
Abstract
Ubiquitination by Nedd4 (neuronally expressed developmentally downregulated 4) family of HECT type E3 ligases plays a key role in degrading misfolded and damaged proteins, and its disruption leads to neurodegeneration. Parkinson's disease-causing protein α-Synuclein (α-Syn) is ubiquitinated by the Nedd4 family and degraded by endosomes. Nedd4l is the only Nedd4 homolog that showed upregulation in post-stroke surviving cortical neurons where it correlated with neuroprotection. We tested the role of Nedd4l after stroke by subjecting the Nedd4l−/− mice to transient middle cerebral artery occlusion. Focal ischemia significantly increased Nedd4l expression and poly-ubiquitinated α-Syn levels, and knockout of Nedd4l reduced post-ischemic poly-ubiquitinated α-Syn that is majorly located in the peri-infarct neurons. Co-immunoprecipitation further shows that focal ischemia enhances the α-Syn-Nedd4l interaction resulting in increased ubiquitination of α-Syn. Nedd4l knockout mice (n = 7 mice/group) showed exacerbated post-ischemic motor dysfunction manifested by decreased time on the rotarod and increased number of foot faults, and significantly increased ischemic brain damage. This suggests that Nedd4l might be a potential therapeutic target to minimize α-Syn-mediated toxicity after cerebral ischemia.
Introduction
Protein aggregation is a pathological hallmark of various chronic neurodegenerative disorders such as Alzheimer’s disease and Parkinson’s disease (PD). 1 Even acute injuries to CNS like an ischemic stroke can promote cerebral protein aggregation.1,2 In addition to specific disease-associated proteins such as β-amyloid and α-synuclein (α-Syn), several other proteins like ubiquitin also localize to the aggregates. Ubiquitination plays a crucial role in the degradation of abnormal proteins.3,4 Once ubiquitinated, proteins are degraded either by the ubiquitin-proteasome pathway or by the endosomal–lysosomal pathway. 5 Disruption or overloading of these mechanisms leads to the accumulation of damaged, oxidized, and misfolded proteins that were implicated in neurodegenerative conditions.
Nedd4 (neuronally expressed developmentally downregulated 4) is a HECT-type E3 ubiquitin ligase family protein. 6 The nine members of the Nedd4 family including the archetypal homologs Nedd4 and Nedd4l share the modular structure with a conserved catalytic HECT domain. 6 α-Syn, which is implicated in the pathogenesis of PD, is a major substrate for the Nedd4 family and once ubiquitinated will be subjected to ubiquitin-mediated endosomal degradation.7,8 Overexpression of Nedd4 prevents α-Syn-induced locomotor defects, while Nedd4 knockdown increases α-Syn-induced dopaminergic neuronal loss in PD models. 9 This indicates that α-Syn ubiquitination by Nedd4 may limit α-Syn levels and hence its cellular toxicity.
We have recently reported that focal ischemia in rodents induces α-Syn expression and aggregation, and knockdown or knockout of α-Syn decreases infarction and promotes better neurological recovery.10,11 This was partly due to the serine-129 phosphorylation of α-Syn, indicating the role of post-translational modifications in α-Syn toxicity. 10 A recent study indicated that Nedd4l levels, but not other Nedd4 homologs, are upregulated in surviving cortical neurons and correlated with cell survival after focal ischemia. 12 We presently evaluated if Nedd4l homolog is responsible for post-ischemic α-Syn ubiquitination and whether its disruption affects the post-stroke functional outcome.
Materials and methods
All experimental protocols using animals were approved by the University of Wisconsin Research Animal Resources and Care Committee, and animals were cared in accordance with the Guide for the Care and Use of Laboratory Animals, U.S. Department of Health and Human Services Publication Number 86-23 (revised). In all experiments, animals were randomly assigned to groups. Behavioral and histological analyses were performed by an investigator blinded to the study groups. Experiments were conducted in compliance with the “Animal Research: Reporting of In Vivo Experiments (ARRIVE)” guidelines.
Focal ischemia
Adult, male, homozygous Nedd4l−/− mice (BALB/cByJ-Nedd4landi/EiJ) (four to five months old; 24–28 g) and the respective wild-type controls were obtained from Jackson Labs, USA. The Nedd4l−/− knockout mice were normal in appearance without showing differential performance in sensorimotor tests before ischemic insult. Under isoflurane anesthesia, mice were subjected to transient middle cerebral artery occlusion (MCAO) for either 60 min or 90 min, followed by either 24 h or seven days of reperfusion, as described previously. 11 Sham-operated mice served as control. Regional cerebral blood flow and physiological parameters (pH, PaO2, PaCO2, hemoglobin, and blood glucose) were monitored, and rectal temperature was maintained at 37.0 ± 0.5°C during surgery. At 1 h of reperfusion, animals were tested with neurological severity scoring where a score of 0 suggests no neurological deficit (normal), 1 being mild neurological deficit (failure to fully extend right forepaw), 2 being moderate neurological deficit (circling to the right), 3 being severe neurological deficit (falling to the right) and 4 being very severe neurological deficit (no spontaneous movement). Rodents with no evidence of neurological deficit (score 0) were excluded. Upon euthanasia, rodents that showed hemorrhage were also excluded from analyses. 10
Motor function and infarct volume determination
Post-ischemic motor function was evaluated by the rotarod test (3 min on a cylinder rotating at 8 r/min), the beam walk test (number of foot faults while crossing a 5 mm × 60 cm beam for mice) at days 1, 3, 5 and 7 of reperfusion as described earlier. 11 On day 7, animals were euthanized by transcardiac 4% phosphate-buffered paraformaldehyde (PFA) perfusion. Each brain was post-fixed, cryoprotected, and sectioned (coronal; 40 µm thickness at an interval of approximately 400 µm). Serial sections were stained with Cresyl violet and scanned using the NIH ImageJ software. Ischemic infarct volume was estimated by numeric integration of data from six serial coronal sections with respect to the sectional interval as described earlier. 13 Each infarct volume was corrected to account for edema and differential shrinkage during tissue processing using the Swanson formula. 14
Western blotting
The brain tissues collected from the ipsilateral peri-infarct region were homogenized using the T-PER Tissue Protein Extraction Reagent (Cat. #: 78510; Life Technologies, USA) and the Halt Protease Inhibitor Cocktail, EDTA-Free (Cat. #: 87785; Life Technologies, USA). Homogenates were centrifuged at 10,000g for 10 min at 4°C. Loading samples were prepared using the NuPAGE LDS Sample Buffer (Cat. #: NP0008; Life Technologies, USA) then denatured by heating at 70°C for 10 min. Samples (40 µg protein equivalent) were electrophoresed using the Novex system from Life Technologies, USA, transferred to nitrocellulose membranes and probed with antibodies against α-Syn (Cat. #: 4179S; 1:1000; Cell Signaling Technology, USA) and Nedd4l (Cat. #: 4013S; 1:1000; Cell Signaling Technology, USA) followed by HRP-conjugated anti-rabbit IgG (1:3,000; Cell Signaling Technology, USA). Blots were stripped and reprobed with antibodies against β-actin (Cat. #: 4970S; 1:3000; Cell Signaling Technology, USA). Blots were developed using enhanced chemiluminescence (Cat#: 34076; Life Technologies, USA) and quantified with Image Studio software (LI-COR Biotechnology, USA).
For identifying mono- and poly-ubiquitinated α-Syn, lysate samples containing ∼0.15 mg protein per sample were further enriched by Pierce Ubiquitin Enrichment Kit (Cat. #: 89899; Life Technologies, USA) as per the manufacturer’s instructions. Enriched samples containing mono- and poly-ubiquitinated proteins were then processed for standard Western blotting analysis. Non-specific ubiquitin proteins were probed using an antibody included in the Pierce Ubiquitin Enrichment Kit.
Co-immunoprecipitation
Co-immunoprecipitation (Co-IP) experiments were conducted using the Abcam Immunoprecipitation kit (Cat. # ab206996; Abcam, USA) as per the manufacturer's recommendations. Briefly, the brain tissue collected from the ipsilateral peri-infarct region was homogenized in non-denaturing lysis buffer containing protease inhibitor cocktail. Homogenates were centrifuged at 10,000g for 5 min at 4°C. α-Syn (Cat. # 4179S; 1:50; Cell Signaling Technology, USA) antibody was added to each sample (300 µg protein equivalent) and incubated overnight at 4°C on a rotary mixer. Rabbit IgG was used as control. After the antibody binding step, 25 µl of protein A/G Sepharose beads slurry was added and incubated for 1 h at 4°C. The protein-bead complex was washed three times, centrifuged and eluted with 40 µl 2× NuPAGE LDS Sample Buffer (Cat. # NP0008; Life Technologies, USA) for 5 min at 80°C. Immunoblotting was performed as described above.
Immunostaining
Brain sections (30 µm thickness) were immunostained with antibodies against α-Syn (Abcam; Cat#: ab1903; 1:200), NeuN (Millipore; Cat#: MAB377; 1:300), GFAP (Abcam; Cat#: ab10062; 1:200), MAP2 (Abcam; Cat#: ab11267; 1:300), Nedd4l (Cell Signaling Technology; Cat#: 4013S; 1:50) and TMEM119 (Synaptic Systems, Cat#400011; 1:500), as described earlier. 10 To ensure that the homologous areas of injury were used as samples between animals, sections between the coordinates +1 to +1.5 from Bregma were used in all cases. Analyses were performed by an investigator blinded to the study groups. Brain sections were scanned, and images were produced by Keyence BZX fluorescence microscope (Keyence, USA).
Statistical analyses
Normality was assessed by the Shapiro–Wilk test. For analyzing data that were collected repeatedly from the same subject at different time points (e.g. rotarod and beam-walk test), we performed a nonparametric two-way repeated-measures ANOVA with Sidak’s multiple-comparisons test. For comparing two groups (e.g. lesion volume, and Western blots), we used a nonparametric Mann–Whitney U-test. Specific statistics used were specified in the corresponding figure legend. GraphPad Prism software was used for statistical analyses.
Results
Ablation of Nedd4l exacerbated ischemic brain damage
Motor function recovery after transient MCAO assessed with rotarod test (Figure 1(a)) and beam walk test (Figure 1(b)) was significantly worsened between days 5 and 7 of reperfusion in the Nedd4l knockout mice compared with wild-type mice. Nedd4l null mice also showed significantly increased infarct volume at seven days of reperfusion compared with wild-type mice (Figure 1(c) and (d)).
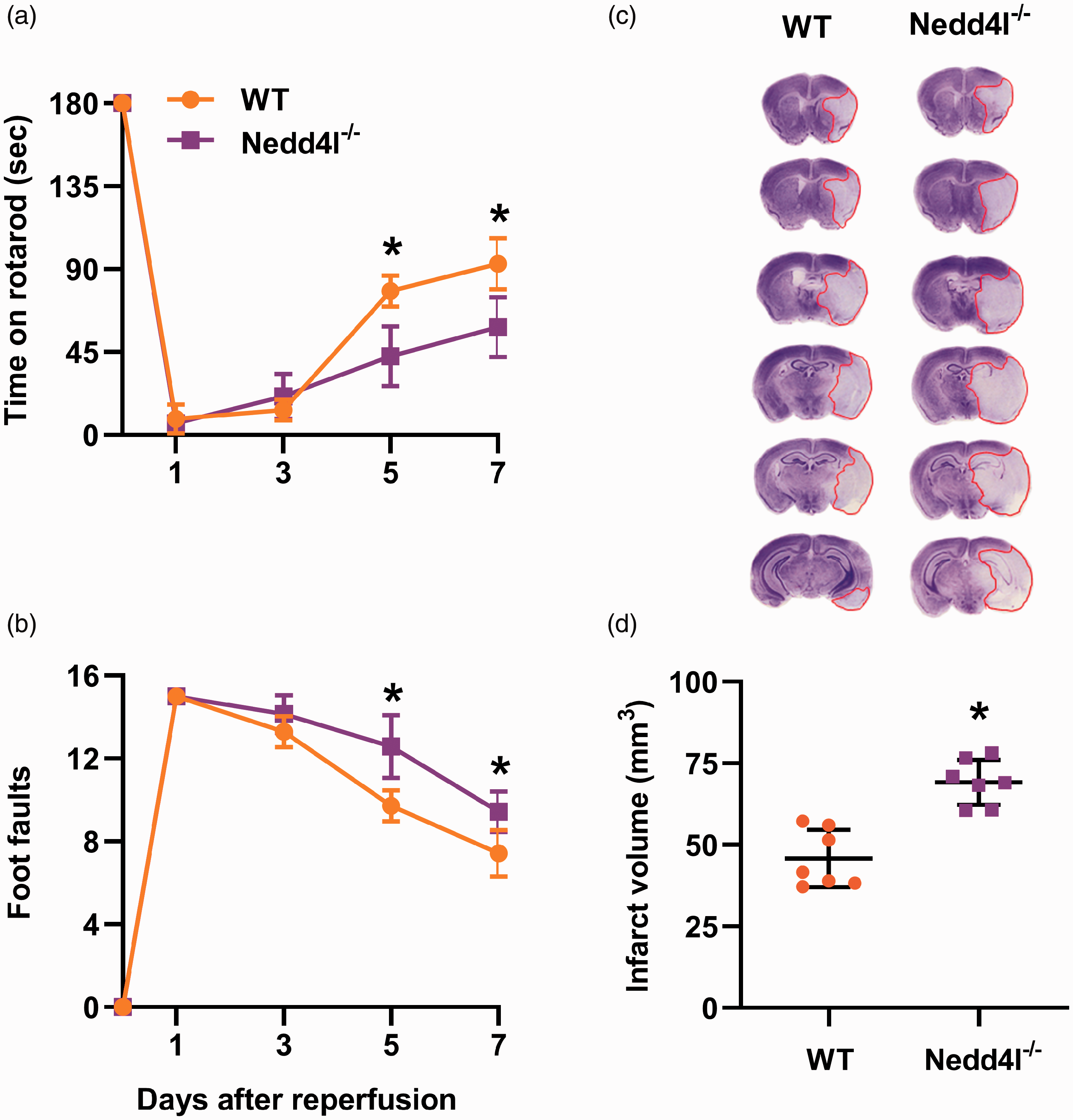
Nedd4l knockout exacerbated motor function recovery and increased infarct volume in young male mice. (a and b) Recovery of functional performance on the rotarod test (a) and beam walk test (b) by wildtype (WT) and Nedd4l knockout (KO) mice that underwent 60 min of transient MCAO. The performance was assessed before treatment (time 0) and on days 1 to 7 of reperfusion. Data are means ± SD (n = 7 mice per group). *p < 0.05 compared with the respective WT group by repeated-measures analysis of variance (ANOVA) followed by Sidak’s multiple comparisons posttest. (c and d) Representative cresyl violet-stained serial sections (c) and quantified infarct volume (d) in brains from WT and Nedd4l KO mice. Infarct volume was measured at seven days of reperfusion. Data are means ± SD (n = 7 mice per group). *p < 0.05 compared with the WT group by Mann–Whitney U test. Mice were randomly assigned to experimental groups, and brains were obtained and analyzed by an investigator blinded to the study groups.
Nedd4l mediates poly-ubiquitination of α-Syn after stroke
Focal ischemia significantly increased non-specific polyubiquitination (Figure 2(a) and (b)) and decreased monoubiquitination (Figure 2(a) and (c)) in the peri-infarct cortex of wild-type mice at one day of reperfusion following transient MCAO compared with sham. Nedd4l knockouts subjected to focal ischemia showed significantly increased non-specific polyubiquitination but not monoubiquitination compared to sham. Nedd4l protein expression was induced significantly (∼2.8 fold; p < 0.05) in the peri-infarct cortex of wild-type mice at one day of reperfusion following transient MCAO compared with sham (Figure 2(d) and (e)). Nedd4l knockouts showed no Nedd4l protein in sham or ischemic cohorts. Following transient MCAO, total α-Syn protein levels significantly increased in wild-type mice (by ∼46%; p < 0.05) compared with sham (Figure 2(d) and (f)), while transient MCAO in Nedd4l knockout mice showed a further increase in total α-Syn protein levels (by ∼41%; p < 0.05) at one day of reperfusion compared with wild-type mice (Figure 2(d) and (f)). Focal ischemia in wild-type mice also significantly increased poly-ubiquitinated α-Syn (by ∼2 fold; p < 0.05) compared to sham (Figure 2(d) and (g)). Nedd4l knockout resulted in a significant reduction (by ∼30%; p < 0.05) of post-ischemic poly-ubiquitinated α-Syn over wild-type (Figure 2(d) and (g)). Transient MCAO had no significant effect on mono-ubiquitinated α-Syn either in wild-type or Nedd4l knockouts (Figure 2(d) and (h)).
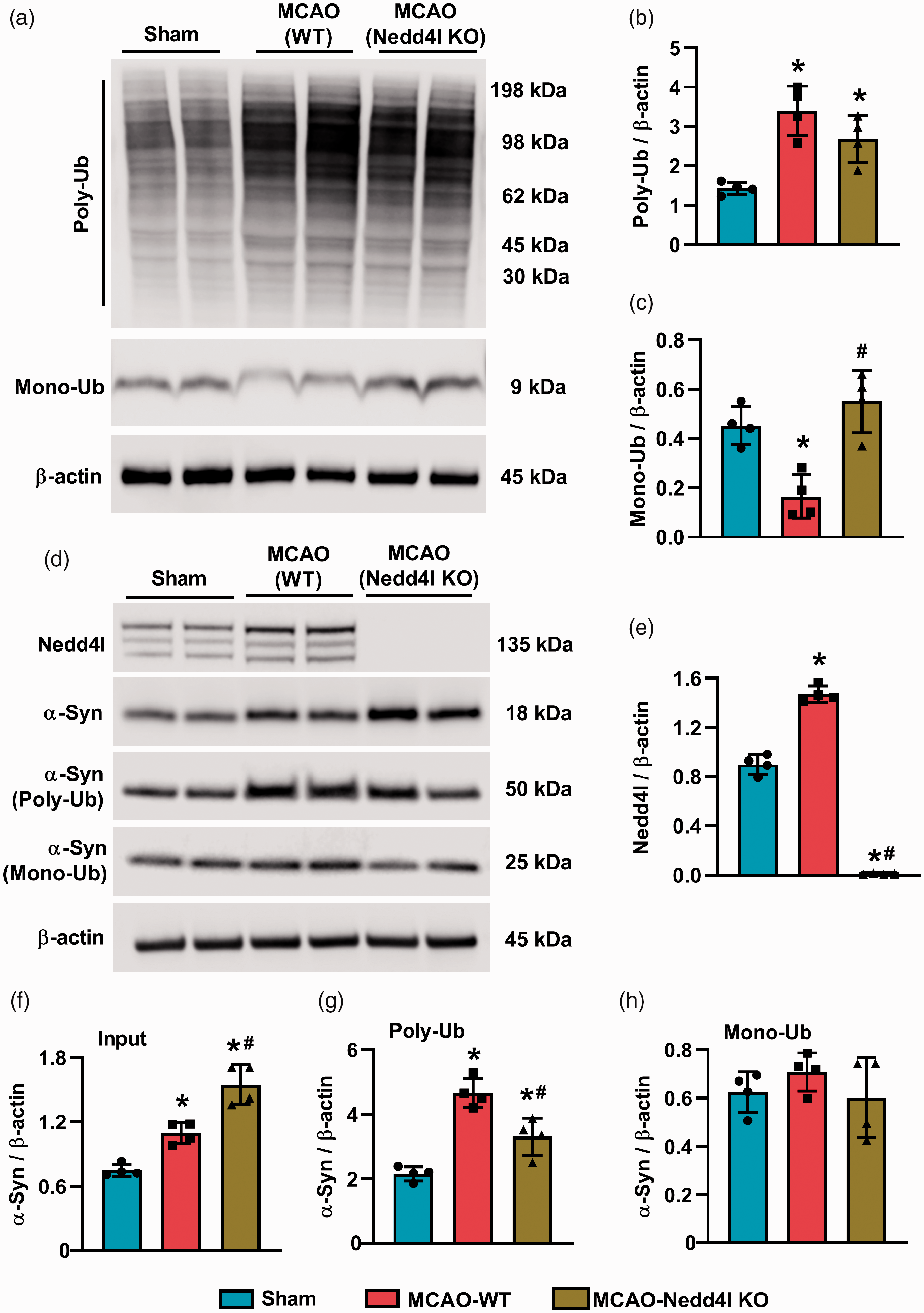
Nedd4l knockout decreased the post-ischemic induction of α-Syn poly-ubiquitination. (a to c) Western blotting and quantification of poly- (a and b) and mono-ubiquitinated proteins (a and c). (d to h) Nedd4l (d and e), total α-Syn (d and f), poly-ubiquitinated α-Syn (d and g), mono-ubiquitinated α-Syn (d and h) in the peri-infarct region of the ipsilateral cortex from sham, MCAO (WT) and MCAO (Nedd4l knockout) mice, assessed at 24 h of reperfusion after 90-min transient MCAO. Note the heavier molecular weight of poly-ubiquitinated α-Syn (∼50 KDa) due to chemical modification by the ubiquitination enrichment kit as Poly-Ubiquitin Affinity Resin binds polymers of ubiquitin containing four or more ubiquitin subunits. Blots are representative of four independent experiments. Data are mean ± SD (n = 4 mice per group). *p < 0.05 compared to sham and #p < 0.05 compared to the wildtype MCAO group, by Mann–Whitney U test.
Focal ischemia enhances α-Syn-Nedd4l interaction and ubiquitination of α-Syn
Following transient MCAO, Nedd4l protein from the ipsilateral peri-infarct cortex was precipitated by the α-Syn antibody but not by a non-specific IgG (Figure 3(a)). Nedd4l protein binding to α-Syn was significantly increased (∼2.5 fold; p < 0.05) in the peri-infarct cortex of wild-type mice at one day of reperfusion following transient MCAO compared with sham (Figure 3(a) to (c)). Post-ischemic samples immunoprecipitated by the α-Syn antibody further showed significantly increased ubiquitination (∼3.0 fold; p < 0.05) compared with sham (Figure 3(d) to (f)).
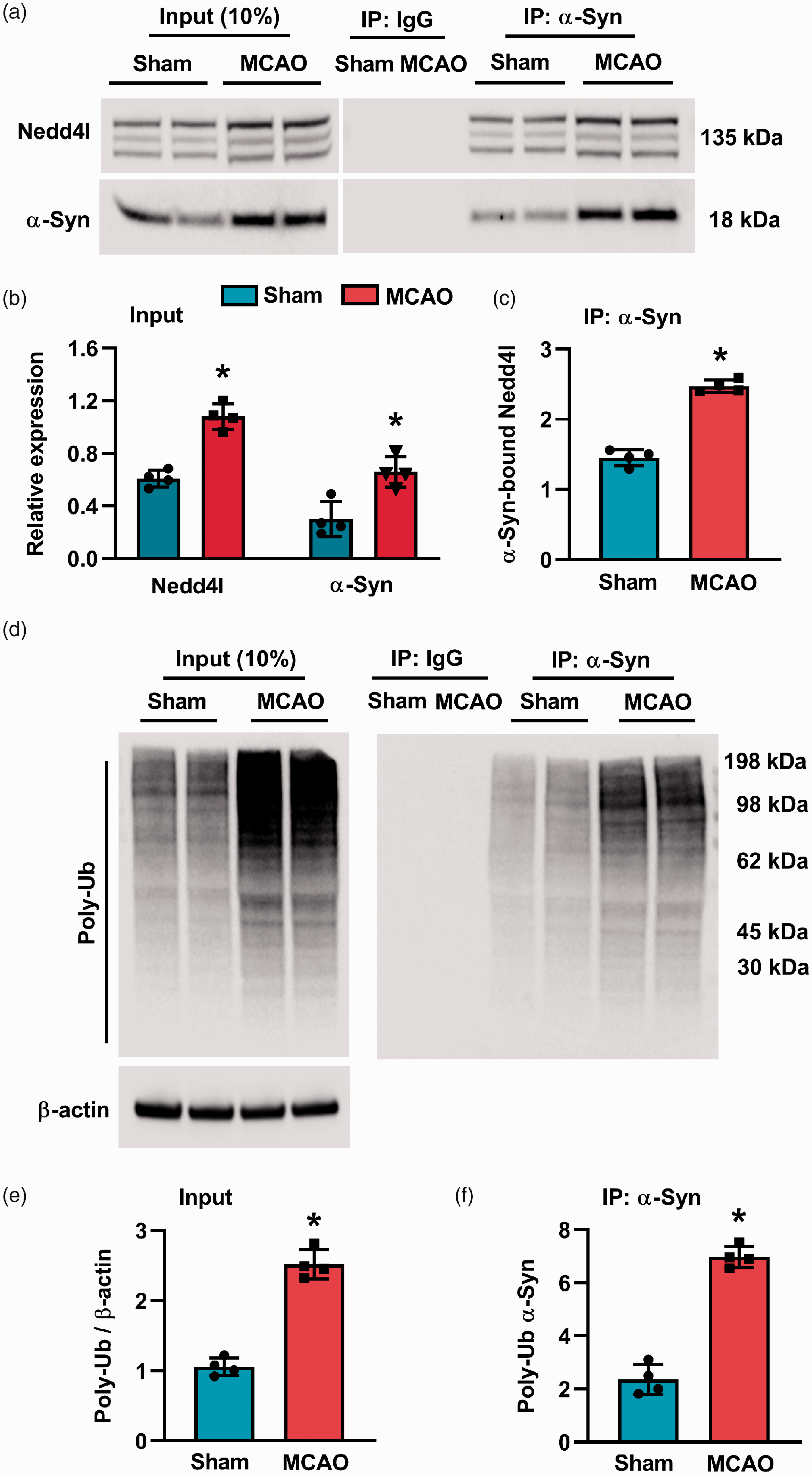
Ischemic stroke increased α-Syn-Nedd4l protein interaction. (a to c) Representative gel images (a) and quantification (b and c) of co-IP showing the effect of ischemic stroke on the α-Syn-Nedd4l association in the peri-infarct region of the ipsilateral cortex from sham and MCAO (WT) mice, assessed at 24 h of reperfusion after 90-min transient MCAO. (d to f) Representative gel images (d) and quantification (e and f) of co-IP showing the effect of ischemic stroke on the α-Syn-ubiquitin association in the peri-infarct region of the ipsilateral cortex from sham and MCAO (WT) mice, assessed at 24 h of reperfusion after 90-min transient MCAO. Blots are representative of four independent experiments. Data are mean ± SD (n = 4/group). *p < 0.05 compared to sham by Mann-Whitney U test.
Nedd4l is co-localized with α-Syn
Increased Nedd4l immunostaining was observed in both NeuN+ (Figure 4(a)) and MAP2+ (Figure 4(b)) cells in the peri-infarct cortex of wild-type mice at 1 day of reperfusion following transient MCAO compared with sham. Nedd4l did not co-localize with GFAP-positive astrocytes (Figure 4(c)) and TMEM119-positive microglia (Figure 4(d)) in the ischemic brain. Following transient MCAO, Nedd4l was also co-localized with α-Syn (Figure 4(c)).
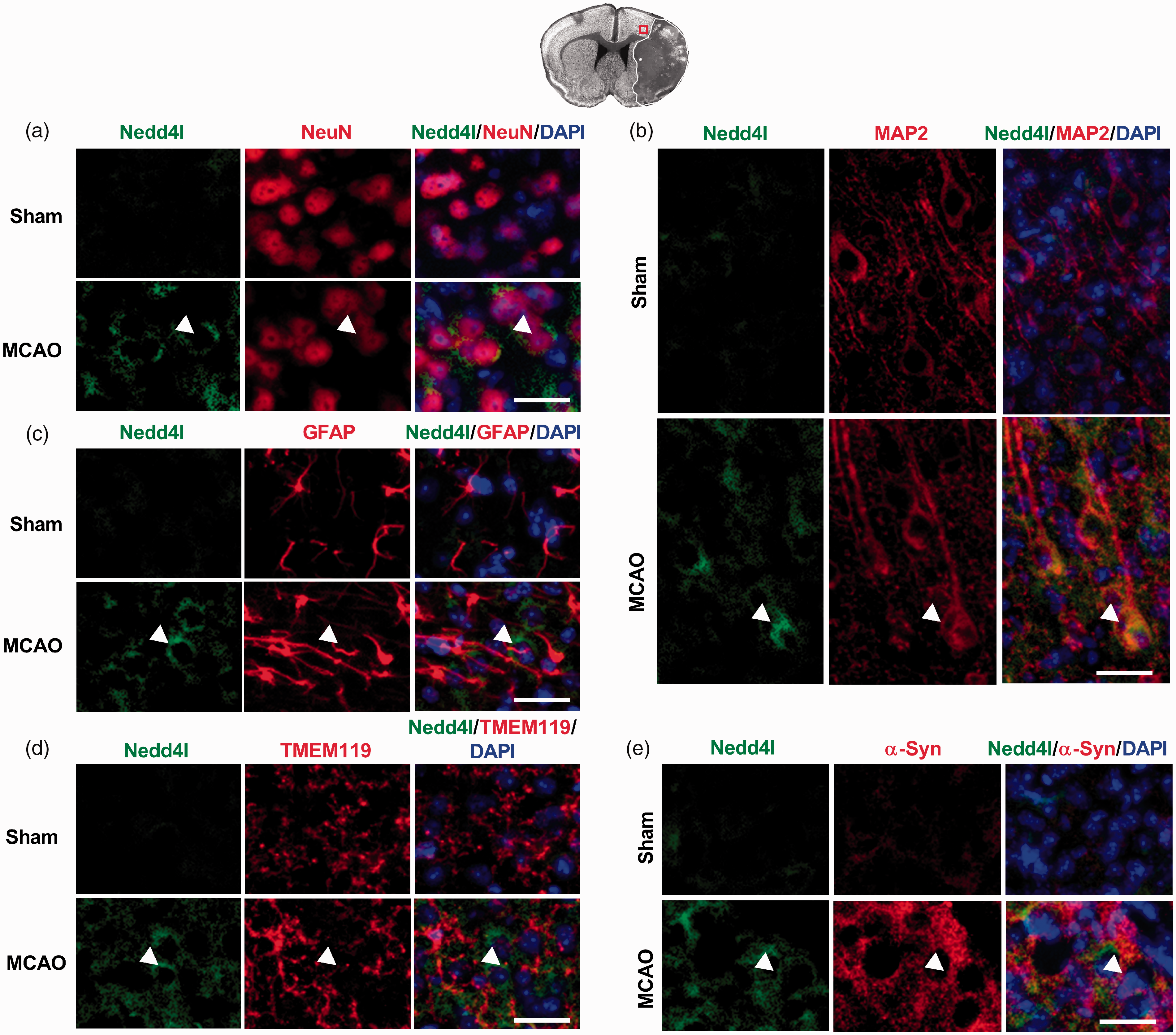
Localization of Nedd4l in the post-ischemic brain. (a to d) Immunostaining of Nedd4l in the NeuN+ (a), MAP2+ (b) GFAP+ (c), and TMEM119+ (d) cells. (e) Co-immunostaining of Nedd4l and α-Syn. Images were taken from the ipsilateral cortical peri-infarct region of Sham or ischemic mice assessed at 24 h of reperfusion after 60-min transient MCAO. Representative immunofluorescence images were taken from the cortical peri-infarct region, where neurons were still relatively intact and viable, as shown in the brain section at the top. Scale bar, 30 µm.
Discussion
In brief, focal ischemia increased Nedd4l and poly-ubiquitinated α-Syn which colocalized mainly in neurons in the peri-infarct area. Genetic deletion of Nedd4l in mice reduced post-ischemic α-Syn poly-ubiquitination, exacerbated motor function deficits, and increased infarction. Both chronic neurodegenerative disorders and acute neurological injuries result in elevated levels of cerebral ubiquitin-protein conjugates which might be an endogenous protective effort to clear the increased load of damaged/misfolded proteins. In particular, α-Syn is highly susceptible to ubiquitination, and dysfunction in α-Syn ubiquitination contributes to chronic synucleinopathies. 15 α-Syn can be ubiquitinated by several ligases that inhibit α-Syn pathology.7,9,16 However, α-Syn ubiquitination by some ligases was found to exacerbate neurotoxicity. 17 Furthermore, there are contradictory findings that Nedd4-mediated ubiquitination promotes α-Syn toxicity and neurodegeneration by ligating heat shock transcription factor 1 (HSF1). 18 Although stroke is known to induce α-Syn expression, we presently show that α-Syn ubiquitination also increases after focal ischemia, which might in part mediated by Nedd4l induction. Nedd4l knockout exacerbated functional recovery and brain damage, indicating a neuroprotective role of Nedd4-mediated α-Syn ubiquitination after stroke.
The Nedd4 family of HECT ubiquitin ligases comprises nine members encoded by unique genes in humans, defined by a shared modular structure. In addition, the Nedd4 family has been implicated in the regulation of a number of substrates post-translationally, including ion channels and membrane receptors, endocytic machinery components, and the tumor suppressor genes. 6 Given its diverse substrates and multiple members within the same family, we were only able to observe partial suppression of α-Syn ubiquitination in Nedd4l knockouts after focal ischemia. However, a worsened functional outcome and bigger infarcts in Nedd4l knockouts indicate that Nedd4l ligase activity is vital in protecting the post-ischemic brain from α-Syn toxicity.
Nedd4 levels were found to be decreased in nigral neurons of human PD brains where it negatively regulates the pro-apoptotic protein RTP801 by conjugating K63-polyubiquitin chains for degradation. 19 In addition, Nedd4l was shown to mediate ubiquitination of glutamate transporters in 1-methyl-4-phenylpyridium (MPP+)-treated astrocytes and in the midbrain of the PD mouse model. 20 Nedd4l knockdown decreased ubiquitination of glutamate transporter, leading to increased uptake, curtailed astrogliosis and microgliosis leading to ameliorated movement disorders in PD mice. 20
Following endothelin-1-induced focal ischemia in rats, Nedd4l was shown to be upregulated and correlated with the number of surviving cortical neurons. 12 The present study also observed increased Nedd4l and α-Syn polyubiquitination in the ischemic peri-infarct area where neurons survive after transient MCAO. By demonstrating that Nedd4l knockouts show diminished α-Syn polyubiquitination and exacerbated brain damage, we further demonstrated the neuroprotective efficacy of this mechanism. Future studies are warranted to elucidate a direct causal mechanistic link between Nedd4l-mediated ubiquitination of α-Syn in mediating α-Syn-induced ischemic brain injury.
Supplemental Material
sj-pdf-1-jcb-10.1177_0271678X20943804 - Supplemental material for Deletion of ubiquitin ligase Nedd4l exacerbates ischemic brain damage
Supplemental material, sj-pdf-1-jcb-10.1177_0271678X20943804 for Deletion of ubiquitin ligase Nedd4l exacerbates ischemic brain damage by TaeHee Kim, Anil K Chokkalla and Raghu Vemuganti in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported, in part, by the US Department of Veterans Affairs Merit Review Grant I01 BX002985, NIH grants RO1 NS101960.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
TK and RV contributed to the conception and design of the study; TK and AKC contributed to the acquisition and analysis of data; TK and RV contributed to drafting the text.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.