic acute phase reactants and body temperature
The acute phase response comprises a variety of systemic changes in response to tissue injury, infection, and inflammation, and is cytokine mediated, mainly by IL-6 and IL-1 (Ramadori and Christ, 1999). Classic “positive” acute phase reactants that are elevated in patients with acute cerebral infarction include the plasma proteins CRP, serum amyloid A protein, and fibrinogen (Syrjänen et al., 1989).
Erythrocyte sedimentation rate and fibrinogen
The erythrocyte sedimentation rate (ESR) is the rate of fall of erythrocytes in a column of blood and is a measure of the acute phase response. A raised ESR reflects an increase in plasma concentration of large proteins such as fibrinogen and immunoglobulins, which promote erythrocyte aggregation, thereby causing them to fall more rapidly. Fibrinogen itself is an independent risk factor for stroke (Wilhelmsen et al., 1984), and significant hyperfibrinogenemia occurs in patients with acute cerebral infarction in association with leukocytosis and an increase in leukocyte aggregation (D'Erasmo et al., 1991; Belch et al., 1998). Elevated ESR in patients with acute ischemic stroke is an independent predictor of poor short-term outcome within the first month (Chamorro et al., 1995; Balestrino et al., 1998) and early stroke recurrence (Chamorro et al., 1997).
Imaging central nervous system inflammation in stroke
Imaging the inflammatory components in clinical stroke in vivo is important because it permits the identification of associations between neurologic outcome and brain tissue damage. Such imaging facilitates the study of inflammation as a dynamic process, and is likely to assist in the development of therapeutic interventions. Various techniques have been used to image leukocyte infiltration in ischemic stroke, including gamma camera imaging of reinjected 111indium-labeled leukocytes (Pozzilli et al., 1985b) and technetium-99m hexamethylpropyleneamine oxime (99mTc HMPAO)-labeled leukocyte brain single-photon emission computed tomography (Wang et al., 1993). Neutrophil invasion can be demonstrated in acute cerebral infarcts within 24 hours of symptom onset using 111indium-labeled autologous neutrophils (Fig. 2) (Price et al., 2002). 99mTc HMPAO-labeled leukocyte single-photon emission computed tomography of the brain has also shown neutrophils to intensively accumulate in cerebral infarctions. This accumulation correlates with the severity of brain tissue damage and poor neurologic outcome (Akopov et al., 1996).
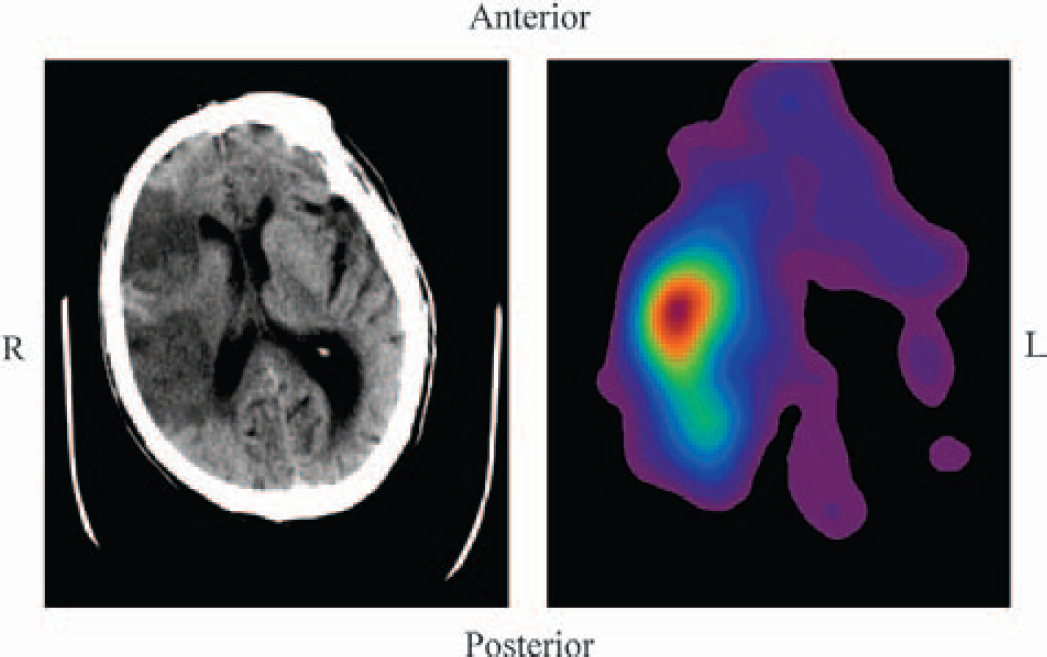
Corresponding trans-axial non-enhanced computed tomogram
PK11195 is a specific ligand for the peripheral benzodiazepine binding site. After cerebral ischemia in rodents, increased binding of PK11195 is colocalized with invading cells of mononuclear–phagocytic lineage, in and around infarcted tissue. Activated microglia, however, are responsible for increased PK11195 binding in lesions where the blood–brain barrier is intact (Banati et al., 1997). In patients with ischemic stroke, [11C]PK11195 positron emission tomography has demonstrated activated microglia in both middle cerebral artery or posterior cerebral artery territory infarctions (Gerhard et al., 2000) and in secondary ipsilateral thalamic lesions in patients with middle cerebral artery territory infarction (Fig. 3) (Pappata et al., 2000). Such microglial activation is present for up to several weeks after the onset of cerebral infarction.
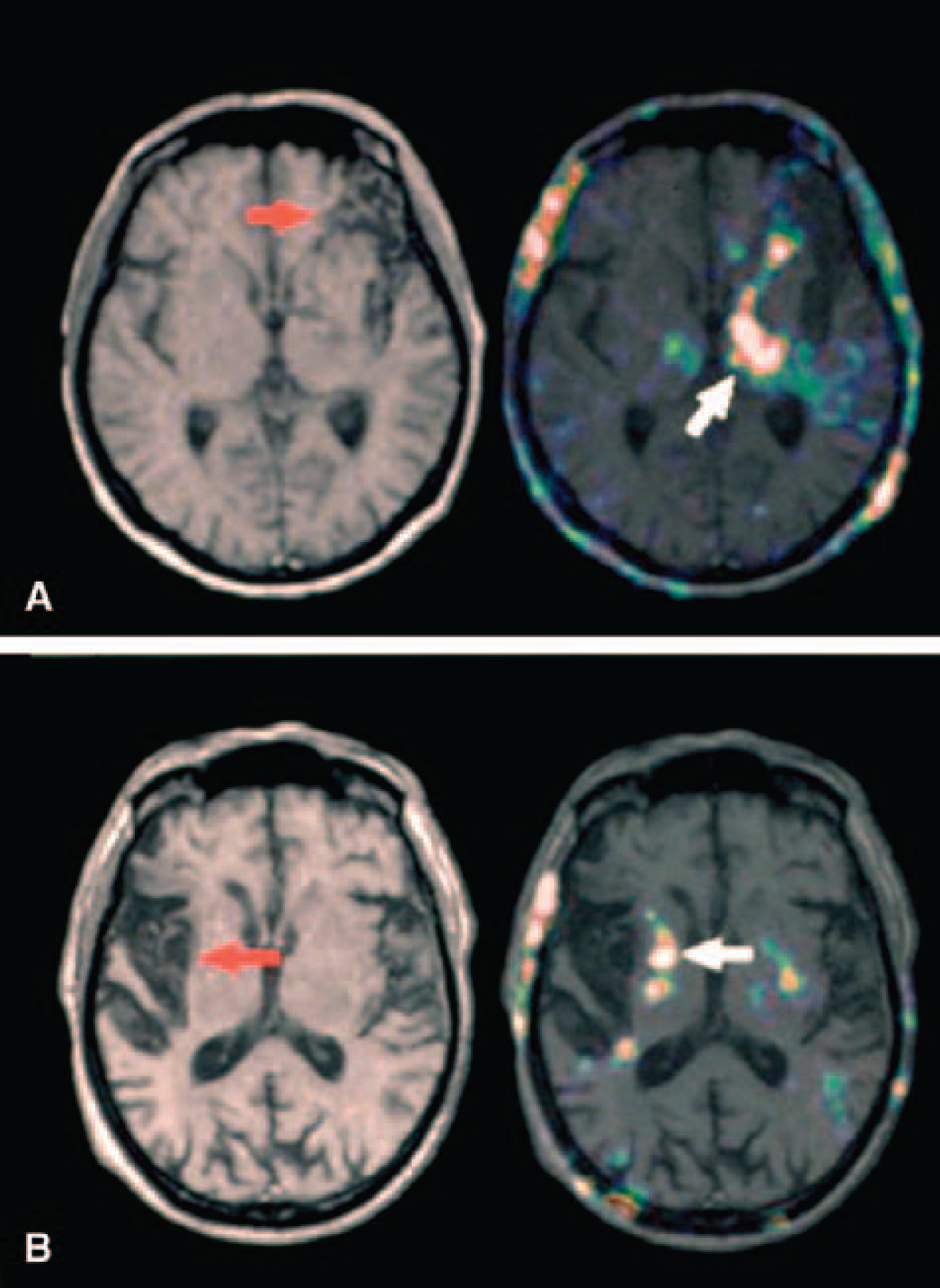
Transverse sections of the magnetic resonance images
Novel magnetic resonance imaging techniques such as perfusion-weighted imaging and diffusion-weighted imaging, when used in combination, have been proposed as a method by which tissue in the ischemic core can be differentiated from penumbral tissue (Heiss et al., 2001a). This might be useful in the identification and monitoring of patients suitable for thrombolytic and/or neuroprotective therapies. Magnetic resonance imaging has been used in experimental cerebral ischemia to monitor evolving cellular responses associated with inflammation (Schroeter et al., 2001), and it is possible that this technique may have clinical applications.
Future developments in the imaging of inflammatory components in stroke, as in many other conditions, may include the use of cytokines to assist with the molecular characterization of immune processes and the development of new therapeutic strategies (Signore et al., 2000).
INFLAMMATORY CONDITIONS ASSOCIATED WITH STROKE
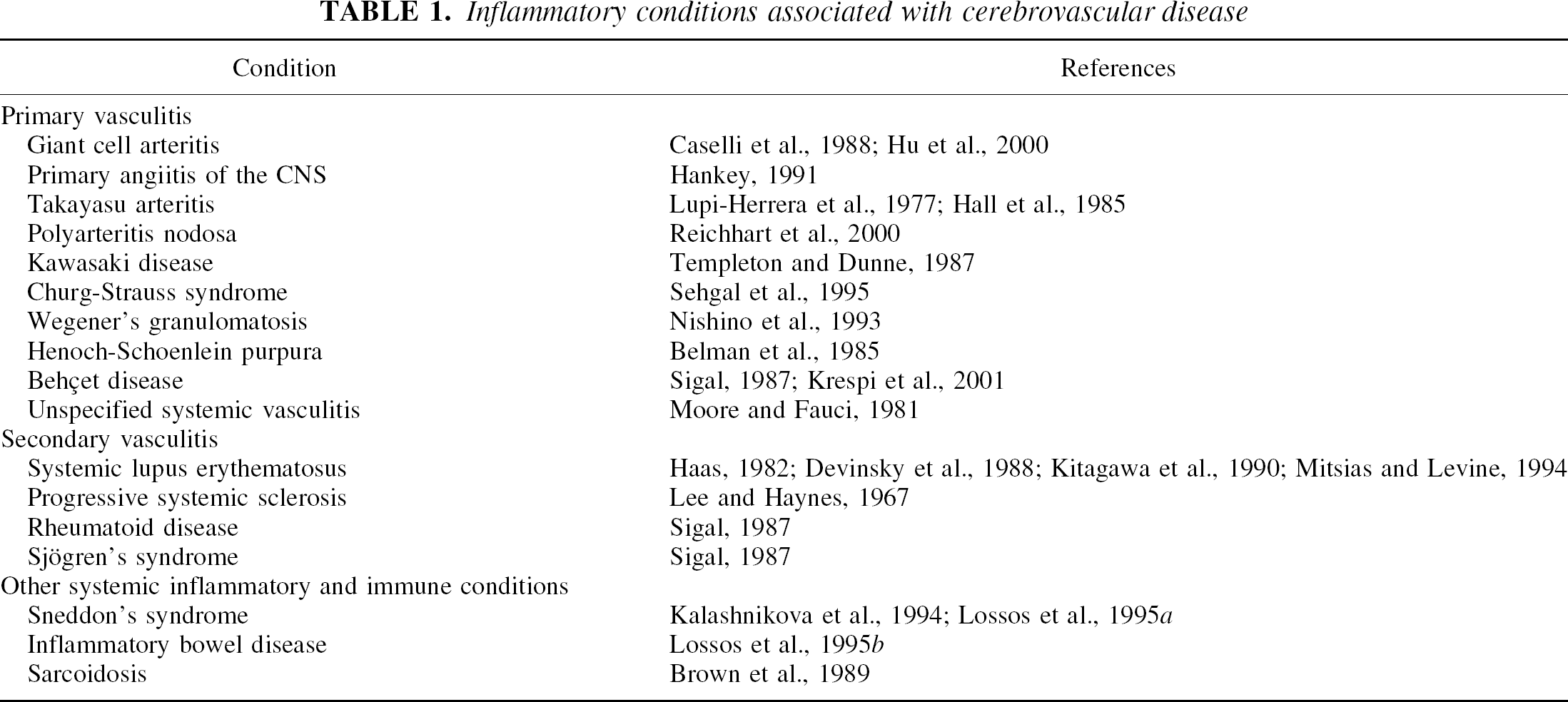
Stroke has been reported to occur in a wide range of systemic inflammatory conditions (Table 1), but it must be borne in mind that the majority of these conditions are rare, and stroke is an unusual complication in most cases. Stroke has been largely ascribed to an associated inflammatory vasculitis in these conditions. It is now recognized, however, that mechanisms other than vasculitis are also important in the pathogenesis of stroke in this setting, some of which are common to both inflammation and infection-associated stroke, and will be discussed later.
Inflammatory conditions associated with cerebrovascular disease
Of the primary inflammatory vascular disorders, giant cell arteritis (temporal arteritis) is probably the most common cause of ischemic stroke. It is a multisystem vasculitis of the elderly, most commonly affecting the branches of the external carotid artery, associated with fever, anemia, headache, and elevated ESR. Transient ischemic attack, cerebral infarction, and cerebral hemorrhage may all occur. The frequency of TIA/stroke in temporal artery biopsy-proven disease varies from 3% to 7% (Caselli et al., 1988; Hu et al., 2000).
Systemic lupus erythematosus (SLE), a chronic autoimmune multisystem connective tissue disease, may be complicated by stroke, or occasionally present with stroke as an early manifestation (Haas, 1982). Both cerebral infarcts and hemorrhages may occur. Although vasculitis had previously been considered to be a frequent cause of stroke in SLE, other mechanisms such as hypertension mediated by immunologic abnormalities, anticardiolipin antibodies, coagulopathy, and cardiac emboli from Libman-Sacks endocarditis appear to be more important (Devinsky et al., 1988; Khamashta et al., 1990; Kitagawa et al., 1990; Mitsias and Levine, 1994; Roldan et al., 1996).
Elevated anticardiolipin antibody titers in patients with ischemic or hemorrhagic stroke vary in frequency between 1% and 38% (Montalbán et al., 1994; Muir et al., 1994; Tuhrim et al., 1999), and may represent an independent stroke risk factor (Tuhrim et al., 1999). Very few patients have the features comprising the antiphospholipid syndrome, which include recurrent venous and arterial thrombosis and recurrent miscarriage in the presence of anticardiolipin antibodies. Antiphospholipid antibodies of other specificities such as those to β2-glycoprotein 1, phosphatidyl serine, or phosphatidyl inositol may be equally or more important (Tanne et al., 1998).
In addition to these systemic vasculitides and inflammatory conditions, various rare primary CNS vasculitides, including granulomatous primary angiitis of the CNS, have been associated with cerebrovascular syndromes (Hankey, 1991). Isolated angiitis of the CNS with cerebral infarction has also been described in children (Lanthier et al., 2001).
INFECTION AND STROKE
Acute infection and stroke
Infections observed to precede stroke have most often been bacterial in origin, although a wide range of organisms and infections are associated with stroke (Table 2). Stroke is well known to occur in infections such as neurosyphilis, bacterial meningitis, and infective endocarditis. The latter condition is complicated by cerebral infarction in approximately 20% of cases (Valtonen et al., 1993). Perhaps less well appreciated, but important from a clinical perspective, many strokes are associated with common infections. The prevalence of infection in the month preceding ischemic stroke has been estimated to be at least 20% (Grau et al., 1995a). Acute infection, mostly respiratory and of bacterial origin, during the preceding month is a risk factor for cerebral infarction (Syrjänen et al., 1988). This association remains significant after controlling for the effects of other risk factors. Several other studies support this observation, particularly in the preceding week (Ameriso et al., 1991; Bova et al., 1996; Macko et al., 1996a; Grau et al., 1998a), and suggest antecedent infection as a risk factor for stroke in all age groups (Grau et al., 1995a). Patients with recent infection tend to present with a more severe neurologic deficit than patients without infection (Grau et al., 1995b), although not all studies accord with this observation (Bova et al., 1996). Numerous problems face clinical investigators examining the role of preceding infection. Consistent identification and classification of infections can be particularly difficult when done retrospectively.
Infections associated with cerebrovascular disease

Chronic or recurrent infection and stroke
A positive association between elevated C. pneumoniae antibodies indicative of chronic infection and stroke has been reported in several studies (Wimmer et al., 1996; Cook et al., 1998; Fagerberg et al., 1999). These observations have been extended to an elderly, multiethnic population (Elkind et al., 2000). In addition, acute recrudescence of infection (diagnosed by specific immunoglobulin M titers) is apparently associated with acute stroke and TIAs (Cook et al., 1998). Increased serum levels of immune complexes containing chlamydial lipopolysaccharide (and anti-cytomegalovirus antibodies) have also been associated with increased stroke incidence and worse clinical outcome (Tarnacka et al., 2002). Two other studies have failed to provide convincing support for an association between C. pneumoniae infection and stroke (Glader et al., 1999; Heuschmann et al., 2001). The role of chronic C. pneumoniae infection as a risk factor for stroke remains unclear.
Recurrent or chronic respiratory infection, independent from recent acute respiratory infection, may also be associated with cerebral infarction or TIA (Grau et al., 1997). Although other microorganisms such as herpes viruses have been identified in carotid atherosclerotic plaque (Chiu et al., 1997), immunoglobulin G antibodies to herpes simplex virus or cytomegalovirus in apparently healthy men do not predict future stroke risk (Ridker et al., 1998).
Periodontal disease (gingivitis or periodontitis) caused by gram-negative bacteria is common in adults. An association between dental infection and risk of cerebral infarction has been suggested, independent from other risk factors (Syrjänen et al., 1989; Grau et al., 1997). Two more recent studies have produced conflicting results. Whereas one study supported periodontal disease as an independent risk factor for cerebrovascular disease (Wu et al., 2000), particularly ischemic stroke, another failed to confirm the association (Howell et al., 2001). In the latter study, self-reported periodontal disease did not independently predict subsequent cardiovascular disease, including nonfatal stroke.
A wide range of neurologic complications can occur in chronic meningitis. In one recent clinical series, cerebral infarction occurred in 47% of cases of tuberculous meningitis, and 32% of cases of cryptococcal meningitis, commonly in the internal capsule and basal ganglia (Lan et al., 2001).
Although neurologic complications of human immunodeficiency virus infection are common, it has been unclear whether any association exists between the acquired immunodeficiency syndrome and stroke (Pinto, 1996). The prevalence of cerebral infarction in patients with acquired immunodeficiency syndrome ranges from 4% to 29% in autopsy series, but cerebral infarcts in human immunodeficiency virus-infected patients without non–human immunodeficiency virus CNS infection are uncommon (Connor et al., 2000).
The potential association between chronic infection and coronary artery disease has been examined in greater detail than for stroke, but the link also remains uncertain (Danesh et al., 1997). The parallels with stroke are evident, and problems such as confounding by causal risk factors and uncertainty over the temporal sequence of infection and stroke may only be resolved, particularly in the case of C. pneumoniae, by randomized intervention studies.
MECHANISMS OF INFLAMMATION AND INFECTION-ASSOCIATED STROKE
A wide range of mechanisms have been suggested to link inflammation and infection to the pathophysiology of stroke. These various pathophysiologic pathways, together with the events occurring after the onset of acute cerebral ischemia, are summarized in Fig. 4.
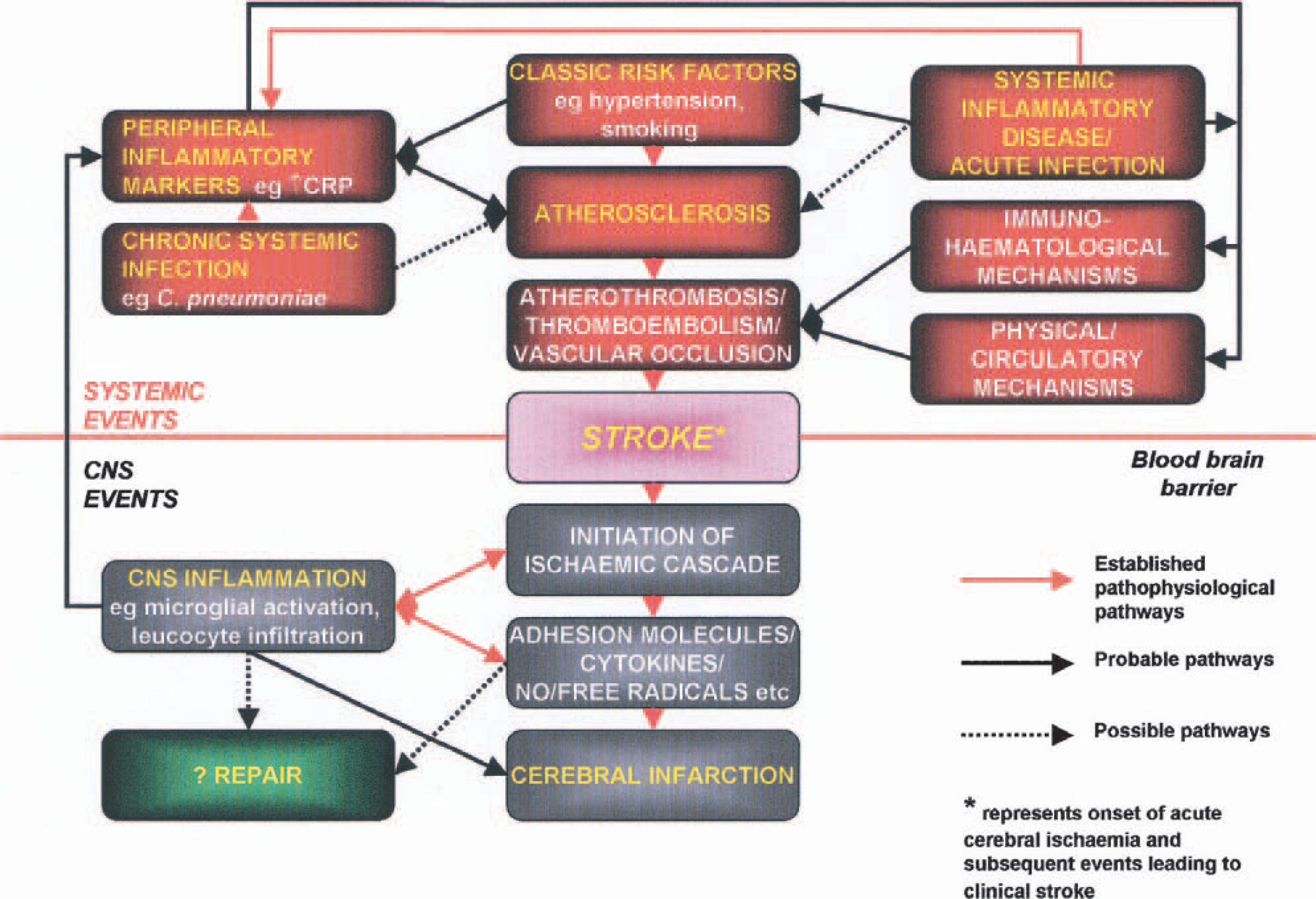
Proposed interactions in the pathophysiology of inflammation and infection-associated stroke.
Atherosclerosis, inflammation, infection and vascular risk factors
Atherosclerosis is an inflammatory disease (Ross, 1999), and inflammation contributes to the risk of plaque rupture (Blake and Ridker, 2001), one of the key events in the occurrence of acute atherothromboembolic clinical events such as stroke. It is likely that traditional risk factors interact with inflammatory mechanisms in a more complex manner than has been envisaged previously. For example, increased blood pressure is significantly associated with elevated levels of sICAM-1 and IL-6 in apparently healthy men, and may support a role for hypertension as a proinflammatory stimulus (Chae et al., 2001). C-reactive protein concentrations are associated with various cardiovascular risk factors including increasing age, smoking, body mass index, lipid levels, and blood pressure. Furthermore, higher CRP levels are seen in individuals with periodontal disease or evidence of C. pneumoniae or H. pylori infection (Mendall et al., 1996; de Maat and Kluft, 2001). Hypertension is an important factor in the development of cerebrovascular disease in SLE, where there is renal involvement mediated by immunologic abnormalities with high anti-DNA antibody titers (Kitagawa et al., 1990), as well as in unusual primary vasculitides such as Takayasu arteritis, also owing to renal involvement (Lupi-Herrera et al., 1977).
Multiple mechanisms have now been proposed to link inflammatory and infective stimuli to the development and progression of atherosclerotic plaque. These include a direct proinflammatory effect of CRP on human arterial endothelium (Pasceri et al., 2000), causing the induction of adhesion molecule expression with the subsequent recruitment of monocytes and other immune cells to the arterial wall. Monocyte-derived macrophages and activated T cells release various growth factors and cytokines including TNF-α and IL-1 (Barath et al., 1990; Galea et al., 1996). It is under the influence of these molecules that endothelial and smooth muscle cells release further growth factors, resulting in smooth muscle cell proliferation and fibrous plaque formation characteristic of advanced atherosclerotic lesions. As mentioned earlier, C. pneumoniae and other microorganisms are possible inflammatory triggers of atherogenesis. In addition, the progression of atherosclerosis may be accelerated by the treatment of certain systemic inflammatory conditions with corticosteroids, as in SLE (Bulkley and Roberts, 1975). Significant correlations have also been reported between peripheral differential leukocyte counts and the severity of carotid atherosclerosis in male nonsmokers (Huang et al., 2001).
Immunohematologic mechanisms
Disturbances in immune function and alterations in the coagulation system have been described in inflammation and infection-associated stroke. A prothrombotic state has been suggested to predispose to large-vessel atherothrombosis and ischemic stroke. In addition, microvascular obstruction may also contribute to a reduction in tissue perfusion and worsen cerebral ischemia.
Reduced circulating antithrombotic activated protein C, elevated C4b-binding protein, and a low ratio of tissue plasminogen activator to plasminogen activator inhibitor are examples of components of the coagulation system that are modified in inflammation or infection-associated stroke (Macko et al., 1996b). Increased fibrin D-dimer levels, cardiolipin immunoreactivity, and fibrinogen levels also occur in patients with acute ischemic stroke with antecedent infection (Ameriso et al., 1991). Seasonal variations in clotting factors may be important, because elevated winter levels of plasma fibrinogen and factor VII clotting activity have been observed (Woodhouse et al., 1994). Increased von Willebrand factor levels also occur in patients with acute stroke for up to 3 months and in subjects with carotid atherosclerosis, perhaps reflecting continued endothelial damage or repair (Blann et al., 1999). Not all studies concur, however, because a range of markers of coagulation and fibrinolysis, including antithrombin III, plasminogen, plasminogen activator inhibitor-1, thrombin-antithrombin complexes, prothrombin fragment F1+2, fibrin monomer or D-dimer, factor VII and factor VIII, von Willebrand factor, C4b-binding protein activity, protein S, anticardiolipin antibodies, and thrombomodulin have been reported not to differ between patients with stroke with and without infection (Grau et al., 1995b; Grau et al., 1998a).
Increased levels of CRP and proinflammatory cytokines can contribute to a procoagulant state. C-reactive protein may promote localized coagulation, and therefore thrombosis, by stimulating monocytes to produce tissue factor, a membrane-bound glycoprotein that initiates the extrinsic pathway of coagulation (Cermak et al., 1993). Greater circulating IL-6 levels in patients with acute ischemic stroke have been associated with decreased levels of free protein S, leading to the suggestion that IL-6 may partly modulate this anticoagulant pathway (Vila et al., 2000b).
In SLE, antiphospholipid antibodies such as anticardiolipin antibodies and lupus anticoagulant may well be important in the development of occlusive cerebrovascular disease via inhibition of prostacyclin formation, causing platelet aggregation. Several other effects of these antibodies have been suggested, including inhibition of prekallikrein, alterations of antithrombin III, decreased fibrinolysis, decreased release of plasminogen activator, inhibition of protein C activation, and the induction of endothelial injury (Kitagawa et al., 1990). Thrombotic thrombocytopenic purpura has also been implicated in SLE patients with cerebral infarction (Devinksy et al., 1988). In inflammatory bowel disease, the cerebral arterial and venous circulation may be affected by hypercoagulability-related thrombosis, vasculitis, and consumption coagulopathy leading to hemorrhagic events (Lossos et al., 1995b).
In patients with bacteremic infections, thromboembolic complications are relatively common, occurring in approximately 20% of patients with infective endocarditis and 10% of septicemia cases (Valtonen et al., 1993). Numerous mechanisms may be responsible, many of which involve activation of the coagulation system. These include reduced levels of antithrombin III, proteins C and S, increased platelet aggregation and adhesion, impaired fibrinolysis, and antiphospholipid antibody formation. In addition, endotoxins, other bacterial toxins, and proinflammatory cytokines such as IL-1 and TNF-α may well contribute to thrombosis via effects on endothelial function.
Physical and circulatory mechanisms
As already discussed, inflammation and infection are likely to play a part in the event that causes at least 50% of ischemic strokes and transient ischemic attacks, acute arterial obstruction. This is usually caused either by atherothrombosis or atherothromboembolism. Less frequently, low-flow distal to a severely narrowed or occluded artery will be responsible for clinical events.
Valvular heart disease is common in SLE and is associated with the presence of antiphospholipid antibodies (Khamashta et al., 1990). Cardioembolism is an important cause of stroke in such patients (Roldan et al., 1996). Cerebral vasculitis as a cause of stroke is reported in a range of vasculitides and inflammatory processes, including giant cell arteritis (Caselli et al., 1988), Takayasu arteritis (Lupi-Herrera et al., 1977) and Wegener granulomatosis (Nishino et al., 1993).
Virally induced inflammatory vasculopathies have been associated with thrombotic occlusion of the internal carotid and cerebral arteries (Eidelberg et al., 1986; Leber et al., 1995; Grau et al., 1998b). Focal middle cerebral artery vasculitis has also been implicated in stroke associated with Mycoplasma pneumoniae infection (Fu et al., 1998). Intraneuronal migration of the varicella zoster virus from the trigeminal ganglion along the trigeminal nerve to the cerebral arteries has been proposed as a reasonable mechanism for varicella-associated ischemic stroke (Askalan et al., 2001). Varicella zoster virus is present within the media of affected large cerebral arteries in adults with herpes zoster ophthalmicus and delayed cerebral infarction (Eidelberg et al., 1986; Melanson et al., 1996). Varicella lesions have also been suggested to cause stroke via a local irritant effect in the region of the superior cervical ganglion, causing sympathetic stimulation (Ganesan and Kirkham, 1997). An inflammatory carotid arteritis with intimal damage, peripheral embolization, and cerebral lesions ranging from transient ischemia to infarction has been described in children with preceding throat infection (Bickerstaff, 1964).
In cerebrovascular neurosyphilis, there is meningovascular inflammation resulting in progressive vascular insufficiency predominantly affecting the middle cerebral artery territory (Dalal and Dalal, 1989). A basal exudate in neurotuberculosis entraps the circle of Willis with involvement of lenticulostriate branches from the middle cerebral artery mainstem (Dalal and Dalal, 1989).
Cervical artery dissection is another important cause of stroke and TIA, particularly in young and middle-aged patients, and a significant association has been reported between recent infection and cervical artery dissection in this age group (Grau et al., 1999). It has been tentatively suggested that preexisting abnormalities of extracellular matrix proteins may increase susceptibility to infection-associated injury of the arterial wall.
IMPLICATIONS FOR TREATMENT
Current approaches to the primary and secondary prevention of ischemic stroke focus on modifiable vascular risk factors such as hypertension, smoking, carotid stenosis, atrial fibrillation, physical inactivity, diabetes mellitus, and dyslipidemia. Commonly used drugs include antiplatelet agents, antihypertensive drugs, lipid-lowering agents, and anticoagulant drugs. At the present time, the only approved treatment for acute ischemic stroke is intravenous tissue plasminogen activator (NINDS, 1995), but only a minority of patients are eligible for such therapy, and as mentioned earlier, even the efficacy of thrombolysis may be restricted by reperfusion-associated inflammation. A clearer understanding of the mechanisms underlying inflammation and infection-associated stroke will pave the way for novel therapeutic approaches, in addition to providing new insights into current strategies.
Antiinflammatory effects of current treatments
It is conceivable that a range of the current stroke treatments possess antiinflammatory effects in addition to their traditionally accepted actions. The discussion here will focus mainly on the possible antiinflammatory mechanisms of the antiplatelet agent acetylsalicylic acid (aspirin) and the lipid-lowering agents β-hydroxy-β-methylglutaryl coenzyme A reductase inhibitors (statins).
Novel therapeutic strategies
Subjects vaccinated against influenza are less prone to viral (influenza) infections and subsequent secondary bacterial infections. A recent study found a negative association between influenza vaccination and brain infarction, particularly in patients younger than age 75 (Lavallée et al., 2002). Whether this protection is attributable to reduced infections or the identification of a subgroup with low stroke risk because of a better lifestyle, is open to debate. The use of vaccination is a potentially promising approach to stroke prevention.
Other, experimental antileukocyte interventions in cerebral ischemia such as the use of other monoclonal antibodies to block the neutrophil–endothelial cell interaction, or antineutrophil serum and antineoplastic agents to achieve neutrophil depletion, have also been suggested (Härtl et al., 1996). The suppression of microglial function has also been put forward as a potential alternative or adjunctive therapeutic avenue (Wood, 1995).
In contrast, IL-1 is uniquely placed as a therapeutic target. As discussed above, a number of studies have demonstrated that inhibiting the release or actions of IL-1 markedly reduces ischemic cerebral damage. Interleukin-1 has little role in normal brain function, and therefore its blockade should not be associated with significant adverse effects within the CNS. Interleukin-1ra is the most advanced approach to modify IL-1 action, and is safe and beneficial in the treatment of patients with rheumatoid arthritis (Bresnihan et al., 1998). A phase II clinical trial of intravenous IL-1ra compared with placebo in patients with acute stroke is under way (Smith et al., 2002).
Inducible or immunologic NOS inhibitors may have some value in patients ineligible for early thrombolysis because these agents have demonstrated an extended therapeutic window of up to 24 hours experimentally (del Zoppo et al., 2000). Further insights into the therapeutic potential of nitric oxide donors in patients with acute stroke should be provided by ongoing studies (Bath, 2002). The situation with COX-2 inhibition is less clear because COX-2, although probably deleterious in ischemia, may also possess regenerative properties (del Zoppo et al., 2000). Matrix metalloproteinases may offer another potential therapeutic target once their beneficial and deleterious effects in stroke have been better defined (Mun-Bryce and Rosenberg, 1998).
Induced hypothermia is another potential therapy currently under investigation. The rationale for cooling in clinical stroke is based on experimental findings showing that hypothermia decreases the final cerebral infarct volume and extends the duration of ischemia the brain is able to withstand before the occurrence of permanent damage. A pilot study of induced hypothermia has shown that this approach appears to be feasible and safe (Krieger et al., 2001), but optimal treatment conditions and clinical efficacy remain to be established in larger studies. Paracetamol has been shown to decrease body temperature in patients with acute ischemic stroke (Dippel et al., 2001), but further studies are required to establish whether antipyretic agents lead to improved outcome.
CONCLUSIONS
Inflammation and infection are increasingly recognized as key elements in the pathogenesis and outcome of ischemic stroke. It is now possible, using laboratory and imaging techniques in carefully selected clinical populations, to extend experimental approaches to human studies. Greater understanding of the role and potential for modulation of the inflammatory response may have profound implications for patient management.
Footnotes
Acknowledgments:
The authors thank Professor N. J. Rothwell, School of Biological Sciences, University of Manchester, Dr. S. J. Hopkins, North West Injury Research Collaboration, Hope Hospital, Salford, and Dr. C. J. Smith and Miss R. F. Drennan, University of Manchester and Stroke Services, Hope Hospital, Salford, for their advice during the writing of this review. The authors also thank the following people for their contribution of figures: Dr. I. W. Turnbull, Consultant Neuroradiologist, Hope Hospital; Dr. C. J. Price, University of Cambridge; and Dr. R. B. Banati, Hammersmith Hospital, London.