
Abstract
After stroke, the blood–brain barrier is transiently disrupted, allowing leukocytes to enter the brain and brain antigens to enter the peripheral circulation. This encounter of normally sequestered brain antigens by the systemic immune system could therefore present an opportunity for an autoimmune response to brain to occur after stroke. In this study, we assessed the immune response to myelin basic protein (MBP) in animals subjected to middle cerebral artery occlusion (MCAO). Some animals received an intraperitoneal injection of lipopolysaccharide (LPS; 1 mg/kg) at reperfusion to stimulate a systemic inflammatory response. At 1 month after MCAO, animals exposed to LPS were more likely to be sensitized to MBP (66.7% versus 22.2%; P = 0.007) and had more profound and persistent neurologic deficits than non-LPS-treated animals. Exposure to LPS was associated with increased expression of the costimulatory molecule B7.1 early after stroke onset (P = 0.009) and increased brain atrophy 1 month after MCAO (P = 0.03). These data suggest that animals subjected to a systemic inflammatory insult at the time of stroke are predisposed to develop an autoimmune response to brain, and that this response is associated with worse outcome. These data may partially explain why patients who become infected after stroke experience increased morbidity.
Introduction
After stroke, the integrity of the blood–brain barrier (BBB) is compromised and cells of the immune system encounter novel central nervous system (CNS) antigens in both the brain and in the systemic circulation. The concentration of circulating CNS antigens after stroke reflects the severity of cell injury and predicts the outcome (Herrmann et al, 2000; Wunderlich et al, 1999). If these antigens were presented to lymphocytes in the proper context, an autoimmune response to brain could develop. Indeed, CNS autoreactive cells and CNS-specific immunoglobulins are seen in patients with a history of stroke (Bornstein et al, 2001; Dambinova et al, 2003; Kallen et al, 1977; Wang et al, 1992; Youngchaiyud et al, 1974). The clinical consequences of this CNS autoimmune response are unknown.
Infection in the poststroke period is associated with poor outcome (Grau et al, 1999; Johnston et al, 1998). There are several plausible mechanisms by which infection could worsen ischemic brain injury, but definitive mechanistic data are lacking. Encounter of antigen by the immune system can lead to a T
Activation of the immune system during a systemic inflammatory response alters the microenvironment of the brain in a way that could promote sensitization to brain antigens during episodes of cerebral ischemia (Kissler et al, 2001). Since Gram-negative bacteria are the predominant organisms causing infection after stroke, we modeled the systemic inflammatory response to infection with lipopolysaccharide (LPS), a component of the Gram-negative bacterial cell wall (Hilker et al, 2003; Puri et al, 2002). The aim of these experiments was to determine whether LPS-induced inflammation during stroke would increase the likelihood of developing an immune response to brain antigens.
Materials and methods
Animals
Experiments were approved by the Institution's Animal Care and Use Committee. Male Lewis rats (250 to 300 g) were used for all studies.
Middle Cerebral Artery Occlusion (MCAO)
Anesthesia was induced with 5% and maintained with 1.5% halothane. After midline neck incision, the right common carotid, internal carotid, and pterygopalantine arteries were ligated. A monofilament suture (4.0) was inserted into the common carotid artery and advanced into the internal carotid artery (Zea-Longa et al, 1989). Animals were maintained at normothermia during surgery. Reperfusion was performed 3 h after MCAO and animals killed at various time points thereafter. Rectal temperature and body weight were assessed at set time intervals. In sham-operated animals, the suture was inserted into the carotid but not advanced.
Lipopolysaccharide Administration
In a subset of animals, LPS (1 mg/kg) (Sigma, St Louis, MO, USA; serotype 026:B6) was injected intraperitoneally at reperfusion (3 h after MCAO or sham surgery). Lipopolysaccharide-treated animals are henceforth referred to as LPS(+), non-LPS-treated animals as LPS(−).
Neurologic Outcome
Neurologic status was assessed using a modification of Bederson scale and an adaptation of the ‘sticky tape test’ (Bederson et al, 1986; Schallert and Whishaw, 1984).
Isolation of Mononuclear Cells
Mononuclear cells (MNCs) were isolated from the brain and spleen using previously described methods (Becker et al, 2003).
ELISPOT Assay
MNCs were cultured (1 × 105 cells/well) for 48 h in 96-well plates (MultiScreen®-IP; Millipore) in media alone or in media supplemented with bovine myelin basic protein (MBP) (25 μg/mL). Experiments were performed in triplicate. Interferon-γ or TGF-β1 capture antibodies were used and the reaction product developed with alkaline phosphatase. Spots were independently counted under a dissecting microscope by two individuals masked to treatment status. The difference between the number of MBP-stimulated and unstimulated spots was considered indicative of an antigen-specific response. Secretion of IFN-γ was used to indicate sensitization (T
Histology
On killing, brains were frozen and stored at −80°C. To determine the infarct size, brains were sectioned (10 μm) at 6 predetermined levels (bregma +2.40, +1.00, −0.40, −1.80, −3.20, and −4.40) and stained with cresyl violet; infarct volume was calculated and corrected for edema (Swanson et al, 1990). Quantitative immunocytochemistry (ICC) was performed on sections at the level of the anterior commissure. Sections were fixed in acetone and methanol and stained with antibodies to CD4 (clone W3/25), CD8 (clone MRC OX-8), B7.1 (CD80; clone 3H5), B7.2 (CD86; clone 24F), MHC I (OX-18), and MHC II (OX-6). OX-18 and OX-6 antibodies were obtained from Pharmingen, San Diego, CA, USA; all other antibodies were obtained from Serotec, Raleigh, NC, USA. Sections were developed using peroxidase and 3,3'-diaminobenzidine (Vector) and counterstained with cresyl violet. The NeuroTACS™ kit (R&D Systems) was used to detect apoptosis. Sections were examined at × 100; the numbers of labeled cells within six different high-power fields in six different brain regions (Figure 1) were counted by two independent investigators masked to treatment status. The phenotype of cells expressing B7.1 was determined using double-label ICC for astrocytes (glial fibrillary acidic protein (GFAP); Sigma), microglia (OX-42; Serotec), and neurons (Neurotrace Nissl stain; Molecular Probes, Carlsbad, CA, USA).
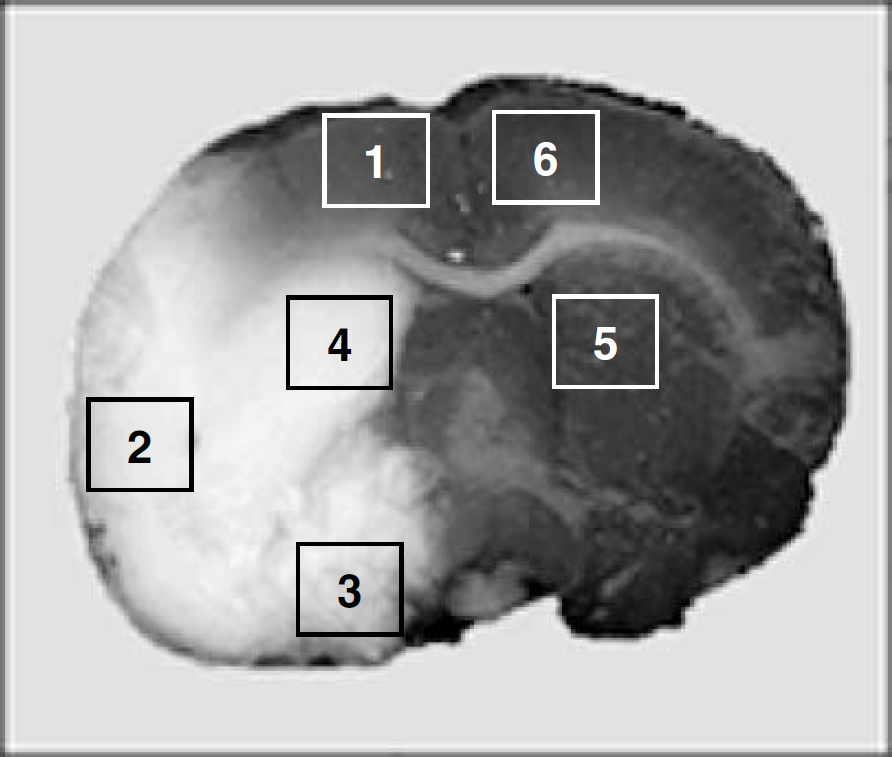
Regions of the brain evaluated for quantitative ICC; the ischemic hemisphere areas 1 to 4 are in the ischemic or right hemisphere; areas 5 and 6 in the nonischemic or left hemisphere.
Statistics
Responses over time were compared using one-way analysis of variance. Data from different treatment groups were compared using the t-test or the Mann–Whitney U-test, and expressed as mean ± standard deviation (s.d.) or median ± s.d. as appropriate. Categorical data were evaluated using the χ2-test statistic. Correlations were performed using Spearman's rho. Significance was set at P ≤ 0.05.
Results
Sensitization to Brain Antigens
Lymphocytes specific for MBP were enriched in brain as very few MBP-reactive cells were seen in the spleen (data not shown). Based on the cytokine profile of MBP-reactive MNCs from the ischemic hemisphere of brain 1 month after MCAO, LPS(+) animals were more likely to develop a T
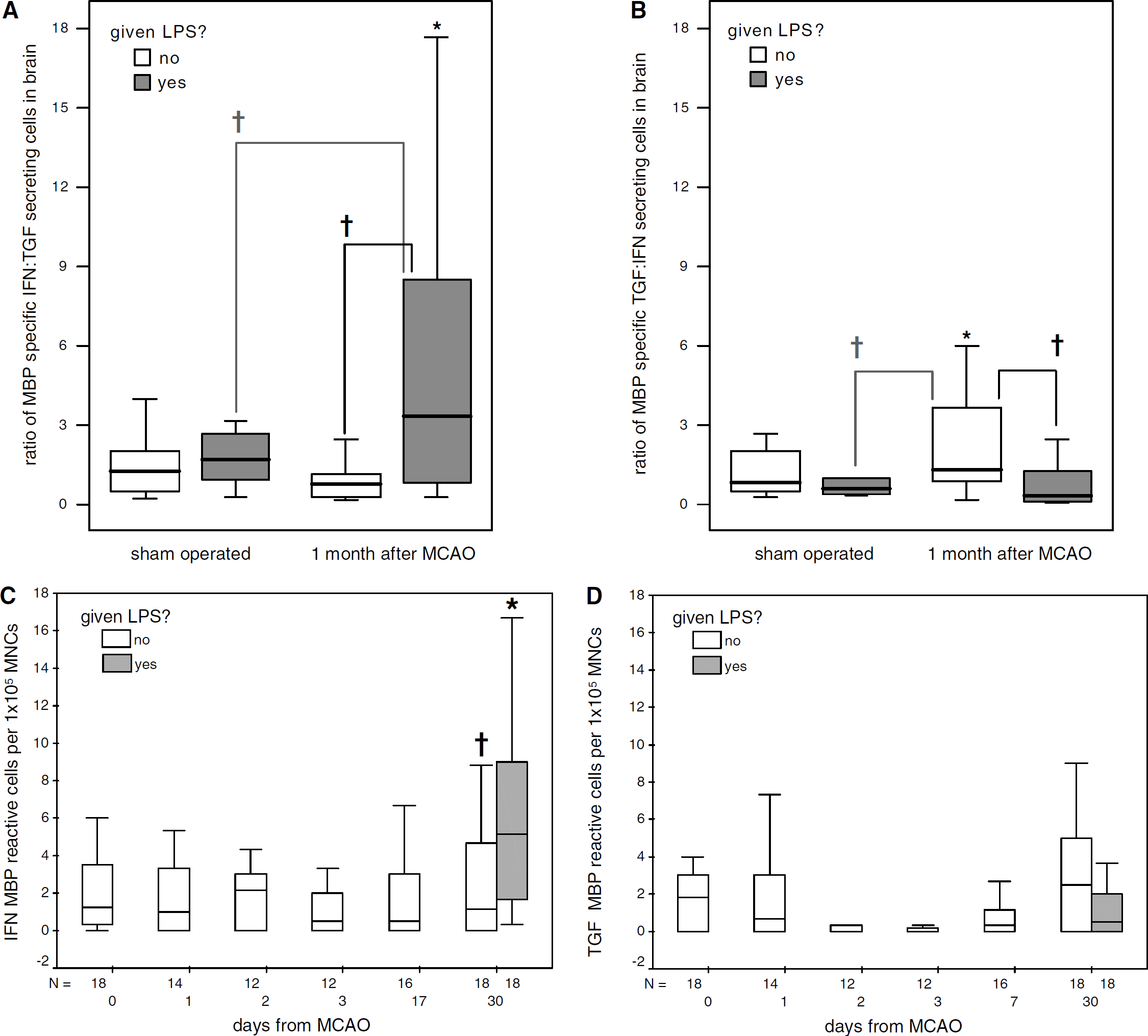
MNCs extracted from the brains of LPS(+) animals 30 days after MCAO evidence a Th1 response to MBP, which is more robust than sham-operated animals and LPS(−) animals at the same time point (
Physiologic and Neurobehavioral Outcome
There was no mortality in LPS(+) or LPS(−) sham-operated animals and no difference in mortality among LPS(+) and LPS(−) animals undergoing MCAO: 7/38 (18.4%) versus 8/81 (9.9%), respectively. Death occurred almost exclusively in the first 24 h after MCAO. All animals undergoing MCAO developed hyperthermia; the highest recorded temperatures occurred 6 h after stroke onset: 38.4°C in LPS(+) and 38.6°C in LPS(−) animals. The temperatures of LPS(+) and LPS(−) animals differed only at 72 h after MCAO (36.9°C ± 0.8°C versus 37.6°C ± 0.6°C, respectively; P = 0.04). Similarly, the temperatures of T
Neurologic scores were higher (i.e. neurologic dysfunction greater) in both LPS(+) and T
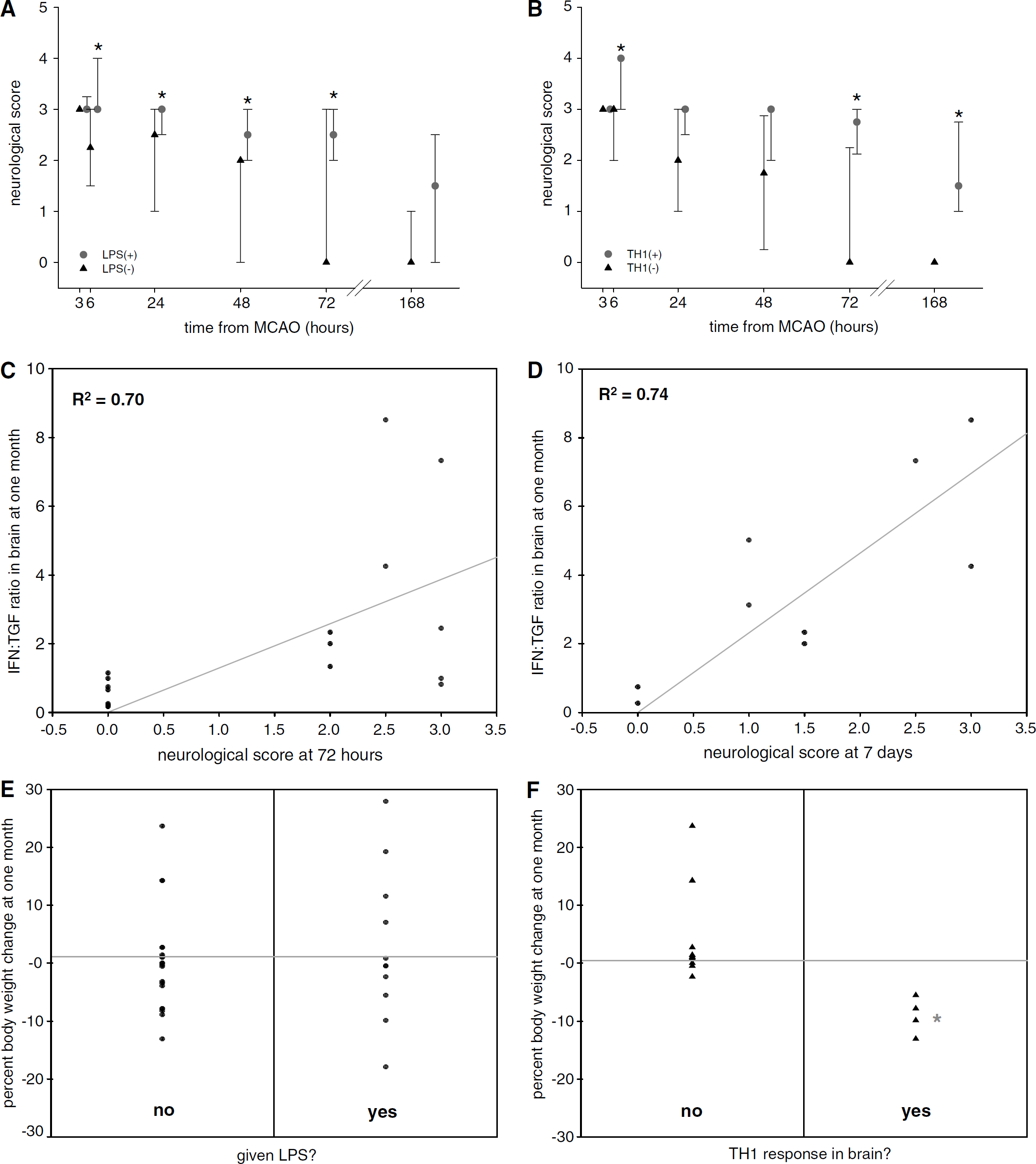
Neurologic scores were higher in LPS(+) animals after MCAO (
All animals experienced a significant decrease in body weight after MCAO; the degree of weight loss did not differ between LPS(+) and LPS(−) animals. Animals that developed a T
Cellular Infiltrate
More CD4+ cells were present in the ischemic hemisphere of LPS(+) animals 24 h after MCAO (12.4 ± 11.1 versus 3.4 ± 2.6; P = 0.002) (Figures 4A and 4B). CD8+ cells, however, were present in similar numbers in LPS(+) and LPS(−) animals until 1 month after MCAO, when the number of CD8+ cells was greater in both the ischemic (16.4 ± 7.7 versus 4.2 ± 4.3; P = 0.02) and nonischemic (9.9 ± 4.6 versus 4.2 ± 4.3; P = 0.005) hemispheres of LPS(+) animals (Figures 4C and 4D). More CD8+ cells were seen in LPS(+) animals undergoing MCAO also 1 month after MCAO than in sham-operated animals 1 month after LPS administration (P = 0.02).
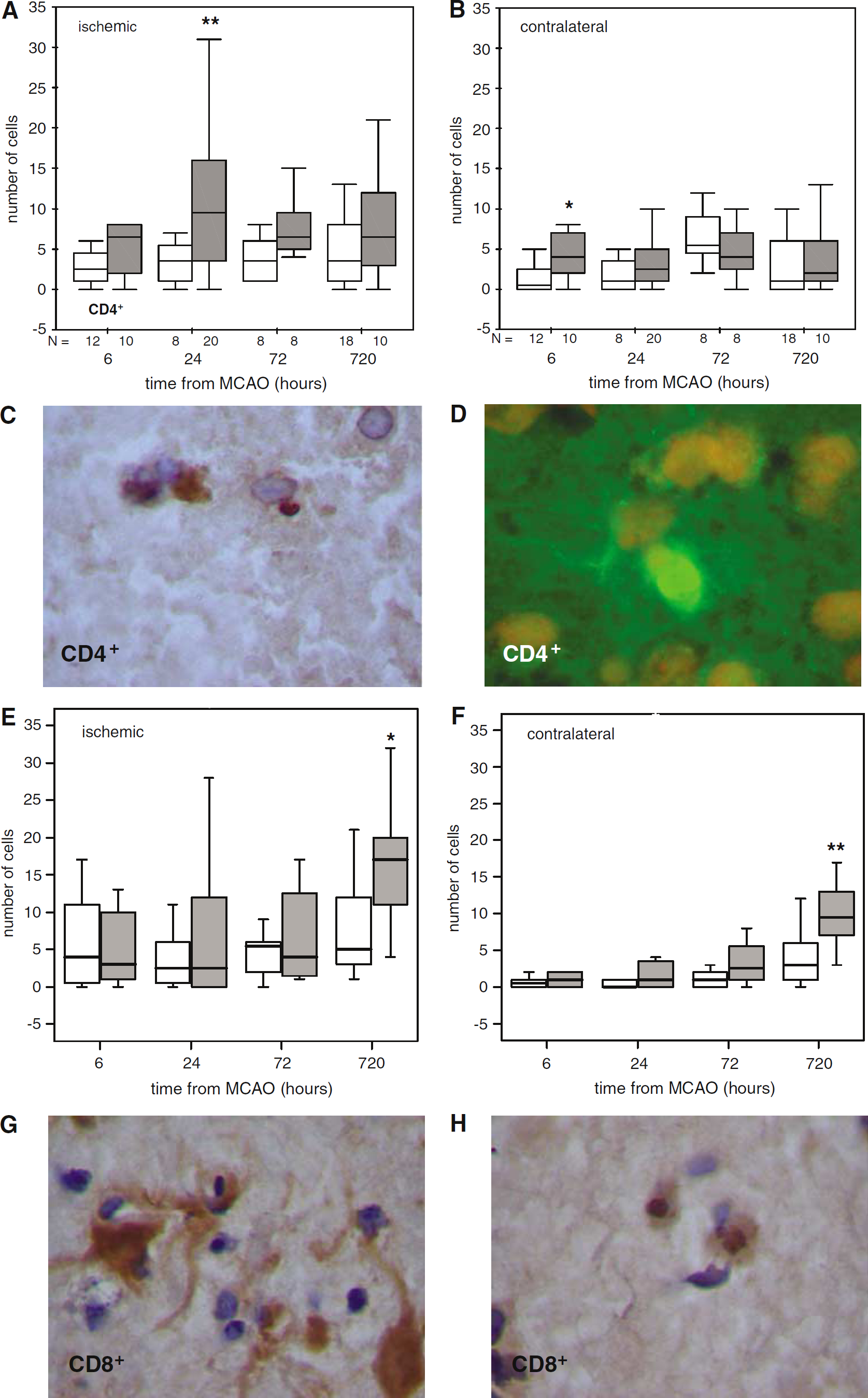
More CD4+ cells are present in both the ischemic (
Costimulatory Molecules
By 24 h after MCAO, more cells expressed B7.1 in both the ischemic and nonischemic hemispheres of LPS(+) animals (21.4 ± 19.0 versus 8.0 ± 7.4; P =0.01 and 11.0 ± 9.5 versus 4.8 ± 4.1; P = 0.02, respectively). This difference persisted to 72 h (Figures 5A and 5B). Lipopolysaccharide(+) animals undergoing MCAO also expressed more B7.1 than sham-operated animals 24 h after LPS administration (P = 0.003). Double-label studies confirmed that cells expressing B7.1 were microglia (Figure 5C). B7.2 expression, on the other hand, was increased in the nonischemic hemispheres of LPS(−) animals 720 h after MCAO (11.6 ± 12.9 versus 3.0 ± 4.5; P = 0.02) (Figures 5D and 5E). Middle cerebral artery occlusion did not alter the expression of B7.2 among LPS-treated animals, although MCAO was associated with an increase in B7.2 expression 720 h after MCAO in LPS(−) animals (P = 0.01). More cells expressed MHC I and MHC II in LPS(+) animals early after MCAO (Figures 5E and 5F). The morphology of cells expressing MHC II was largely that of infiltrating leukocytes, although some cells with the morphology of microglia also appeared to express MHC II (Figure 5G).
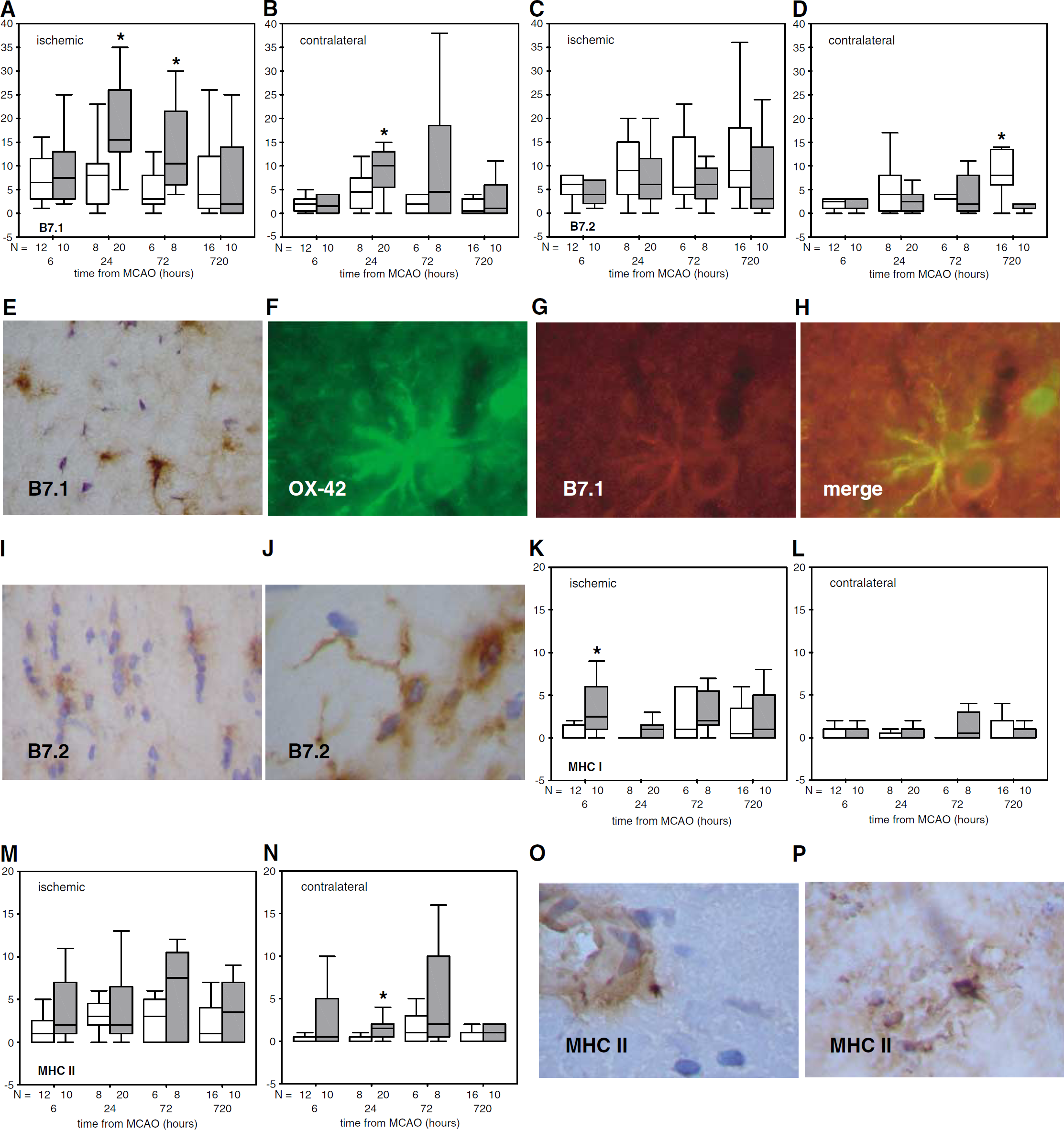
Lipopolysaccharide significantly increases the expression of B7.1 early after MCAO (
Infarct Size
Infarct volume did not differ between LPS(+) and LPS(−) animals at any time point (Figure 6A), and the amount of cerebral edema (volume increase of ischemic compared with nonischemic hemisphere) was similar in LPS(+) and LPS(−) animals from 6 to 72 h after MCAO. At 1 month after MCAO, however, LPS(+) animals evidenced increased atrophy of the ischemic hemisphere (total volume of ischemic hemisphere/total volume of nonischemic hemisphere; 80.9% ± 6.8% versus 89.8% ± 6.6%; P < 0.05) (Figure 6B). The hemispheric atrophy might be a reflection of the fact that there were more apoptotic neurons in both the ischemic (65.8 ± 28.6 versus 23.3 ± 28.7; P = 0.03) and nonischemic (26.0 ± 17.8 versus 8.4 ± 10.7; P < 0.05) hemispheres of LPS(+) animals 1 month after stroke (Figures 6C to 6E). The percentage of apoptotic to total neurons was also significantly greater in LPS(+) animals in the ischemic (16.4% ± 6.8% versus 5.1% ± 6.7%, P = 0.02) and nonischemic (14.7% ± 9.2% versus 3.8% ± 5.0%; P = 0.02) hemispheres. Sham-operated animals treated with LPS (N = 4) had no evidence of neuronal apoptosis 1 month after surgery/LPS administration.
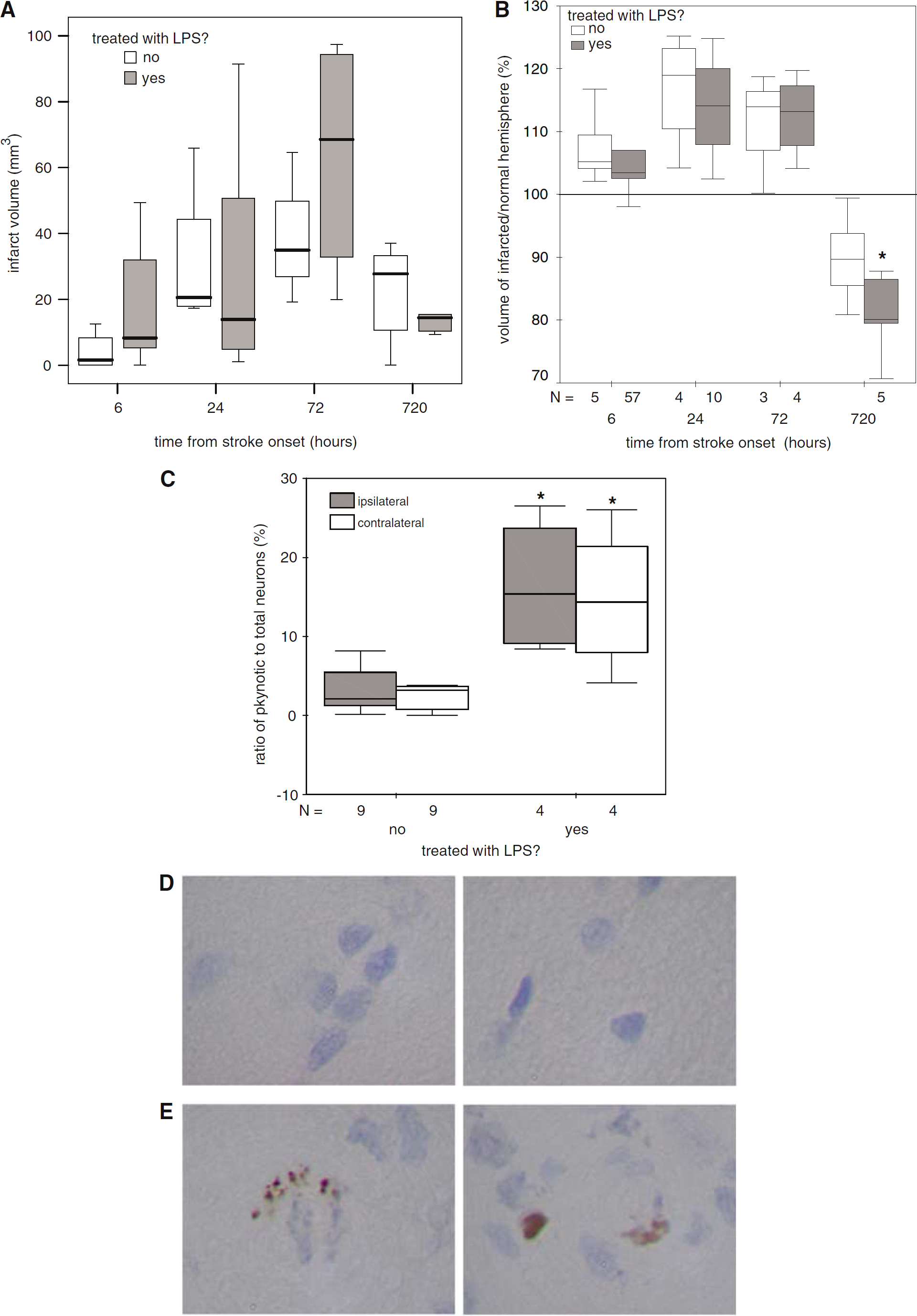
Animals treated with LPS have more brain atrophy after stroke. Infarct volume did not differ between LPS(+) and LPS(−) animals at any time point after MCAO (
Discussion
In these experiments, we show that an autoimmune response to brain does not generally occur after stroke; in fact, animals that experience cerebral ischemia seem predisposed to develop tolerance (a T
The fact that animals become sensitized to brain antigens after stroke would merely be an interesting observation unless there was a clinical correlate to the immune response. It could be argued that the differences in the neurobehavioral outcome of LPS(+) and LPS(−) animals were related directly to LPS, but it seems unlikely that LPS could account for the higher neurologic scores seen 1 week after administration or worse performance on the ‘sticky tape test’ 1 month after administration (Dantzer et al, 1998; Larson et al, 1996). In fact, LPS(−) animals that were T
Infection in the poststroke period is common and is associated with worse outcome (Enlimomab Acute Stroke Trial Investigators, 2001; Davenport et al, 1996; Grau et al, 1999; Johnston et al, 1998). The intent of LPS administration in these experiments was to mimic the inflammatory response associated with systemic infection. The dose of LPS used in these experiments is relatively high and would be expected to compromise the BBB (Singh and Jiang 2004). Our data, however, argue that it is the combination of a cerebral insult and LPS that leads to the CNS autoimmune response. Given that there was no mortality in LPS(+) sham-operated animals and no significant changes in body temperature or weight, the plasma levels of LPS achieved in these animals seem clinically relevant. As such, our data may offer an explanation as to why infection during stroke is associated with worse outcome from stroke. Because Gram-negative bacteria are the predominant organisms causing infection after stroke, the use of LPS in these experiments is clinically relevant (Hilker et al, 2003; Puri et al, 2002). Future studies will need to address whether infection with other pathogens after stroke would similarly predispose to CNS autoimmunity.
Observational studies do not address the issue of whether infection worsens outcome from stroke or is just a marker for persons with more severe strokes; there are, however, several lines of evidence to suggest that infection is deleterious. Firstly, induction of an aseptic systemic inflammatory response after stroke is associated with increased morbidity and mortality (Enlimomab Acute Stroke Trial Investigators, 2001). Secondly, fever, a cardinal feature of the immune response in infection, is associated with worse neurologic outcome after stroke (Azzimondi et al, 1995; Reith et al, 1996). Thirdly, the cytokines secreted by leukocytes during the effector phase of an immune response might be toxic to neurons and glia (Barone et al, 1997; Hanisch et al, 1996; Touzani et al, 1999). Finally, because the bacterial products associated with infection (i.e. LPS) and the biologic mediators of the immune response (cytokines) can alter the microenvironment of the brain and peripheral lymphoid organs, lymphocytes could become sensitized to brain antigens in the context of an infection and contribute to cerebral ischemic injury.
Fever does not appear to be a primary mechanism by which LPS worsens outcome from MCAO, because body temperature was not elevated in LPS(+) or T
The long-term clinical consequences of the CNS autoimmune response after stroke are unknown. When the BBB is compromised by recurrent stroke or systemic inflammation, CNS autoreactive T cells could transit into brain and cause injury. This potential is illustrated by the fact that transgenic animals in which >95% of CD4+ T cells express receptors specific for MBP experience less recovery after spinal cord injury than wild-type controls and that animals immunized to MBP are more likely to die after MCAO (Becker et al, 1997; Jones et al, 2002). Moreover, T lymphocytes obtained from animals shortly after spinal cord injury precipitate histopathologic changes similar to experimental allergic encephalomyelitis (EAE) when injected into naïve animals (Popovich et al, 1996). While the absolute number of MBP-sensitized MNCs isolated from the brains of LPS(+) animals after MCAO in our study is only a fraction of the total number of inflammatory cells in the brain, it is similar to that seen in animals with EAE (where the numbers of MBP-reactive IFN-γ-secreting cells range from 10 to 20 per 1 × 105 MNCs) (Di Rosa et al, 2000; Li et al, 1998; Targoni et al, 2001). It is therefore conceivable that, in the appropriate clinical setting, MBP-specific MNCs generated after stroke could be of clinical consequence.
In summary, we showed that animals treated with LPS during MCAO are more likely to become sensitized to MBP. Furthermore, animals that develop a T