
Abstract
Ischemic stroke is a major cause of mortality and long-term disability with limited treatment options, and a greater understanding of the gene regulatory mechanisms underlying ischemic stroke-associated neuroinflammation is required for new therapies. To study ischemic stroke in vivo, mice were subjected to sustained ischemia by intraluminal filament-induced middle cerebral artery occlusion (MCAo) for 24 h without reperfusion or transient ischemia for 30 min followed by 23.5 h reperfusion, and brain miRNA and mRNA expression changes were quantified by TaqMan OpenArrays and gene (mRNA) expression arrays, respectively. Sustained ischemia resulted in 18 significantly altered miRNAs and 392 altered mRNAs in mouse brains compared to Sham controls; however, the transient ischemic condition was found to impact only 6 miRNAs and 126 mRNAs. miR-367-3p was found to be significantly decreased in brain homogenates with sustained ischemia. G protein-coupled receptor, family C, group 5, member A (Gprc5a), a miR-367-3p target gene, was found to be significantly increased with sustained ischemia. In primary neurons, inhibition of endogenous miR-367-3p resulted in a significant increase in Gprc5a expression. Moreover, miR-367-3p was found to be co-expressed with GPRC5A in human neurons. Results suggest that loss of miR-367-3p suppression of GPRC5A may contribute to neuroinflammation associated with ischemic stroke.
Introduction
Ischemic stroke is a debilitating neurovascular event and a serious health and financial burden to society. However, there are few treatment strategies aimed at limiting ischemic damage, particularly therapies aimed at reducing neuroinflammation. The pathophysiology of ischemic stroke involves interconnected processes of inflammation,1–4, neurotoxicity, 5 and apoptosis 6 across multiple cell types, including neurons, macrophages, glial cells, and astrocytes. The identification of new mechanisms that underlie these ischemia-induced processes and the key driver genes are required for new therapies. Neuroinflammation is a central hallmark of ischemic stroke and drives many of the gene expression changes in response to ischemia in the brain. The interplay between inflammatory cells and brain cells (e.g. neurons) in ischemic stroke is complex and extensive with both detrimental and beneficial outcomes. 7 Although neuroinflammation and ischemic stroke have been widely studied, new regulators of ischemia-induced inflammation have emerged. Further investigation of this relationship may advance our understanding of neuroprotection and neurotoxicity.
miRNAs are a promising new class of gene regulators in neuroinflammation associated with ischemic stroke. 8 miRNAs post-transcriptionally regulate gene expression through complementary binding to mRNA targets at sites enriched within the 3′ untranslated region (3′ UTR).9–12 miRNAs suppress gene expression through inhibition of protein translation and mRNA degradation.9–11 In the brain, miRNAs likely contribute to most, if not all, neuroinflammatory processes in some way, including the generation of reactive oxygen species, apoptosis, loss of blood-brain barrier integrity, leukocyte migration and activation, and edema. 8 For example, miR-155 was found to be a transcriptional target of p53 in microglial cells and induced by cytokines. 13 Moreover, inhibition of miR-155 in vivo altered temporal outcomes and cytokine expression in a mouse model of ischemic stroke. 14 The most studied miRNA in ischemic stroke is miR-181b-5p, which is decreased in ischemic stroke and serves to promote neuroprotection.15,16 In addition, miR-181b-5p inhibition in vivo was found to limit brain damage and improve outcomes in ischemic stroke. 16 Evidence clearly indicates that brain miRNAs are altered in ischemic stroke and these changes result in significant alterations to gene expression underlying neuroinflammation.17,18 Most importantly, brain miRNAs represent a new class of therapeutic targets to treat ischemic stroke, prevent neurodegeneration and brain damage, and improve long-term recovery. Nonetheless, miRNA-mediated gene regulatory mechanisms in ischemic stroke and neuroinflammation have not been fully explored but hold great potential for new discovery.
Here, we identified multiple potential brain miRNA regulatory modules, i.e. miRNA-gene (mRNA) target regulatory networks, that are associated with sustained and transient ischemia. We sought to compare the impact of sustained ischemia with no reperfusion to a model of transient ischemia to gain a greater understanding of the impact of the initial hypoxic stress (within 30 min) and reperfusion on gene expression at the 24-h time-point post-surgery. Integration of miRNA and mRNA datasets, pathway analyses, and co-expression networks identified many putative miRNA gene regulatory modules in inflammatory pathways. We report that middle cerebral artery occlusion (MCAo)-induced brain ischemia suppresses the expression of multiple miRNAs, including miR-367-3p. Furthermore, sustained ischemia without reperfusion resulted in a significant increase in many genes, including the G protein-coupled receptor, family C, group 5, member A (Gprc5a), a novel miR-367-3p target gene and factor in neuroinflammation. Results from this study suggest the hypothesis that increased pro-inflammatory gene expression in ischemic stroke is mediated through suppression of key miRNA regulatory modules.
Materials and methods
Animals
This study was approved by Monash University Animal Ethics Committee (Project MARP/2011/112) and performed in accordance with National Health and Medical Research Council of Australia (NHMRC) guidelines for the care and use of animals in research, and is in compliance with the ARRIVE guidelines (Animal Research: Reporting in Vivo Experiments) for how to REPORT animal experiments. For cerebral ischemia experiments in vivo, 6- to 10-week-old male C57BL/6 mice were studied (weight = 26 ± 3 g). The mice had free access to water and food pellets before and after surgery. Some mice were excluded from the study due to either: incomplete flushing of the brain with phosphate-buffered saline (PBS) during tissue collection 19 ; plasma contamination by red cells 20 ; or less than 75% drop in rCBF occurred following insertion of monofilament. There were no mortalities. Eight pregnant C57Bl/6 females were used to obtain cortical neurons from E17 pups.
Focal cerebral ischemia
Mice underwent intraluminal filament-induced MCAo, a model of focal cerebral ischemia similar to that described previously.21–23,21 Sustained ischemia with no reperfusion (InoR) was achieved by occlusion for 24 h, and transient ischemia with reperfusion (IR) was completed in mice with 30 min of MCAo ischemia followed by 23.5 h of reperfusion (Figure 1(a)). This approach will generate a model of sustained ischemia in the absence of reperfusion; however, the duration of the ischemia was limited to 24 h and thus, this animal strategy does not inherently represent a long-term model of ischemia. Mice were anesthetized with a mixture of ketamine (80 mg/kg intraperitoneal injection [i.p.]) and xylazine (10 mg/kg i.p.). Body temperature was monitored and maintained at 37.0 ± 0.5℃ with a heat lamp throughout the procedure and until mice regained consciousness. A midline neck incision was made, and the right external carotid (ECA) and pterygopalatine arteries were isolated and cauterized. The right internal carotid artery (ICA) was lifted and occluded distal to the bifurcation of the common carotid artery (CCA). Focal cerebral ischemia was induced by intraluminal occlusion of the right MCA for 0.5 h using a 6–0 nylon monofilament with a silicone-coated tip (0.20–0.22 mm, Doccol Co.). Severe (>75%) reduction in rCBF was confirmed using trans-cranial laser-Doppler flowmetry (Perimed) in the area of cerebral cortex supplied by the MCA (approximately 2 mm posterior and 5 mm lateral to bregma). For InoR group, the filament was tied in place and the occlusion was maintained for 24 h. To model transient ischemia, the monofilament was retracted after 0.5 h to allow reperfusion for 23.5 h. Sham-operated mice were anesthetized and the right CCA was visualized, dissected free from surrounding connective tissue, but no monofilaments were inserted. When animals regained consciousness, they were injected subcutaneously with 1 mL saline to reduce post-operative dehydration, and they were returned to their cage. Neurological deficit score (using a five-point scoring system: 0, no deficit; 1, failure to extend right paw; 2, circling to the right; 3, falling to the right; and 4, unable to walk spontaneously) was evaluated in a blinded fashion, as described previously.21–24
Study design and mouse model of ischemic stroke. (a) Study design. (b) Diagram representing the mouse intraluminal filament-induced middle cerebral artery occlusion (MCAo) model. InoR: ischemia no reperfusion; IR: ischemia-reperfusion. (c) Regional cerebral blood flow (rCBF) recorded during and after 1 h of MCAo (n = 17–27). (d) Hanging wire tests (time). (n = 26–27). (e) Neurological deficit at 24 h (n = 26–27). Data presented as mean ± SD.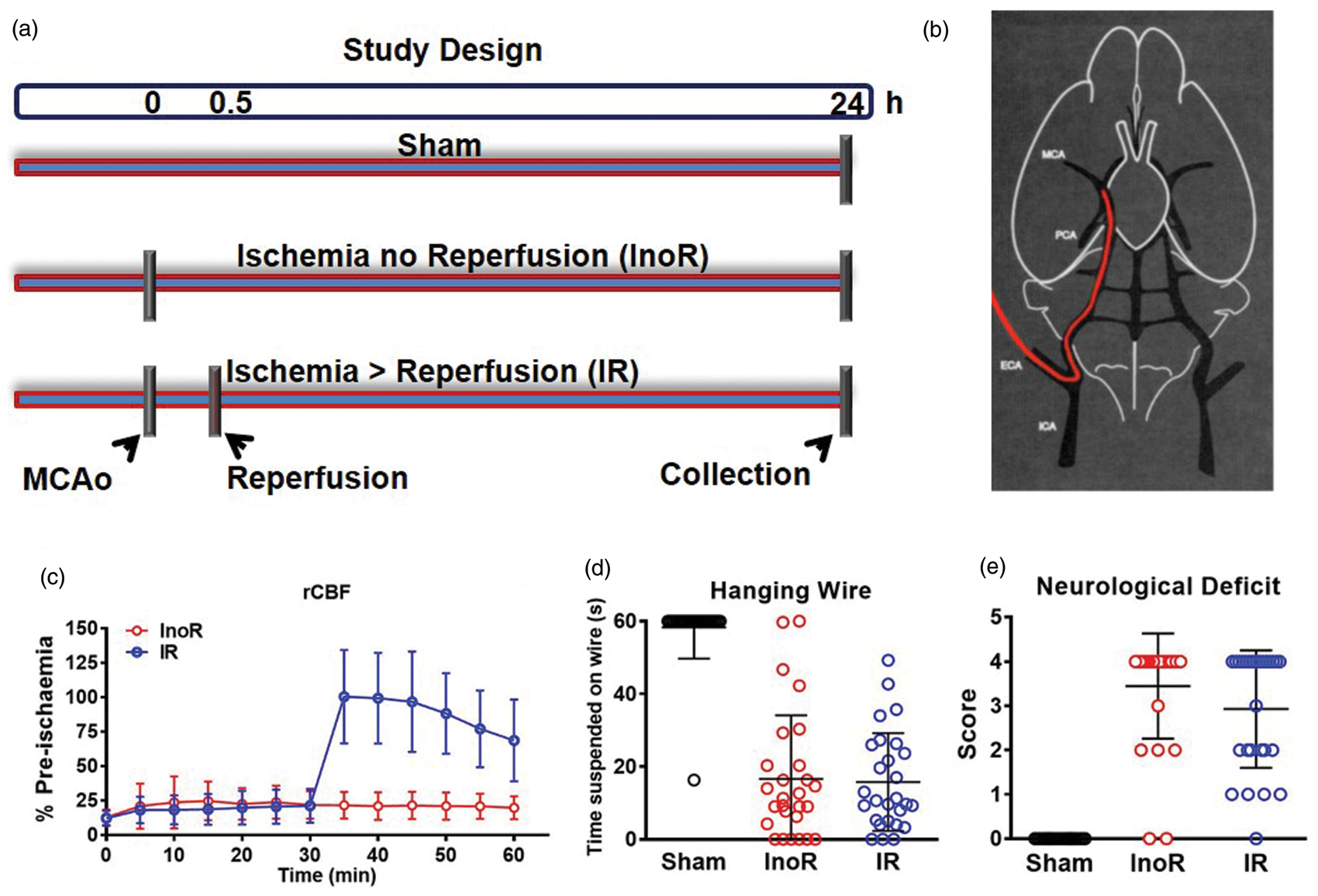
Cell culture
Cortical neuronal cultures were prepared from E17 embryos, as previously described. 22 Briefly, dissociated neurons were plated on poly-d-lysine (Sigma) coated 12-well plates (NUNC) in neurobasal medium (NBM) containing L-glutamine (2 mmol/L), gentamicin (5 mg/L), and B-27 supplements (Invitrogen) pH 7.2. Cells were maintained at 37℃ in a humidified atmosphere of 5% CO2 in air. On day 9, primary neurons were transfected overnight with miRCURY LNA power inhibitor (100 nmol) against miR-367-3p or scrambled control sequence (Exiqon) in DharmaFECT 4 Transfection Reagent (Thermo Scientific). The next day (10 d in vitro), the medium was changed with NBM (+supplements) and incubated in humidified atmosphere of 5% CO2 in air at 37℃ for 24 h. For glucose deprivation, transfected neurons (10 d in vitro) were incubated in glucose-free Locke's buffer solution containing (in mmol/L) NaCl 154.0, KCL 5.6, CaCl22H2O 2.3, MgCl26H2O 1.0, NaHCO3 3.6, HEPES 5.0, pH 7.2, supplemented with gentamicin (5 mg/L) at 37℃ in humidified atmosphere of 5% CO2 in air for 24 h. At 11 d in vitro, cells were washed with PBS, scraped off the plates and collected in QIAzol solution (Qiagen). HCN-2, CCF-STTG1, and M059K cells were purchased (ATCC). HCN-2 neurons were cultured in DMEM media with 10% FBS and 5% Penicillin-Streptomycin (Pen-Strep). CCF-STTG1 astrocytes were cultured in RPMI-1640 media with 10% FBS and 5% Pen-Strep. M059K glial cells were cultured in DMEM/F12 media with 10% FBS and 5% Pen-Strep, and supplemented with L-glutamine (2.5 mM), HEPES (15 mM), sodium pyruvate (0.5 mM), sodium bicarbonate (1.2 g/L), and non-essential amino acids (0.05 mM). Human monocytic THP-1 cells were stimulated with PMA (50 ng/mL) for three days to differentiate cells to a macrophage phenotype.
Transcriptomics
Immediately after euthanasia, the brains were excised, placed in ice-cold PBS and cleaned of connective tissue. Brains were snap-frozen in liquid nitrogen and homogenized. As miRNA gene expression in response to hypoxic stroke could be different in different parts of the brain, total RNA was isolated from whole mouse brains for consistency using miRNAEasy kits, as per manufacturer’s protocol. To complete miRNA profiling of mouse brains, total RNA was prepared for TaqMan OpenArray microRNA Panels as per manufacturer’s protocol. Briefly, total RNA was reverse transcribed using Megaplex RT primers for pools A only. cDNA was amplified using Megaplex PreAmp pool A primers and PreAmp Mastermix for 12 cycles (Life Technologies). PCR reaction mix was prepared and loaded onto QuantStudio Rodent OpenArrays using the AccuFill System (Life Technologies). For mRNA profiling, total RNA was reverse transcribed and hybridized onto Affymetrix Exon Expression Chips (Affymetrix.ExonExprChip.MoGene-1_0-st-v1_na32_mm9_2011-10-07), as per manufacturer’s instructions (Affymetrix).
Informatics
For miRNA analyses, raw data from QuantStudio was processed and organized using ExpressionSuite software (Life Technologies). Global normalization was used to determine relative quantitative value (RQV) for each miRNA, 2-ΔCRT mean, which was loaded as generic expression data into GeneSpring GX 12.1 (Agilent) with no baseline transformation. Samples were grouped and averaged over replicates for high-level analyses. For differential expression analyses, an unpaired t-test was used, and significance was defined as p-value < 0.05 and absolute fold change >1.5-fold. For mRNA analyses, raw data (.CEL) files were uploaded and process using GeneSpringGX14.9 (Agilent) software. Briefly, data were summarized using ExonRMA16 and quantile normalized. Data were baseline transformed to the median of all samples. Genes were filtered based on expression values of a percentile cut-off of 20%, and genes that met these criteria in at least 1 out of 15 samples were kept for further analysis. For differential expression analyses, an unpaired t-test was used with Benjamini-Hochberg correction, and significance was defined as corrected p-value < 0.05 and absolute fold change >1.5-fold. Putative targets for altered miRNAs were identified using TargetScan in R.
PCR
Total RNA was isolated from cells using miRNEasy mini kits (Qiagen). For miRNA individual TaqMan Assays, total RNA (100–300 ng) was reverse transcribed using the TaqMan microRNA reverse transcription kit (Applied Biosystems) for miR-181b-5p, miR-224-5p, miR-296-5p, miR-302b-3p, miR-367-3p and miR-489-3p according to the manufacturer’s protocol. Subsequently, 6–9 µl of the reverse transcription product was used for detecting miRNA expression by real-time PCR using TaqMan miRNA Assay Kits (Applied Biosystems) for the specific miRNAs. Values were normalized to a mouse mature miRNA control, U6, and expressed as 2—(CT[microRNA]—CT[U6]). For mRNA analyses, RNA samples were reverse transcribed using 100–300 ng of total RNA and real-time PCR was performed with iQ SYBER Green Supermix (Bio-Rad) and the MyiQ single color real-time PCR detection system. Relative expression levels for candidate mRNAs were normalized to mouse β-actin. Primer sequences are shown in Table S1.
Statistics
Data are presented as mean ± SD. When comparing two groups, Mann–Whitney non-parametric tests were used. For multiple comparisons, a one-way ANOVA with Tukey’s multiple comparisons tests were used. A value of p < 0.05 was considered to be significant.
Results
Brain miRNAs are suppressed in stroke
To model focal cerebral ischemic stroke in mice, MCAo was performed as previously described.
22
Control mice were subjected to sham surgery. MCAo for 24 h was used as a model of sustained ischemia (Ischemia no Reperfusion, InoR). MCAo for 30 min followed by reperfusion for 23.5 h was used as a model of transient ischemia (Ischemia-Reperfusion, IR) (Figures 1(a) and (b)). Regional cerebral blood flow (rCBF) was equally compromised in both InoR and IR groups at 30 min post MCAo, but was restored in the IR group with reperfusion (Figure 1(c)). We found no statistical difference between InoR and IR groups for wire suspension (time) or neurological deficit scores (Figure 1(d) and (e)). To quantify the impact of ischemic stroke on post-transcriptional gene regulation in the brain, miRNAs were quantified by real-time PCR-based TaqMan OpenArrays in brain homogenates from InoR, IR, and sham mice. Remarkably, 18 miRNAs (10 up and 8 down) were found to be significantly altered in InoR compared to sham control brains (InoR/Sham; Figure 2(a), Table 1). These include mmu-miR-302b-3p (−1.87-fold, p = 2.24 × 10−2), mmu-miR-489-3p (−2.47-fold, p = 5.79 × 10−3), mmu-miR-296-5p (−2.54-fold, p = 2.88 × 10−2), and mmu-miR-224-5p (−3.47-fold, p = 2.01 × 10−2) (Table 1). Most interestingly, reperfusion after 30 min of ischemia substantially reduced the number of miRNA changes at 24 h. Only 6 miRNAs were found to be significantly upregulated in brains from IR mice compared to sham mice (IR/Sham; Figure 2(b), Table 1). Of note, the significant miRNA changes associated with sustained ischemia (InoR/Sham) were not apparent in the model of transient ischemia (IR/Sham), suggesting that either these changes occur in response to ischemia for >30 min or these changes are corrected by reperfusion (Table 1). Moreover, the levels of 24 miRNAs (12 up, 4 down) were found to be significantly altered between IR and InoR treated mice, thus confirming that the miRNA profile in the brain is significantly different in mice with sustained ischemia compared to transient ischemia (Figure 2(c), Table 1).
Ischemic stroke alters brain miRNAs. (a–c) Volcano plots of significant differentially expressed miRNAs. Red, significantly increased; Blue, significantly decreased miRNAs (≥1.5-absolute fold change, p < 0.05; n = 5). InoR: ischemia no reperfusion; IR: ischemia-reperfusion. (a) InoR/Sham. (b) IR/Sham. (c) IR/InoR. (D) Real-time PCR quantification of candidate miRNAs (n = 7–8). One-way ANOVA Tukey’s multiple comparisons tests. Data presented as mean ± SD (*p < 0.05). Groups indicated by * without accompanied bar linking two groups indicates that the group with the * was compared to the sham control group. Significantly altered mouse brain miRNAs in ischemia and reperfusion.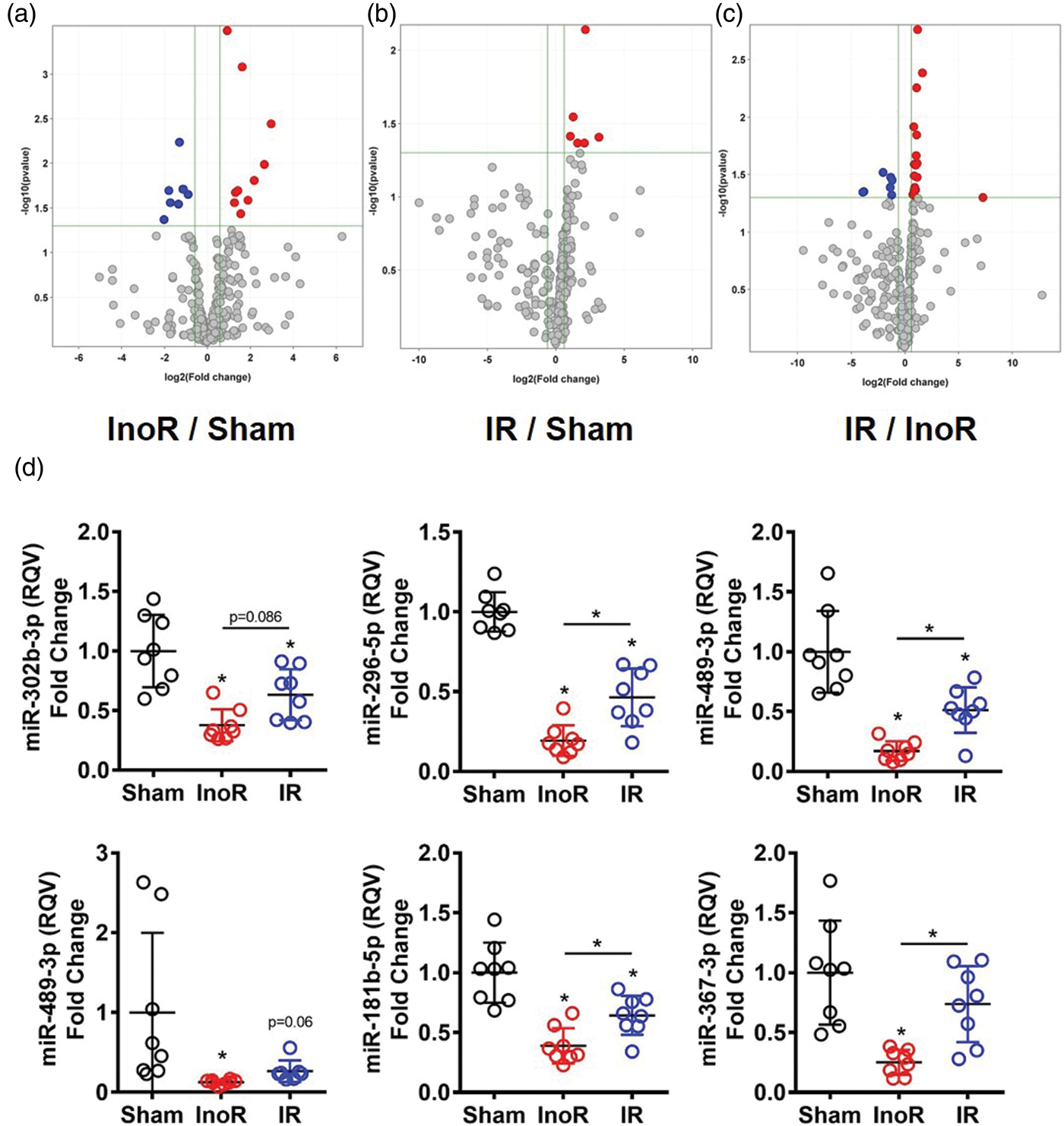

To validate these miRNA changes, individual real-time PCR assays were used to quantify candidate miRNAs in brain homogenates from treated mice. miR-302b-3p, miR-296-5p, miR-489-3p, and miR-224-5p were all significantly reduced in brains of InoR mice with sustained ischemia, compared to sham control mice (Figure 2(d)). Real-time PCR assays were also used to quantify the levels of miR-181b-5p, a brain miRNA previously reported to be altered in stroke that was not included in our initial OpenArray screen.15,16,25,26 We found that miR-181b-5p levels were also significantly decreased in brain homogenates from mice with sustained ischemia (InoR/Sham) (Figure 2(d)). Real-time PCR assays were used to quantify miR-367-3p levels. This miRNA has previously been linked to intracerebral hemorrhagic (ICH) stroke. 27 miR-367-3p was decreased by 10.73-fold (InoR/Sham) in our OpenArray analysis, although these changes were not statistically significant (p = 2.53 × 10−1). However, miR-367-3p levels were significantly reduced in brain homogenates from InoR mice compared to sham control mice by individual PCR assay (Figure 2(d)). According to individual real-time PCR assays, reperfusion after 30 min ischemia attenuated ischemia-associated repression of multiple miRNAs, with miR-296-5p, miR-489-3p, miR-181b-5p, and miR-367-3p all being significantly increased in brains from IR compared to InoR-treated mice (Figure 2(D)). Collectively, these data suggest that brain miRNAs are highly sensitive to both ischemia and reperfusion and that many gene regulatory networks and biological pathways in the brain are affected by ischemic stroke.
Brain miRNA-targets are increased in stroke
Gene expression in the brain is greatly affected by ischemic stroke,
28
and miRNAs are likely contribute to those changes.15,25,26,29,30 To determine if the changes in brain miRNAs coincided with changes to mRNA target genes, and thus identify new miRNA-mediated gene regulatory modules associated with ischemia and reperfusion, gene mRNA levels were quantified by microarray (Affymetrix) in brain homogenates isolated from InoR, IR, and sham-treated mice. First, we compared mouse brain mRNAs at the full transcriptome (coding gene) level. Principal Coordinate Analyses (PCoA) were performed on expression values for each sample. Each group displayed distinct clustering patterns, suggesting that the gene expression signatures for each condition were unique (Figure 3(a)). To quantify differences in the homogeneity of the multivariable distributions between groups, PERMANOVA tests were performed with 999 permutations. We found that InoR and IR profiles were significantly different from the control (Sham) profiles. However, there was no difference in the gene distributions between InoR and IR signatures using PERMANOVA tests (Table S2). Nevertheless, we did find significant differences in the data dispersions between the IR and InoR profiles, as determined by beta-dispersion tests between groups (Table S2). To identify changes in individual mRNAs between groups, differential expression analyses were performed by GeneSpringGX (Agilent). Ischemia for 24 h led to many significant differentially expressed genes in the brain (Benjamini-Hochberg False-Discovery Rate (FDR) corrected p-value ≤ 0.05; absolute fold change ≥1.5). A total of 392 (167 up, 225 down) non-redundant genes were significantly altered in brains from mice with sustained ischemia compared to sham control mice (Figure 3(b), Table S3). By contrast, transient ischemia for 30 min followed by reperfusion for 23.5 h (IR) reduced the number of significantly altered non-redundant genes to only 126 (IR/Sham; 74 up, 52 down) (Figure 3(c), Table S4). We failed to detect any significantly altered genes in brain homogenates between IR and InoR treatments (data not shown). Despite that reperfusion limited the number of significant gene changes in the transient ischemia-treated mice, as compared to sustained ischemia, 59 genes were significantly increased and 34 genes were significantly decreased in both datasets compared to sham controls (Figure 3(D)). This indicates that some genes are altered within 30 min of ischemia and these changes persist to 24 h despite reperfusion. Conversely, other ischemia-associated gene changes are protected by limiting ischemia time or corrected in response to reperfusion.
Ischemic stroke alter brain gene (mRNA) expression. (a) Principal Coordinate Analysis (PCoA) of mouse brain transcriptomes (n = 5). InoR: Ischemia no reperfusion (blue); IR: ischemia-reperfusion (red); and sham control (green). NMDS: Non-metric multidimensional Scaling. (b, c) Volcano plots of significant differentially expressed mRNAs. Red, significantly increased; Blue, significantly decreased miRNAs (≥1.5- absolute fold change, Benjamini–Hochberg-corrected p < 0.05; n = 5). Presented as Log2 fold change vs. −Log10 corrected p-values. (b) InoR/Sham (n = 5). (c) IR/Sham (n = 5). (d) Venn diagram of overlapping gene (mRNA) expression changes in response to sustained (InoR) and transient (IR) ischemia in the brain.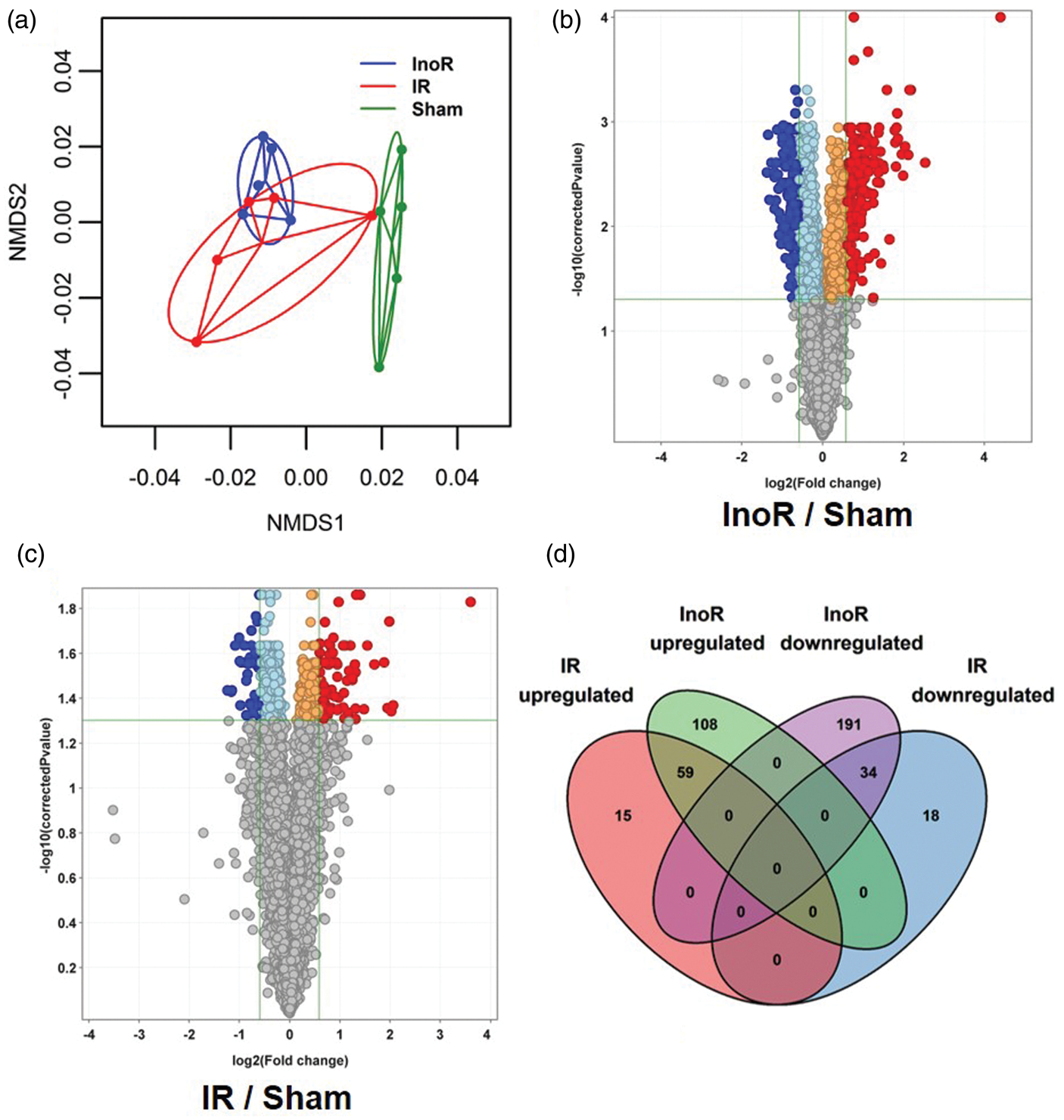
To confirm the impact of our ischemic model on brain gene expression, we filtered significantly increased genes for transcriptional targets of hypoxia-inducible factor 1 α (HIF1-α) and found that 30 unique genes were significantly increased with sustained ischemia (Table S5), consistent with the MCAo treatments inducing cerebral ischemia in these mice. Inflammation is a critical component of both beneficial (e.g. repair) and detrimental (e.g. neurotoxicity) processes in ischemic stroke. 31 Pathway analysis of gene changes with sustained ischemia identified many significantly altered inflammatory immune response pathways in mouse brains at 24 h post-MCAo (Table S6). Within these immune pathways, we identified many inflammatory genes that were significantly increased, including tumor necrosis factor-α (Tnf, 2.19-fold, Cp = 4.94E-03), chemokine (C-C motif) ligand 3 (Ccl3, 21.18-fold, Cp = 2.65E-05), chemokine (C-X-C motif) ligands 1 and 2 (Cxcl1, 1.7-fold, Cp = 2.65E-03; Cxcl2, 3.64-fold, Cp = 1.46E-03) and chemokine (C-C motif) receptor-like 2 (Ccrl2, 3.55-fold, Cp = 1.14E-03) (Tables S3, S6). In addition to these chemokines and cytokines, multiple inflammation-linked transcription factors were altered at the mRNA level, including ATF-3 (activating transcription factor 3, Atf3, 4.55-fold, Cp = 4.99E-04), AP-1 (Fos, 1.74-fold, Cp = 9.11E-03; Fosb, 2.15-fold, Cp = 1.41E-03; Jun, 1.8-fold, Cp = 2.49E-03), and NF-κB (Rel, 2.11-fold, Cp = 1.55E-03) (Tables S3, S6). Other candidate genes that were significantly increased at 24 h post-MCAo were Gprc5a (2.39-fold, Cp = 4.94E-03), zinc finger protein 36 homolog (Zfp36, 4.42-fold, Cp = 4.99E-04), prostaglandin-endoperoxide synthase 2 (Ptgs2, 2.21-fold, Cp = 4.78E-03) and suppressor of cytokine signaling 3 (Socs3; 2.08-fold, Cp = 2.48E-03) (Tables S3, S6). To determine the impact of ischemia followed by reperfusion on brain gene expression at the pathway level, pathway analyses were performed using MetaCore. The most significantly altered pathway for genes in IR brains, compared to sham control brains, were chemokines in inflammation, transcriptional targets of HIF-1, hepatitis C-regulated hepatocellular carcinoma, T cell generation in chronic obstructive pulmonary disease, and immune cell response to interleukin 4 (IL-4) (Table S7). We found that the pathway with the most genes in the active dataset (IR/Sham) was the HIF-1 pathway, despite 23.5 h of reperfusion after only 30 min of ischemia (Table S7). These results suggest that hypoxia-induced genes changes are maintained after the initial response when the stress has been removed (Table S7). One gene, CCL2, was identified as being significantly altered in many of the key pathways suggesting it may contribute more than other genes to the observed phenotype associated with ischemia/reperfusion, e.g. CCL2 was classified to be in 28 of the top 50 significantly altered pathways for our model of ischemia reperfusion (IR/Sham) which was more than that observed for our model of ischemia only (InoR/Sham) where CCL2 was classified in 23 pathways (Tables S6,7).
To identify potential post-transcriptional gene regulatory networks that may contribute to the observed changes in gene expression associated with ischemic stroke, in silico predictions based on TargetScan were used to link significantly altered miRNAs to their inversely altered mRNA targets for sustained and transient ischemia.
32
As depicted by circos plots, 15 significantly altered brain miRNAs in InoR-treated mice were predicted to target 245 significantly and inversely altered mRNAs (15:245) (Figure 4(a), Table S8). For IR-treated mice, 5 brain miRNAs that were significantly increased compared to sham controls were linked to 33 significantly decreased mRNA targets (5:33) (Figure 4(B), Table S8). These data suggest that limiting ischemia (e.g. <30 min MCAo in mice) dramatically decreases potential brain miRNA regulatory networks associated with sustained ischemia. In silico prediction studies identified several putative target genes that were significantly increased for the miRNAs that were significantly decreased: miR-367-3p, 6 target genes; miR-302b-3p, 6 target genes; and miR-181b-5p, 1 target gene (Table S8).
miRNA regulatory modules are significantly altered in ischemic stroke. (a, b) Circos plots linking significantly altered miRNAs and their predicted inversely altered mRNA targets. Purple, miRNAs increased and mRNA decreased. Blue, miRNAs decreased and mRNAs increased. Expression signal (fold change) represented by gray bars in inner Circos ring. InoR: Ischemia no Reperfusion; IR: ischemia-reperfusion; and Sham control. (a) InoR/Sham (n = 5). (B) IR/Sham (n = 5). (C) Validation of candidate gene (mRNA) expression by real-time PCR for Tnf, Ccl3, Atf3, Cxcl2, Cxcl1 Socs3, Ccrl2, Gprc5a, Zfp36, Ptgs2, Fosb, Jun, Bcl2, Rarres2, Serpinb2, and Fos (n = 8). One-way ANOVA with Tukey’s multiple comparisons test. Data are presented as mean ± SD (*p < 0.05). Groups indicated by * without accompanied bar linking two groups indicates that the group with the * was compared to the sham control group.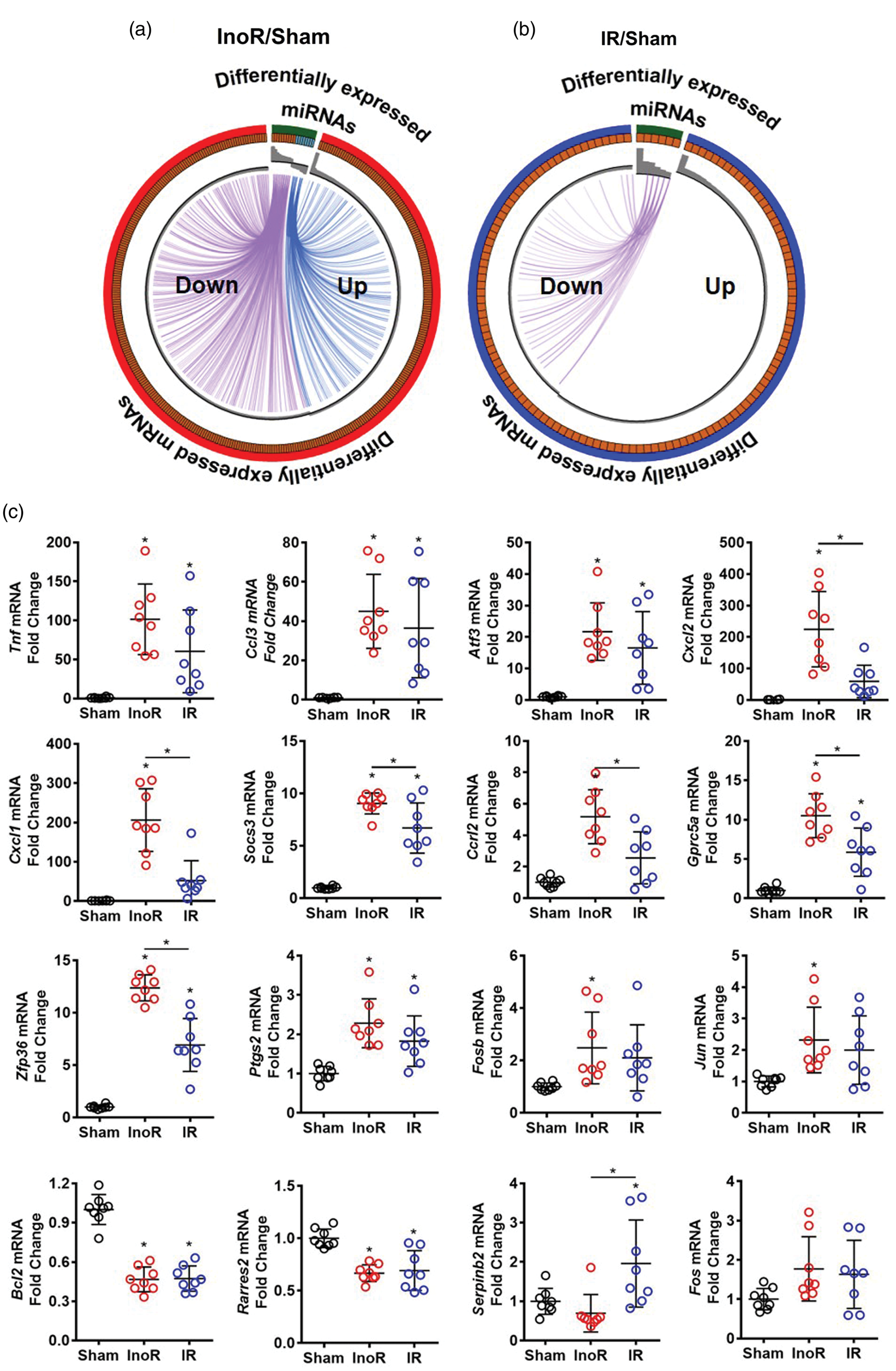
To validate the observed gene expression changes with microarrays, real-time PCR was used to quantify candidate gene expression at the mRNA level. Tnf, Ccl3, Atf3, Cxcl2, Cxcl1, Socs3, Ccrl2, Gprc5a, Zfp36a, Ptgs2, Fosb and Jun were significantly increased in brain homogenates from the InoR-treated mice compared to sham-treated mice (p < 0.05 for all) (Figure 4(c)). Conversely, Bcl2 and Rarres2 mRNA levels were significantly decreased. Reperfusion after 30 min limited the magnitude of the induction of multiple candidate genes that were significantly increased in response to ischemia, with Cxcl2, Cxcl1, Socs3, Ccrl2, Gprc5a, and Zpf36 mRNA levels being significantly decreased in brain homogenates from IR mice compared to InoR mice (Figure 4(c)). Conversely, Serpinb2 mRNA levels were significantly increased in brain homogenates from IR mice compared to InoR mice (Figure 4(c)). These results suggest that ischemic stroke causes massive changes to gene expression in the brain at 24 h, and many of these genes are predicted targets of inversely altered miRNAs. Moreover, limiting the time of ischemia and/or reperfusion prevented or corrected these ischemia-induced gene changes. Many of these genes are well-known factors in immune processes; however, this study also identified many new candidate genes that have previously not been reported to be associated with ischemic stroke or neuroinflammation, including Gprc5a which was significantly increased in ischemia and decreased with reperfusion.
miR-367-3p regulates GPCR5A in neurons
miR-367-3p was selected for further investigation based on the number and impact of predicted target genes that were altered in sustained ischemia (Table S9, Figure 4(c)). Suppression of miR-367-3p in the brain may serve as a critical post-transcriptional regulatory mechanism in response to ischemic stroke, as approximately 19% (32/167) of the genes that were significantly increased in the brain following sustained ischemia were predicted target genes of this miRNA. miR-367-3 is predicted to target GPRC5A in both humans and mice through a single conserved target site in the 3′ UTR (Figure 5(a)). To determine if miR-367-3p and GPRC5A are co-expressed in human brain cells, real-time PCR was performed on RNA isolated from human neurons (HCN-2 cells), glial cells (M059K cells), astrocytes (CCF-STTG1), and phorbol-12-myristate-13-acetate (PMA)-stimulated macrophages (THP-1 cells). Of these cell types, miR-367-3p was found to be present in human cortical neurons (HCN-2 cells) and present at only a low level, or not detected in the other cell types (Figure 5(b)). GPRC5A mRNA levels were also found to be the highest in the neurons (HCN-2). GPRC5A was also expressed in glial cells (M059K) (Figure 5(c)). These results suggest that miR-367-3p is co-expressed with GPRC5A in human neurons. To experimentally validate that miR-367-3p regulates GPRC5A, loss-of-function studies for miR-367-3p were performed in primary neurons ex vivo. Mouse primary neurons were collected and transiently transfected with LNA-based miRNA inhibitors against miR-367-3p (LNA-367-3p) or scrambled control sequence (Scr). Gprc5a mRNA levels were significantly increased by miR-367-3p inhibition (LNA-367-3p) in primary neurons, suggesting that that Gprc5a is endogenously regulated by miR-367-3p in neurons (Figure 5(d)). To quantify the impact of miR-367-3p loss-of-function on neuroinflammation and the expression of inflammatory cytokines in mouse primary neurons, mRNA levels of Tnf, which is expressed and secreted from neurons during neuroinflammation,
33
were quantified by real-time PCR and we found that inhibition of miR-367-3p resulted in a significant increase in Tnf mRNA levels compared to negative control treated cells (Figure 5(e)). These results support a critical role for miR-367-3p, and potentially Gprc5a, in inflammation in neurons and cytokine secretion from neurons in ischemic stroke.
miR-367-3p regulates GPRC5A in neurons. (a) Conserved putative miR-367-3p target sites in the 3′ untranslated regions (3′ UTR) of mouse and human GPRC5A mRNAs. Black, putative seed region for miR-367-3p. (b, c) Real-time PCR quantification of (b) miR-367-3p (n = 6) and (c) GPRC5A mRNA (n = 6) in human brain cells: HCN-2 (cortical neurons), M059K (glial cells), CCF-STTG1 (astrocytes) and THP-1 (PMA-stimulated macrophages). RQV: relative quantitative value. (d, e) Real-time PCR quantification of (d) Gprc5a and (E) Tnf mRNA levels in mouse primary neurons transfected with locked-nucleic acid (LNA) miRNA inhibitors against scrambled control sequence (LNA-Scr) or miR-367-3p (LNA-367) (n = 9–11). Mann–Whitney non-parametric tests. Data are presented as mean ± SD (*p < 0.05).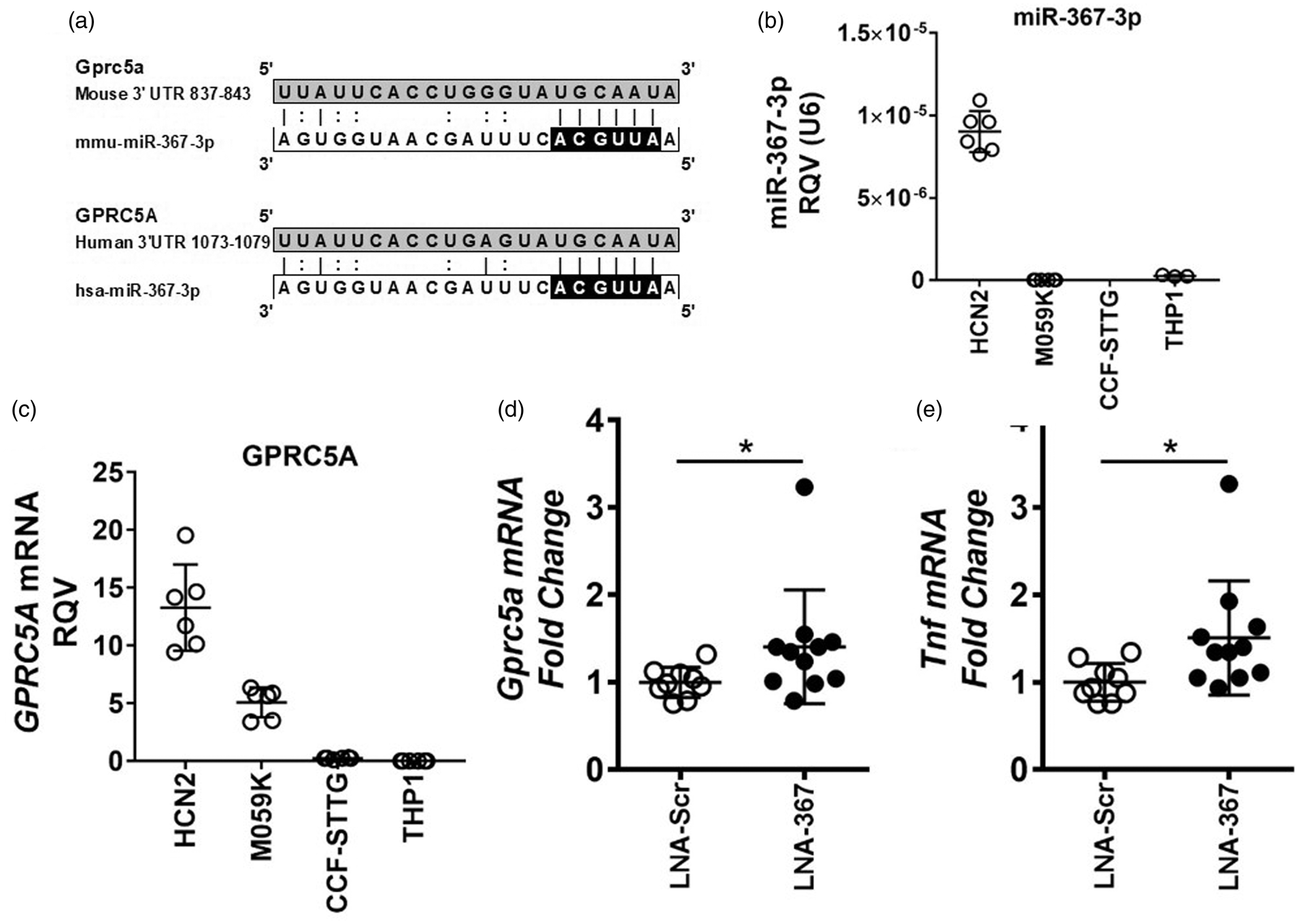
Discussion
miRNAs are critical regulators of inflammatory processes in the brain and are likely altered with ischemic stroke.17,18,25,29,30,34–37 Here, we performed a comprehensive analysis of miRNA changes associated with sustained and transient ischemia and identified novel miRNA regulatory modules associated with inflammation. Using real-time PCR-based TaqMan OpenArrays and individual assays, multiple miRNAs were found to be significantly decreased in response to sustained ischemia in the brain at 24 h post-MCAo. Many of these miRNA changes were prevented when ischemia was limited to 30 min and followed by reperfusion (transient ischemia). Candidate miRNAs that were significantly inhibited with sustained ischemia included miR-367-3p, miR-302b-3p, miR-296-5p, miR-489-3p, miR-224-5p, and miR-181b-5p. Conversely, sustained ischemia resulted in a significant increase of many genes at the mRNA level, several of which were blunted by transient ischemia followed by reperfusion. Strikingly, many of the significantly increased mRNAs were identified as predicted targets of the significantly decreased miRNAs in sustained and transient ischemia conditions, including Gprc5a and Ccrl2. Although Ccrl2 has previously been linked to ischemic brain injury, the functional role(s) of Gprc5a in ischemic stroke or the brain have not been reported. 38
miR-367-3p belongs to the poly-cistronic miRNA cluster harboring both miR-302b and miR-367. This cluster is abundantly expressed in mouse and human embryonic stem cells, 39 and has been reported to be critical in the early stages of embryo development.40,41 Related to this developmental role, the promoter of the primary miR-302/367 transcript is activated by the pluripotent transcription factors Oct4, Sox2 and Nanog.42,43 Like many miRNAs, miR-367-3p has been widely studied in cancer. For example, miR-367-3p levels are reduced in human gastric cancer tissues compared to matched paraneoplastic tissues. Conversely, miR-367-3p overexpression inhibits cellular migration and invasion in gastric cancer cells, likely through regulation of Rab23. 44 The present study established that miR-367-3p was decreased by 75% in mouse brain homogenates after sustained ischemia at 24 h post-MCAo. Although miR-367-3p has not been previously linked to ischemic stroke, miR-367-3p levels are decreased in microglial cells in hemorrhagic stroke. 27 ICH stroke results in inflammatory activation of microglial cells mediated, in part, through activation of nuclear factor-κB (NF-κB). miR-367-3p was found to directly target interleukin 1 (IL-1) receptor-associated kinase 4 (IRAK4), which is a key signaling factor in NF-kB activation in glial cells. Thus, loss of miR-367-3p in microglial cells with ICH may contribute to the observed increase in inflammatory gene expression. Moreover, miR-367-3p was found to regulate expression of the p65 subunit of NF-κB, interleukin 6 (IL-6), interleukin 1β (IL-1β), and tumor necrosis factor α (TNF-α) in both microglial cell cultures and brain tissues after ICH. 27 Intracerebroventricular injection of miR-367-3p mimetics also significantly decrease cerebral water content, neurological severity scores, and pro-inflammatory cytokines after ICH. 27 Although this report clearly established a functional role of miR-367-3p in microglial cells in the brain, we found that miR-367-3p was more abundant in neurons than in glioma cells (M059K), PMA-stimulated THP1 macrophages, or astrocytes (CCF-STTG1). This suggests that miR-367-3p mimetics may be used to restore miR-367-3p levels in neurons, silence pro-inflammatory gene expression in neurons, and attenuate inflammation associated with ischemic stroke.
Based on previous studies, and the present results, miR-367-3p likely contributes to neuroinflammation associated with stroke through direct and indirect mechanisms. 27 One candidate target gene of interest, and a novel gene to the brain and ischemic stroke, is GPRC5A. Also known as retinoic acid-induced gene 3 (RAI3), GPRC5A has been studied extensively as a tumor suppressor of lung cancer.45–49 GPRC5A has also been implicated in multiple other cancers.50,51 GPRC5A is an orphan G-protein coupled receptor and is localized to the plasma membrane, endoplasmic reticulum (ER), Golgi, and cellular vesicles. 49 miR-367-3p is predicted to target both mouse and human GPRC5A at a single conserved target site within the 3′ UTR of the mRNA. GPRC5A has a variety of interesting functions. For example, it binds to the eukaryotic initiation factor 4F (eIF4F) complex and inhibits translation of key mRNAs, including epidermal growth factor receptor (EGFR) in the endoplasmic reticulum. 49 GPRC5A also suppresses signal transducer and activator of transcription 3 (STAT3) activation, with Gprc5A deletion in mice having increased STAT3 phosphorylation and STAT3-mediated transcription of pro-survival genes in lung epithelial cells. 52 Therefore, suppression of miR-367-3p and the subsequent increase in GPRC5A may decrease STAT3-mediated cell survival in neurons in the brain following ischemic stroke. Interestingly, five STAT3 transcriptional target genes, including Bcl2, the critical anti-apoptotic gene in B-cell lymphoma, were significantly decreased in brain homogenates from mice with sustained ischemia. Other significantly decreased STAT3 targets genes included cyclin dependent kinase 4 (Cdk4), early growth response protein 2 (Egr2), lymphocyte antigen 6 A (Ly6a), and cystine/glutamate transporter (Slc7a11). These findings suggest that over-expression of miR-367-3p or silencing of GPRC5A may be a potential strategy for increasing cell survival in brain cells (e.g. neurons) in response to ischemic stroke. One caveat is that GPRC5A has been identified as a repressor of NF-κB transcriptional activity. For example, Gprc5a-deficiency in mice increases basal and lipopolysaccharide-induced NF-κB transcriptional activity in lung epithelial cells, and thus, promoted lung inflammation. 53 Therefore, based on our observation that Gprc5a mRNA levels are significantly increased in ischemic stroke, we would predict that NF-kB activity and transcriptional target genes would be decreased. Therefore, we mined the list of significantly altered genes and found more NF-kB transcriptional target genes to be increased than decreased. For example, we identified 43 NF-kB target genes to be significantly increased (out of 167 genes that were increased) with sustained ischemia, compared to 11 significantly decreased NF-kB target genes (out of 225 genes that were decreased). Moreover, key pro-inflammatory NF-kB target genes were present in the significantly increased gene set, including Tnf, toll-like receptors 2 and 7 (Tlr2 and Tlr7), intercellular adhesion molecule-1 (Icam1), and p65 (Rel). Based on these observations, GPRC5A may not suppress NF-kB activation in the brain in the setting of ischemic stroke. Nonetheless, one limitation of the study is that we only quantified gene expression changes at 24-h post-ischemia (surgery) and many of the gene changes likely differentially occur at distinct intervals. For example, many pro-inflammatory NF-kB target genes may be activated or repressed at different times post-MCAo surgery in response to hypoxia (ischemia) and reperfusion. Therefore, we acknowledge that this pattern of gene expression at 24 h may be different if sampled at earlier or later time-points.
Although GPRC5A has not previously been investigated in the brain, studies in other tissues provide insight into possible mechanisms that regulate and/or influence GPRC5A expression. GPRC5A expression was originally cloned as a retinoic acid (RA)-inducible gene (i.e. RAI3) and the impact of RA on GPRC5A is well established.45,54 GPRC5A expression is also increased by cAMP. 55 In addition to these transcriptional ligands, previous studies have implicated a few other transcriptional networks. For example, GPRC5A is likely a target of mutant p53 involved in cell growth, 56 and it is increased in response to hypoxia in colorectal cancer cells. 57 In colonic epithelial cells, GPRC5A is a transcriptional target of both HIF-1α and HIF-2α. 57 Based on these data, loss of miR-367-3p in ischemic stroke may help support hypoxia-induced GPRC5A transcriptional activation by increasing GPRC5A mRNA stability. In addition to miR-367-3p, GPRC5A has been reported to be a post-transcriptional target of other miRNAs, including miR-204, miR-31, and miR-103a-3p.58–60 Of note, miR-103a-3p binds to the 5′ UTR of GPRC5A mRNA, as opposed to the 3′ UTR where miRNA target sites are normally enriched. 58 These results support the notion that GPRC5A is regulated by both transcriptional and post-transcriptional mechanisms that may have contributed to the observed increase in Gprc5a expression in brains after sustained ischemia. Our results also suggest that the pro-inflammatory miR-367-3p:Gprc5a axis is suppressed by reperfusion or limited by restricting the duration of ischemia.
In addition to Gprc5a, miR-367-3p may regulate other genes that were significantly increased in the brain after ischemic stroke. For example, we found that the expression of the atypical receptor of chemerin (Ccrl2) was significantly increased with sustained ischemia, and was blunted with transient ischemia. miR-367-3p is predicted to target Ccrl2 in mice and the observed decrease in miR-367-3p with sustained ischemia may contribute to the increase in Ccrl2 expression in mouse brain. Chemerin is an agonist of the adipokine CCRL2 which contributes to endocrine-like signaling. 61 Chemerin also recruits inflammatory cells such as dendritic cells and macrophages, to sites of inflammation.62,63 Activation of its primary receptor, chemerin receptor 23 (ChemR23), induces signaling and NF-kB activation and inflammation 64 ; and there is some evidence of an interaction between chemerin’s classic (ChemR23) and atypical (CCRL2) receptors. CCRL2 is not predicted to serve as an agonist recruiter similar to other atypical chemoattractant receptors. However, increased CCLR2 expression and chemerin activation may induce pro-inflammatory signaling in the brain. Although regulation of Ccrl2 expression in the brain is not well understood, Ccrl2 is a possible transcriptional target of NF-kB.65,66 One limitation to this potential mechanism, and the reason we did not investigate it further in this study, is that miR-367-3p is not predicted to target CCRL2 in humans. Nevertheless, the role of chemerin and CCRL2 in human ischemic stroke is a potential area for future study.
In addition to miR-367-3p, other miRNAs were also suppressed in response to sustained ischemia. Many of these miRNA changes are unique to ischemic stroke and the brain. Furthermore, these miRNAs were also predicted to target mRNAs that were significantly increased, including miR-302b-3p, which is harbored on the same poly-cistronic miRNA cluster as miR-367-3p. Other regulated candidate miRNAs included miR-296-5p, miR-489-3p, and miR-224-5p. miR-181b-5p has previously been associated with ischemic stroke,16,25 and recent evidence suggests that miR-181b-5p inhibition improves long-term recovery after stroke. 21 Previous studies have shown that miR-181b-5p is enriched in neurons and microglia cells in the brain67–69 and is altered in stroke.15,26 Here we found that miR-181b-5p levels were significantly decreased in both sustained, and to a lesser extent in transient ischemia in the brain. This confirms previous studies which also reported that miR-181b-5p levels are down-regulated in mouse brains following transient MCAo ischemic stroke.15,34 Based on our results and previous studies, miR-367-3p, miR-181b-5p, and the other miRNAs found to be inhibited with ischemic stroke warrant further investigation as key miRNAs in brain gene regulation.
Conclusions
Our results provide evidence that miR-367-3p is a critical regulator of post-transcriptional circuitry in the brain and that it likely affects ischemia-reperfusion associated neuroinflammation. Although it is well established that sustained ischemia causes many changes to inflammatory gene expression and miRNAs in the brain, this study has identified a new post-transcriptional mechanism for miR-367-3p suppression that underlies some of these changes. Moreover, a new factor in ischemic stroke, GPRC5A, that likely contributes to neuroinflammation has been identified. Due to findings that this miRNA regulatory module is attenuated by limiting the time of ischemia and/or reperfusion, future studies should investigate the targeting of this mechanism to treat ischemic stroke. Specifically, increasing miR-367-3p with mimetics and decreasing GPRC5A expression with antisense oligonucleotides may reduce neuroinflammation associated with ischemic stroke.
Supplemental Material
Supplemental material for microRNA-367-3p regulation of GPRC5A is suppressed in ischemic stroke
Supplemental Material for microRNA-367-3p regulation of GPRC5A is suppressed in ischemic stroke by Fatiha Tabet, Seyoung Lee, Wanying Zhu, Michael G Levin, Cynthia L Toth, Luisa F Cuesta Torres, Antony Vinh, Hyun Ah Kim, Hannah X Chu, Megan A Evans, Meaghan E Kuzmich, Grant R Drummond, Alan T Remaley, Kerry-Anne Rye, Christopher G Sobey and Kasey C Vickers in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by the National Institutes of Health (USA), National Heart, Lung and Blood Institute (NHLBI) extramural research awards and intramural research funds. This study was also supported by awards from the National Health and Medical Research Council of Australia (NHMRC) and the National Heart Foundation of Australia. These studies were supported by Project Grants from the National Health and Medical Research Council of Australia (NHMRC) (APP1041326; APP1064686; APP1062721). FT was supported by the National Heart Foundation of Australia Future Leader Fellowship (100090). We also acknowledge support from NHMRC Senior Research Fellowships (CGS: APP1079467; GRD: APP1006017). KCV was supported by NIH awards K22HL113039, P01HL116263, R01HL128996, and R01HL127173.
Acknowledgement
We would like to thank Andrea DiBiase and Life Technologies for their assistance with this study. The authors would also like to acknowledge Marisol A. Ramirez for assistance with bioinformatics.
Declaration of conflicting interest
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
FT, SL, WZ, MGL, CLT, LFCT, AV, HAK, HXC, MAE, MEK, performed the experiments and analyzed results. FT, SL, GRD, ATR, KAR, CGS, and KCV designed the study, provided technical and funding assistance, and wrote the manuscript.
Supplementary material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.