
Abstract
The ubiquitin-proteasome system (UPS) and autophagy are two major pathways to degrade misfolded proteins that accumulate under pathological conditions. When UPS is overloaded, the degeneration pathway may switch to autophagy to remove excessive misfolded proteins. However, it is still unclear whether and how this switch occurs during cerebral ischemia. In the present study, transient middle cerebral artery occlusion (tMCAO) resulted in accelerated ubiquitin-positive protein aggregation from 0.5 h of reperfusion in mice brain after 10, 30 or 60 min of tMCAO. In contrast, significant reduction of p62 and induction of LC3-II were observed, peaking at 24 h of reperfusion after 30 and 60 min tMCAO. Western blot analyses showed an increase of BAG3 and HDAC6 at 1 or 24 h of reperfusion that was dependent on the ischemic period. In contract, BAG1 decreased at 24 h of reperfusion after 10, 30 or 60 min of tMCAO after double immunofluorescent colocalization of ubiquitin, HSP70, p62 and BAG3. These data suggest that a switch from UPS to autophagy occurred between 10 and 30 min of cerebral ischemia depending on the BAG1/BAG3 ratio and level of HDAC6.
Keywords
Introduction
Quality control of intracellular proteins is essential for cells to maintain their function and survival, so eukaryocytes require a precise control mechanism for protein synthesis, processing and degradation. 1 Misfolded proteins are mainly degraded by the ubiquitin-proteasome system (UPS) or autophagy. 2 UPS consists of 26S proteasome as the major protease complex with two subcomplexes, the 20S catalytic core and the 19S regulatory subunit.3,4 Autophagy mainly targets long-lived proteins, protein aggregates and organelles. 5
UPS and autophagy pathways require HSP70, 6 p62, 7 and LC3-II. 8 Two members of the BAG (Bcl2-associated athanogene) protein family, BAG1 and BAG3, as well as histone deacetylase 6 (HDAC6), also participate in protein quality control mechanisms.5,9,10 Although UPS and autophagy are now recognized as complementary protection systems against neurodegenerative processes, 11 the switch in these processes following cerebral ischemia is still incompletely understood.
In the present study, we thus investigated the alterations of UPS and autophagy activities in relation to the above key proteins after a transient middle cerebral artery occlusion (tMCAO) in mice, and examined whether there was crosstalk between these two pathways after cerebral ischemia.
Materials and methods
Animals
All animal experiments were performed in compliance with a protocol approved by the Animal Committee of the Graduate School of Medicine and Dentistry, Okayama University (Changes in proteasome and autophagy activity in a stroke mice model; OKU-2017-248) and conducted in accordance with ARRIVE guidelines (https://www.nc3rs.org.uk/arrive-guidelines) and the Okayama University guidelines on the Care and Use of the Laboratory Animals. Experiments were performed in male ICR mice (aged eight weeks; mean weight 34.6 g, n = 108). Mice were randomly assigned to the experimental groups as shown in Figure 1(a).
(a) Experimental groups, and (b) representative Nissl staining sections, showing the infarction at different reperfusion times after tMCAO. (c) Quantitative analysis of cerebral infarct volume (*p < 0.05 versus 30 min. Scale bar = 50 µm).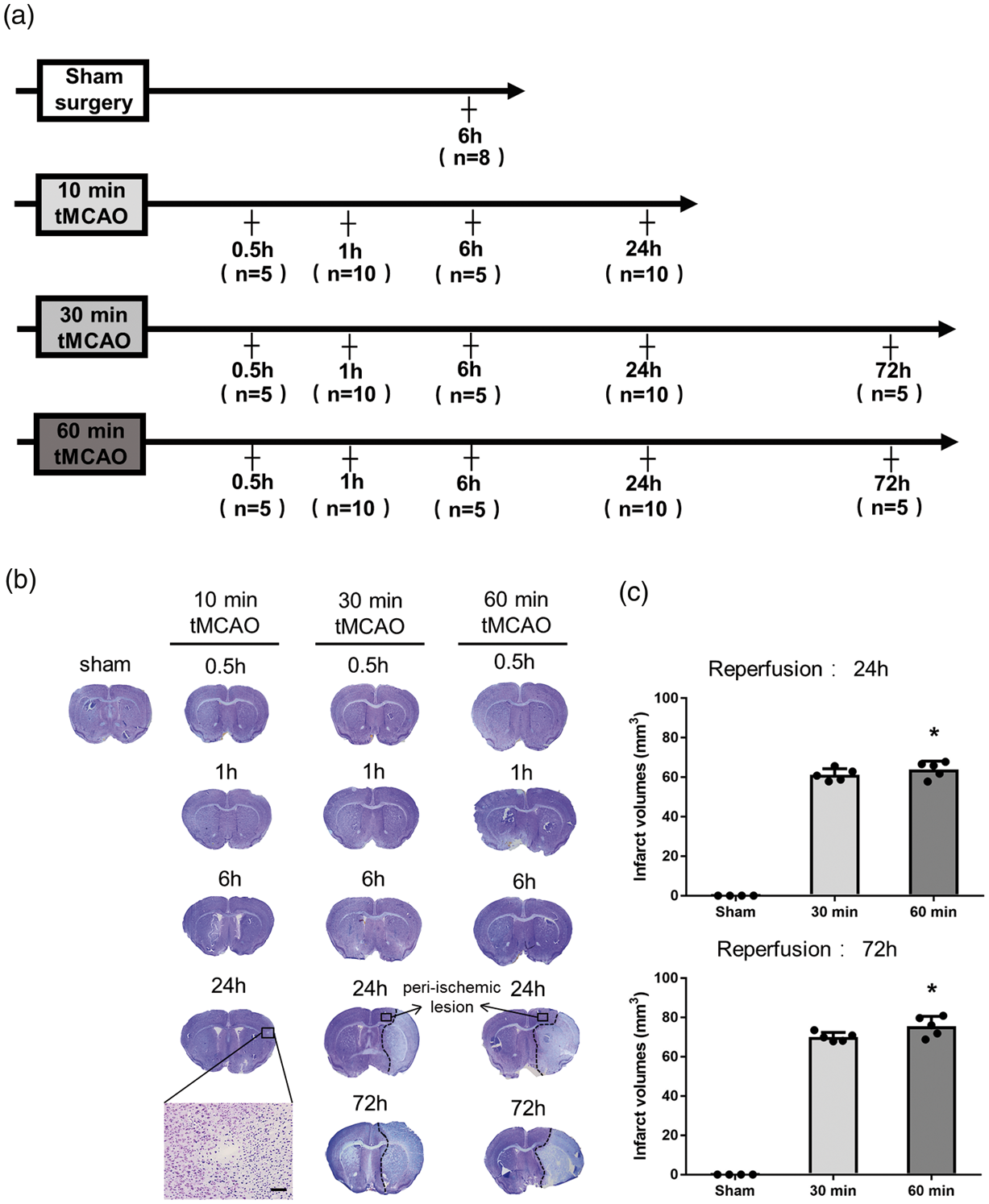
Focal cerebral ischemia
Transient focal cerebral ischemia was induced using the intraluminal filament model of right middle cerebral artery occlusion (MCAO) of mice as described previously.12,13 Briefly, mice were anesthetized with a mixture of nitrous oxide/oxygen/halothane (69:30:1%) during surgery with an inhalation mask and tied to a heating pad to monitor and maintain the body temperature at 37 ± 0.5℃ in a supine position. A silicon-coated 8–0 nylon filament thread was inserted into the right common carotid artery and advanced along the internal carotid artery until it occluded the MCA. The occluding filament remained in position for the ischemic period (10 min, 30 min or 60 min) and was subsequently retracted to induce reperfusion. For sham MCAO, vessels were visualized and cleared of connective tissue, but no further manipulations were made. The successful artery occlusion was evaluated by measuring the decrease in cerebral blood flow (CBF) of the right frontoparietal cortical region by using a Laser-Doppler flowmeter (model ALF21; Advance, Tokyo, Japan). We defined a successful tMCAO as a >85% decrease of the baseline (before ischemia) CBF during ischemia and a >80% increase of baseline CBF during reperfusion. 14
Tissue preparation
The mice for histologic analysis were deeply anesthetized after 0.5 h, 1 h, 6 h, 24 h and 72 h of reperfusion and then perfused with chilled phosphate-buffered saline (PBS, pH 7.4), followed by 4% paraformaldehyde in PBS. After washing with PBS, the brains were incubated in 10, 20 and 30% (wt/vol) sucrose in PBS, each for 24 h at 4℃. Brains were then frozen in liquid nitrogen and stored at −80℃. Successive 20-µm thick coronal sections were cut on a cryostat at −20℃ and mounted on silane-coated glass slides. The mice for Western blot analysis were decapitated under deep anesthesia, and the brains were quickly removed, then frozen in liquid nitrogen and stored at −80℃.
Measurement of infarct volume
Brain sections were stained with cresyl violet and observed under a light microscope (Olympus BX-51; Olympus Optical). Infarct volume was measured in five sections by counting pixels in Photoshop CS6, and then calculating the volume as described previously. 15
Immunohistochemistry
Brain sections were immersed in 0.3% hydrogen peroxide/methanol for 30 min to block non-specific antibody expression, and then incubated with 5% bovine serum albumin (BSA) in PBS with 0.1% triton for 1 h to block non-specific antibody responses. Slides were incubated at 4℃ overnight with the following primary antibodies: mouse anti-NeuN antibody (1:200, Millipore Corporation, Billerica, MA, USA), rabbit anti-c-Fos antibody (1:100, Millipore Corporation), rabbit anti-ubiquitin antibody (1:200, DAKO, Carpinteria, CA, USA) and mouse anti-p62 antibody (1:200, Abcam, Cambridge, MA, USA). After washing with PBS, brain sections were treated with suitable biotinylated secondary antibodies (1:500; Vector Laboratories) at room temperature for 2 h. Sections were incubated with the avidin-biotin-peroxidase complex (VECTASTAIN Elite ABC Kit; Vector Laboratories) for 30 min and visualized with 3,3′-diaminobenzidine (DAB). For the negative control, a set of brain sections were stained in the same manner without the primary antibody. These sections were analyzed under a light microscope (Olympus BX-51, Tokyo, Japan). We analyzed Nissl staining of 30 and 60 min tMCAO groups and set the peri-ischemic lesion to a precise area as shown in Figure 1(b), and all data related to the peri-ischemic lesion were collected from this region in all mice groups for analysis. For semiquantitative analysis, the number of NeuN-, c-Fos-, ubiquitin- and p62-positive cells was calculated in the peri-ischemic lesion of three sections per brain and five randomly selected regions.
Double immunofluorescence histochemistry
Brain sections were incubated with 5% BSA in PBS with 0.1% triton at room temperature for 1 h. The slides were incubated with the following corresponding primary antibodies: rabbit anti-ubiquitin antibody (1:200, DAKO), mouse anti-ubiquitin antibody (1:500, Abcam), mouse anti-p62 antibody (1:200, Abcam), rabbit anti-HSP70 antibody (1:200, Cell Signaling Technology, Beverly, MA, USA), mouse anti-HSP70 antibody (1:200, Enzo Life Sciences, New York, NY, USA) and rabbit anti-BAG3 antibody (1:500, Abcam). The immunoreactions were visualized using fluorescent secondary antibody. All the slides were visualized under a confocal microscope equipped with an argon and HeNe1 laser (LSM-780; Zeiss, Jena, Germany).
Western blot analysis
Tissue samples from the right ischemic hemisphere were homogenized with a homogenizer in ice-cold RIPA buffer (25 mM Tris-HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate and 0.1% SDS) and then centrifuged at 12,000 × g for 15 min at 4℃. The supernatant was collected and separated by SDS-polyacrylamide gel electrophoresis and transferred onto PVDF membranes (Millipore Corporation, USA). The membranes were blocked with 5% skimmed milk for 1 h at room temperature, and then incubated with the corresponding primary antibodies: rabbit anti-LC3 antibody (1:2000, Abcam), rabbit anti-BAG1 antibody (1:500, Abcam), rabbit anti-BAG3 antibody (1:1000, Abcam), rabbit anti-HDAC6 antibody (1:1000, Millipore Corporation) or rabbit anti-β-actin antibody (1:5000 Abcam) overnight at 4℃. Membranes were incubated with peroxidase-conjugated immunoglobulin secondary antibodies at room temperature for 1 h. Protein bands were visualized with enhanced chemiluminescence (ECL) substrate (Pierce, Rockford, USA). Image analysis was performed using Image J (http://imagej.nih.gov/ij/; provided in the public domain by the National Institutes of Health, Bethesda, MD, USA). Briefly, after setting the measurement criteria, the region of interest was specified by the rectangle tool of Image J. We then plotted the same rectangle around all the other bands to quantify the intensity of each band which was normalized with the intensity of the corresponding loading controls.
Statistical analysis
During data collection and analysis, the investigators were blinded to the experimental group. Data are expressed as the mean ± standard deviation (SD). Statistical comparisons were performed using one-way ANOVA analysis followed by the Tukey-Kramer test for intergroup comparisons. Differences were considered to be significant at p < 0.05.
Results
Cerebral infarct volume and neuronal damage after tMCAO
Nissl staining showed only a small spot visible at 24 h of reperfusion after 10 min tMCAO, where neuronal shrinkage was observed (Figure 1(b), magnification). Infarct legions became more evident at 24 and 72 h of reperfusion after 30 and 60 min of tMCAO (Figure 1(b)). The differences in infarct volumes between 30 and 60 min tMCAO were significant (Figure 1(c), *p < 0.05 vs. 30 min group).
Compared to sham controls, NeuN staining showed a time-dependent decrease after tMCAO in the peri-ischemic lesion from 6 to 72 h of reperfusion (Figure 2(a) and (b), *p < 0.05 and **p < 0.01 vs. sham; ##p < 0.01 vs. 0.5 h; $p < 0.05 and $$p < 0.01 vs. 1 h; &&p < 0.05 vs. 6 h).
(a) Immunohistochemical staining of NeuN and (c) c-Fos in the peri-ischemic lesion, with (b), (d) quantitative analyses (*p < 0.05 versus sham, **p < 0.01 versus sham; #p < 0.05 versus 0.5 h, ##p < 0.01 versus 0.5 h; $p < 0.05 versus 1 h, $$p < 0.01 versus 1 h; &p < 0.05 versus 6 h, &&p < 0.01 versus 6 h; §p < 0.01 versus 24 h. Scale bar = 50 µm).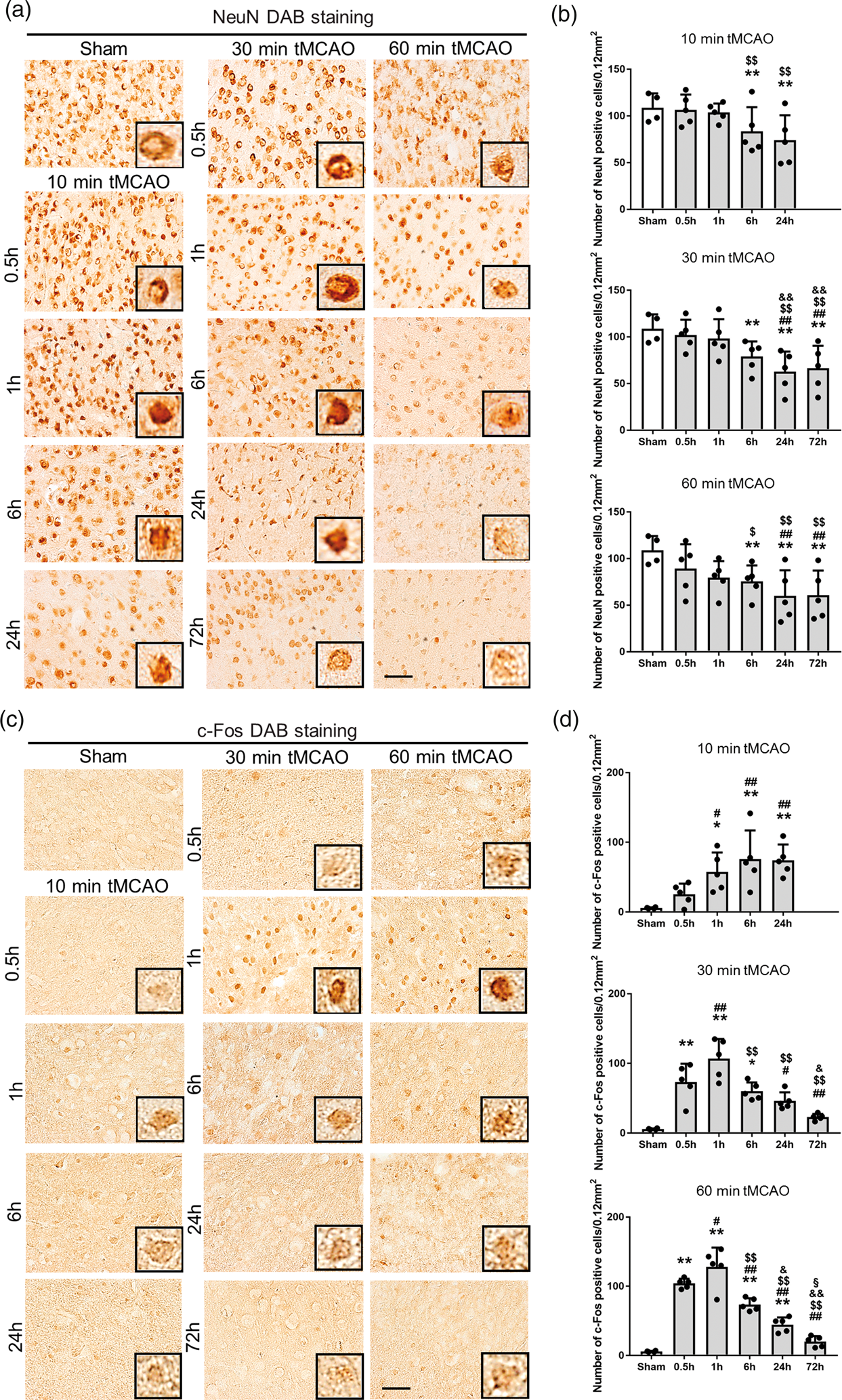
c-Fos was only weakly induced even after 10 min tMCAO, but the number of c-Fos-positive cells increased progressively from 1 to 24 h of reperfusion (Figure 2(c) and (d)). In contrast, c-Fos was strongly induced after 30 and 60 min tMCAO from 0.5 h, peaking at 1 h then decreasing until 72 h (Figure 2(c) and (d), *p < 0.05 and **p < 0.01 vs. sham; #p < 0.05 and ##p < 0.01 vs. 0.5 h; $$p < 0.01 vs. 1 h; &p < 0.05 and &&p < 0.05 vs. 6 h; §p < 0.01 vs. 24 h).
Ubiquitin-positive protein aggregates after tMCAO
As shown in Figure 3(a), ubiquitin-positive protein aggregates were strongly induced as early as 0.5 h of reperfusion in all three tMCAO groups, but peaked earlier at 6 h, 1 h, and 0.5 h after 10, 30 and 60 min tMCAO, respectively (**p < 0.01 vs. sham; #p < 0.05 and ##p < 0.01 vs. 0.5 h; $p < 0.05 and $$p < 0.01 vs. 1 h).
(a) Immunohistochemical staining of ubiquitin in the peri-ischemic lesion, with (b) quantitative analysis (*p < 0.01 versus sham; #p < 0.05 versus 0.5 h, ##p < 0.01 versus 0.5 h; $p < 0.05 versus 1 h, $$p < 0.01 versus 1 h; &p < 0.01 versus 6 h; §p < 0.01 versus 24 h. Scale bar = 50 µm).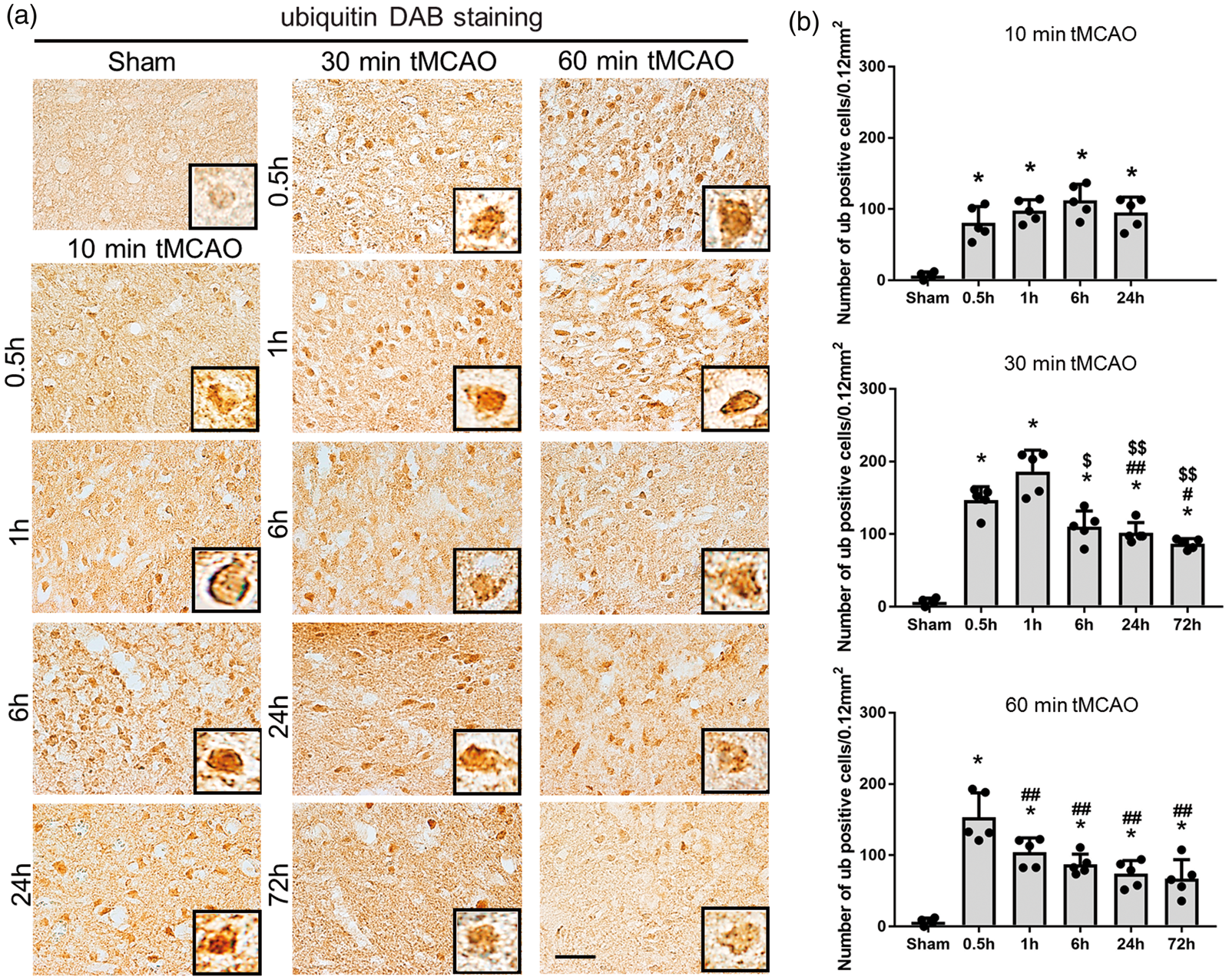
Changes to autophagic markers after tMCAO
p62 expression decreased significantly at 24 and 72 h of reperfusion only after 30 and 60 min tMCAO, but not after 10 min tMCAO (Figure 4(a) and (b), *p < 0.05 vs. sham; #p < 0.05 and ##p < 0.01 vs. 0.5 h; $$p < 0.05 vs. 1 h; &p < 0.05 and &&p < 0.01 vs. 6 h; §p < 0.01 vs. 24 h). Figure 4(c) shows a significant increase in LC3-II at 24 h of reperfusion after 30 min tMCAO and at 1 and 24 h of reperfusion after 60 min tMCAO (Figure 4(c) and (d), *p < 0.05 and **p < 0.01 vs. sham).
(a) Immunohistochemical staining of p62 in the peri-ischemic lesion, with (b) quantitative analysis. (c) Western blot for LC3-I and LC3-II levels. (d) Quantitation of LC3-II/β-actin ratio (*p < 0.05 versus sham, **p < 0.01 versus sham; #p < 0.05 versus 0.5 h, ##p < 0.01 versus 0.5 h; $p < 0.05 versus 1 h, $$p < 0.01 versus 1 h; &p < 0.05 versus 6 h, &&p < 0.01 versus 6 h; §p < 0.01 versus 24 h. Scale bar = 50 µm).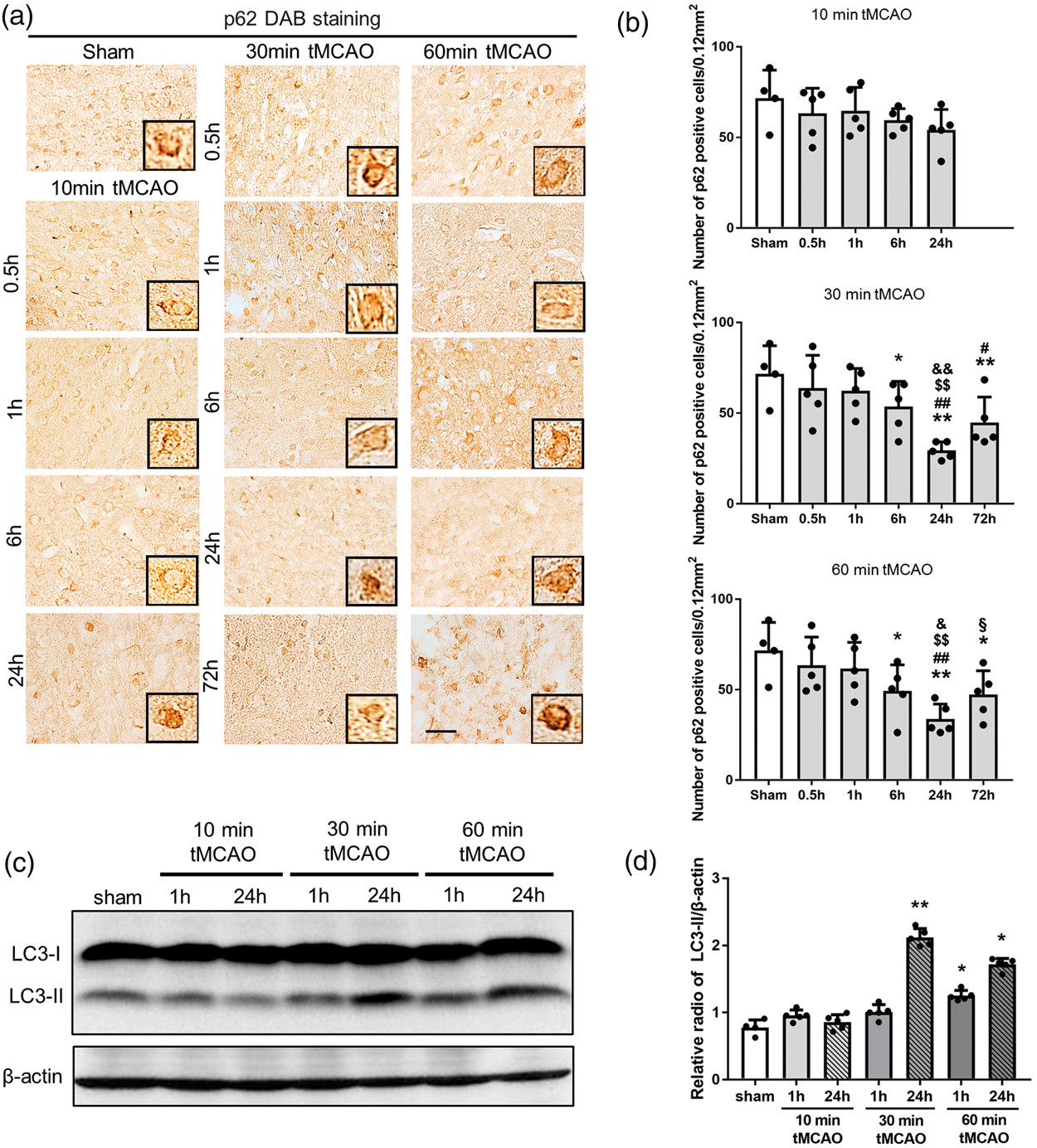
Double immunofluorescent analysis after tMCAO
Among stained sections at all time points (0.5 h–72 h) after 10, 30 and 60 min tMCAO, immunofluorescent analysis showed good colocalization of ubiquitin with Hsp70 and p62 at 1 h of reperfusion after 60 min tMCAO (Figure 5(a) and (b)). p62 also colocalized well with HSP70 (Figure 5(c)) but not with BAG3 (Figure 5(d) at 1 h of reperfusion after 30 min tMCAO. Most BAG3 was colocalized both with ubiquitin and HSP70 at 1 h of reperfusion after 30 min tMCAO (Figure 5(e) and (f)). The rate of colocalization in Figure 5(f) was 44.0% and the average rate of colocalization of BAG with HSP70 was 50.8%.
Double immunofluorescence analysis for ubiquitin (a, b, e), p62 (b, c, d), HSP70 (a, c, f) and BAG3 (d–f) (Scale bar = 50 µm).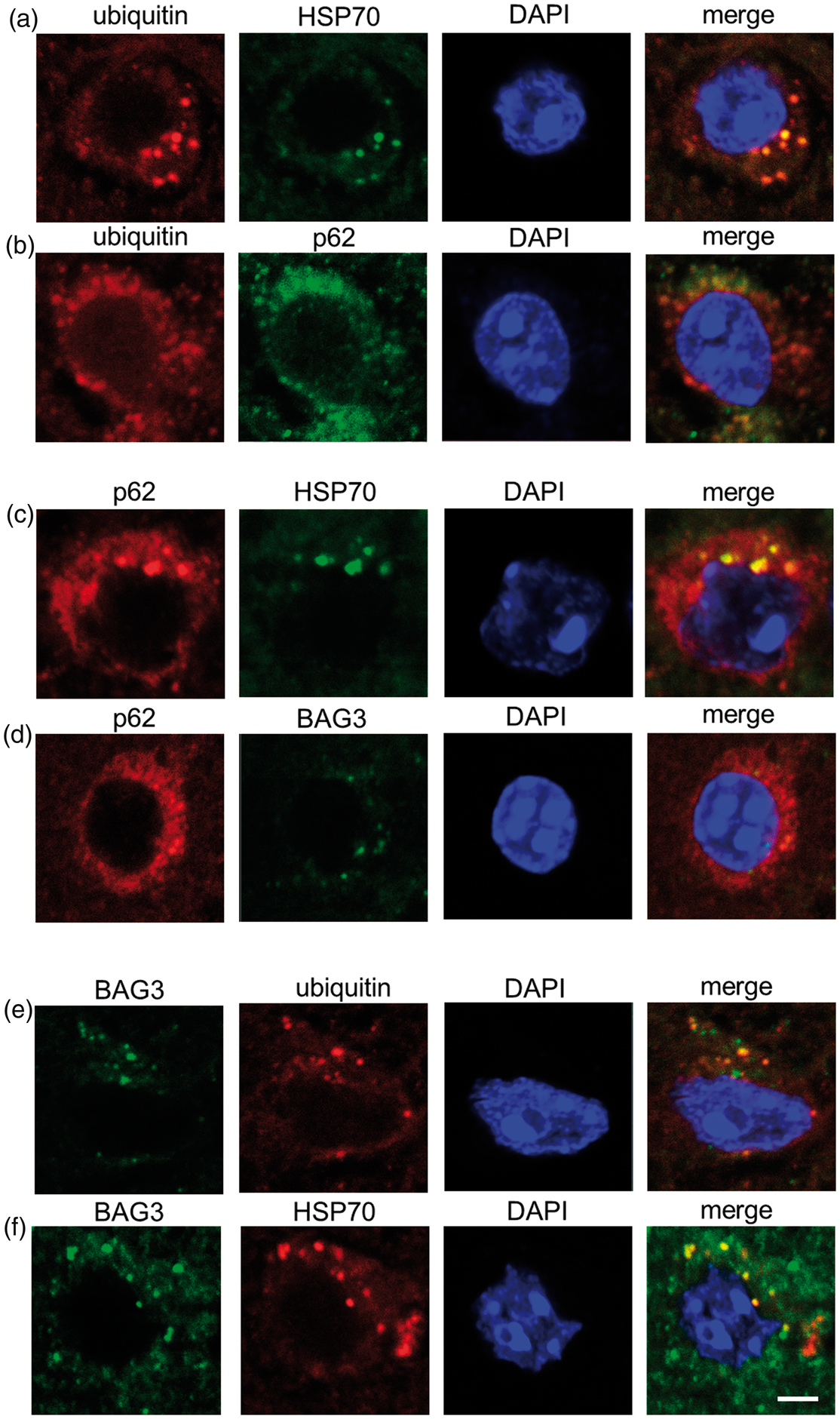
Changes in BAG1, BAG3 and HDAC6 protein levels after tMCAO
Western blot analysis showed a significant decrease of BAG1 at 24 h of reperfusion in three tMCAO groups (Figure 6(a) and (b), *p < 0.05 vs. sham), but not at 1 h of reperfusion. In contrast, BAG3 showed an ischemic-period-dependent increase after 10, 30 and 60 min tMCAO (Figure 6(a) and (c), *p < 0.05 and **p < 0.01 vs. sham). HDAC6 was significantly upregulated only at 24 h of reperfusion after 10 and 30 min tMCAO, but was also upregulated from 1 h of reperfusion after 60 min tMCAO (Figure 6(d) and (e), *p < 0.05 and **p < 0.01 vs. sham).
Changes in the protein expression level of BAG1, BAG3 and HDAC6 in brain tissues. (a) Representative Western blot for BAG1 and BAG3, and (d) HDAC6. The ratio of the relative density of BAG1 (b), BAG3 (c) and HDAC6 (e) normalized with β-actin (*p < 0.05 versus sham, **p < 0.01 versus sham).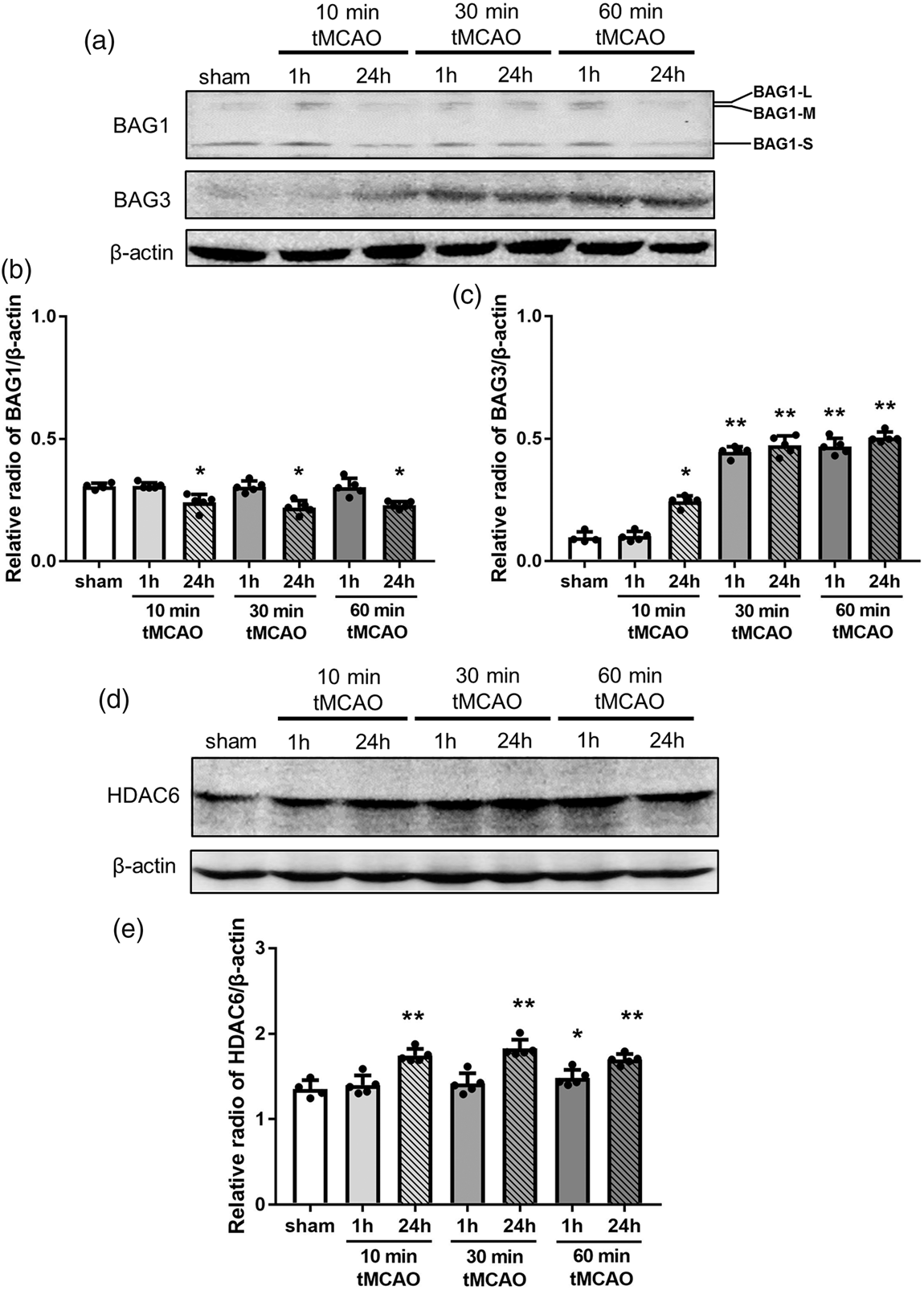
Discussion
In the present study, we found a switch and an interaction between UPS and autophagy in a mouse model of transient focal cerebral ischemia. UPS and autophagy are two major intracellular systems for clearing cellular waste products. 16 UPS usually targets short-lived proteins, 17 while autophagy is restricted to long-lived proteins and organelles. 18 UPS and autophagy are basically two independent proteolytic pathways that may also be linked by specific mechanisms, especially when massive protein degradation occurs such as in cerebral ischemia. 19
Cerebral ischemia causes the rapid and robust production of misfolded proteins due to energy failure, oxidative stress and other mechanisms.20,21 Under such stresses, misfolded proteins are produced from their native folded state by an incorrect folding process. 22 If refolding fails, misfolded proteins will be ubiquitinated and targeted for degradation by UPS. 23 Accumulation of ubiquitin-conjugated proteins burdens the proteasome following ischemic injury, and an overload to UPS then causes intracellular protein aggregation and eventual neuronal death.24,25 Aggregates of misfolded proteins in neurons after cerebral ischemia have been observed by transmission electron microscopy,26,27 but the relationship between the amount of misfolded proteins, UPS overload and activity of autophagy remains unclear. Expression of an immediate early gene c-Fos represents a response to a variety of stimuli such as oxidative stress. 28 In the present study, the results of Nissl, NeuN and c-Fos staining revealed cell injury after ischemia that was dependent on the duration of ischemia (Figures 1 and 2).
The 26S proteasome is made up of subcomplexes of the 20S core proteasome and the 19S regulatory complex.
29
26S proteasome activity is ATP-dependent since ATP is necessary for 19S assembly with the 20S core particle (Figure 7).
30
Thus, in case of severe ATP depletion as a result of cerebral ischemia, UPS may switch to an ATP-independent autophagy pathway. BAG1 protein binds HSP70 family molecular chaperones and regulates diverse pathways involved in cell proliferation, apoptosis, and stress responses (Figure 7).
31
Although a high level of BAG1 stimulates proteasomal degradation,
10
the decrease in BAG1 at 24 h reperfusion in the present study suggests an impairment of UPS (Figure 6(a) and (b)).
A hypothetical diagram showing the crosstalk of UPS and autophagy in mice brains that depends on the duration of cerebral ischemia.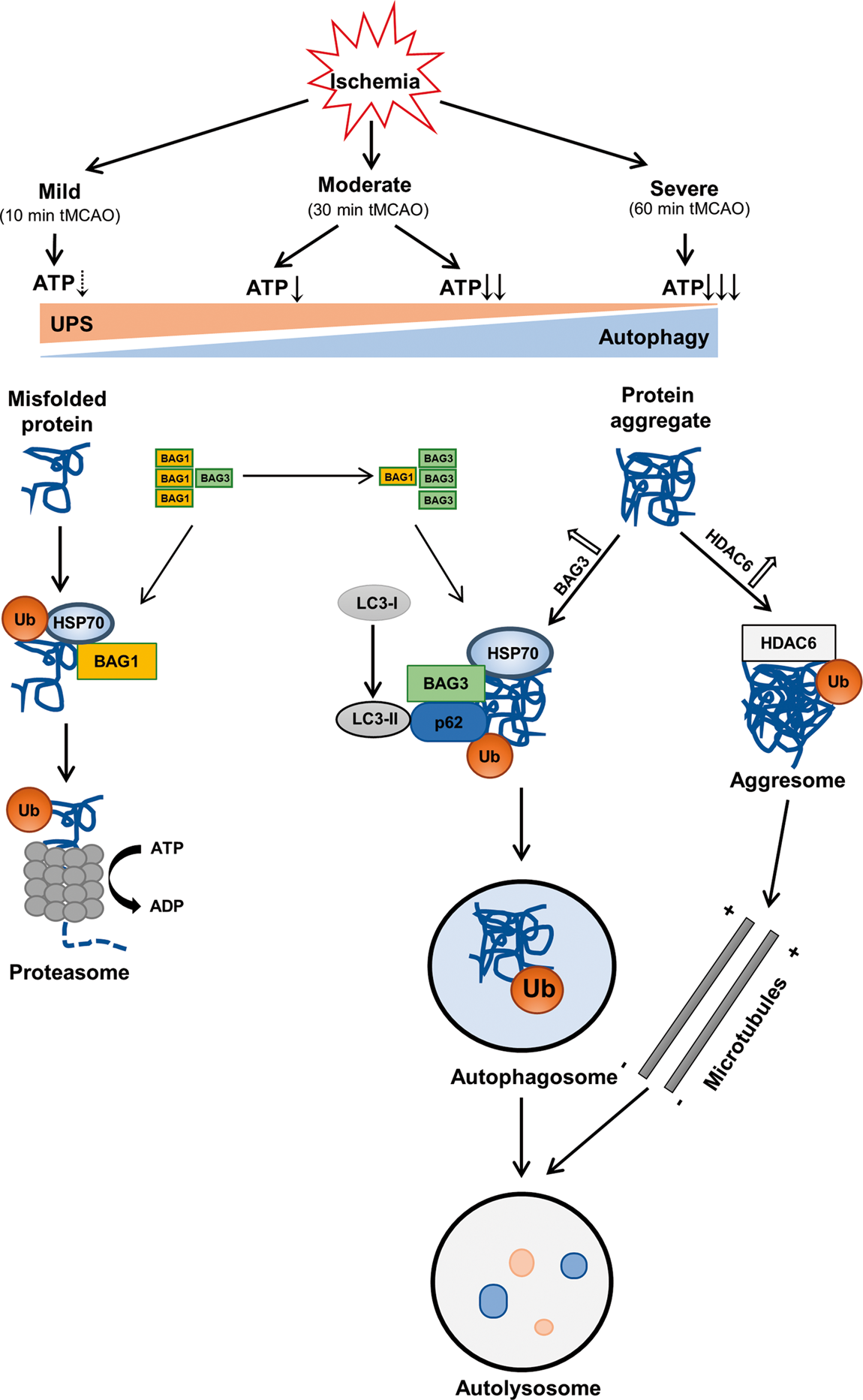
Unlike BAG1, the upregulation of BAG3 after tMCAO suggests increased autophagy (Figure 6(a) and (c)), 10 and a switch from UPS to autophagy (Figure 7). HDAC6, a microtubule-associated deacetylase, is required for the formation of aggresomes and the autophagic degradation of aggregated proteins (Figure 7),32,33 which was activated in an HDAC6-dependent manner to compensate for the impairment of UPS. 34 Our present results also showed an increase of HDAC6 at 24 h after 10 and 30 min tMCAO and at 1 h after 60 min tMCAO when UPS was impaired (Figure 6(d) and (e)). Ubiquitin binds the scaffold protein p62, 35 which then binds HSP70 and BAG3 providing an important substrate for autophagy degradation (Figures 5 and 7). 36
p62 protein is a marker of autophagic flux that is degraded during the autophagic process. 36 Thus, defects in autophagy may increase p62, 37 as was shown by some studies in which autophagy was impaired after ischemia.38,39 We found an increase of p62 at 72 h reperfusion after 30 and 60 min ischemia compared to 24 h reperfusion (Figure 4(a) and (b)), which may indicate the end of the autophagic process in the 72 h reperfusion groups, although this theorem still needs further testing and elucidation.
Thus, the present results provide evidence that mild cerebral ischemia (10 min) only activated UPS but not autophagy. Furthermore, a switch from UPS to the autophagy-mediated protein degradation pathway was turned on at 6 h reperfusion after moderate (30 min) ischemia and 1 h of reperfusion after severe (60 min) cerebral ischemia (Figures 1 and 2), trends that were related to the BAG1/BAG3 ratio (Figures 6 and 7) and HDAC6 (Figure 6). Ubiquitin (Figure 3), p62 (Figure 4) and HSP70 also participated in this crosstalk via binding to BAG3 (Figure 5).
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was partly supported by Challenging Research 15K15527 and Young Research 15K21181.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors' contributions
All authors had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Study concept and design: KA Acquisition of data: XL, JS Analysis and interpretation of data: XL, JS, TY Drafting of the manuscript: XL Critical revision of the manuscript for important intellectual content: TY, KA Statistical analysis: XL, TY Obtained funding: KA Administrative, technical, and material support: XL, TY, JS, XS, RM, YH, KS, MT, NH, YO Study supervision: KA.