
Abstract
Extracellular proton concentration is at 40 nM when pH is 7.4. In disease conditions such as brain ischemia, proton concentration can reach µM range. To respond to this increase in extracellular proton concentration, the mammalian brain expresses at least three classes of proton receptors. Acid-sensing ion channels (ASICs) are the main neuronal cationic proton receptor. The proton-activated chloride channel (PAC), which is also known as (aka) acid-sensitive outwardly rectifying anion channel (ASOR; TMEM206), mediates acid-induced chloride currents. Besides proton-activated channels, GPR4, GPR65 (aka TDAG8, T-cell death-associated gene 8), and GPR68 (aka OGR1, ovarian cancer G protein-coupled receptor 1) function as proton-sensitive G protein-coupled receptors (GPCRs). Though earlier studies on these GPCRs mainly focus on peripheral cells, we and others have recently provided evidence for their functional importance in brain injury. Specifically, GPR4 shows strong expression in brain endothelium, GPR65 is present in a fraction of microglia, while GPR68 exhibits predominant expression in brain neurons. Here, to get a better view of brain acid signaling and its contribution to ischemic injury, we will review the recent findings regarding the differential contribution of proton-sensitive GPCRs to cerebrovascular function, neuroinflammation, and neuronal injury following acidosis and brain ischemia.
Introduction
It has long been known that the brain becomes acidic during and following ischemic stroke.1–3 There have been extensive studies on the multiple mechanisms contributing to the regulation of brain pH homeostasis in health and disease. The data have revealed that carbonic anhydrases, sodium-proton exchanger, different proton, bicarbonate, and lactate transporters all contribute to pH regulation in the brain (for reviews, see4–7) On the other hand, it remains underappreciated regarding the complexity of brain acid signaling or its contribution to ischemia-induced cerebrovascular dysfunction. Protons can modulate the activities of multiple membrane proteins, e.g., inhibiting the NMDA receptors.8,9 Moreover, proton can serve as ligands and signal directly through three classes of proton-sensitive receptors. These include the cationic acid-sensing ion channels (ASICs), 10 the proton-activated chloride channel (PAC)/acid-sensitive outward rectifying anion channel (ASOR),11,12 and the family of proton-activated GPCRs, which include GPR4, GPR65 (aka T cell death associated gene-8, TDAG8), and GPR68 (aka ovarian cancer G-protein coupled receptor 1, OGR1). 13 Section ‘An overview of brain pH regulation’ of this review will summarize what the literature has documented regarding brain pH regulation and pH dynamics in brain ischemia. Next, we will discuss acid-induced signaling through proton-sensitive receptors in the brain, with a focus on recent findings of PAC/ASOR and proton-sensitive GPCRs.
An overview of brain pH regulation
Within the brain parenchyma, the main cell types contributing to brain pH homeostasis include neurons, astrocytes, and microglia.4,5 One universal buffer in these cells is the carbon dioxide (CO2)-bicarbonate system. However, the conversion between carbonic acid and bicarbonate is a very slow reaction on its own. For the CO2/HCO3− buffering system to work efficiently, the presence of functional carbonic anhydrase is a key.6,7 Indeed, the brain expresses eleven of the thirteen functionally active carbonic anhydrases, with some exhibit preferential localization to specific domains (intracellular, membrane bond, or extracellular) or cell types. For example, carbonic anhydrase IV and XIV, which are membrane bond, exert their catalytic function on the extracellular side. For a detailed review on carbonic anhydrases in the brain, see reference. 7
Besides the bicarbonate system, non-bicarbonate buffering also contributes to pH regulation in the brain. While most molecules can act as proton donor and receiver and thus contribute to buffering, lactate is one important molecule for brain pH during neural activity or in anaerobic metabolism.6,14 This is in part because lactate levels exhibit over 10 fold change under anaerobic conditions such as ischemia. 15 The regulation of brain pH apparently depends on the crosstalk between neurons and astrocytes.5,6,14 Under physiological conditions, neurons preferentially utilize aerobic respiration for its ATP production. Astrocytes, on the other hand, tend to go through aerobic glycolysis, which converts glucose to lactate even when there is sufficient oxygen supply. 6 Astrocytes also are the main cells for glucose uptake, especially in response to increased activities. These properties together enable the astrocytes to uptake glucose, convert it to lactate, and then shuttle it to neurons as the energy source for aerobic respiration.6,14
The monocarboxylate transporters (MCT2 in neurons; MCT1 and MCT4 in astrocytes) are responsible for transporting lactate in and out of astrocytes and neurons. 6 Multiple other transporters in neurons and astrocytes are important for pH regulation. These include the sodium-hydrogen exchangers, sodium bicarbonate cotransporter, sodium-driven chloride/bicarbonate exchanger, calcium/proton exchangers. For more information on these topics, see reviews.5,6,14
Brain pH dynamics in ischemia
Protons get buffered fast as essentially all biomolecules can act as proton donors/acceptors. Thus, accurate pH measurement, especially in vivo, is challenging. Typical approaches used in previous studies include direct measurement with pH microelectrode, fluorescent imaging with pH sensitive dyes or reporters, and functional pH imaging. It has long been known that ischemia leads to brain acidosis, both during ischemia and after reperfusion. The exact pH values measured differ between studies. In various studies, the resting or physiological extracellular brain pH is in the range of 7.2–7.4, while that of intracellular pH can be slightly lower.16–19 Based on these data, in our discussion here, we consider extracellular “acidotic or acidosis” starts at ∼pH 7.2.
pH changes during the ischemic phase
During ischemia, the stop of blood flow limits the supple of oxygen to brain tissue, and consequently brain cells transit from aerobic respiration to anaerobic glycolysis. 20 This conversion appears to occur within minutes of ischemia. As a result, neurons quickly deplete ATP, creatine, and phosphor-creatine but build up lactate and protons, and the intracellular pH (pHi) becomes acidic.3,15,20 The subsequent exporting of lactate could contribute in part to extracellular acidification. Other mechanisms which can contribute to interstitial pH reduction include the activation of NHE and the import of bicarbonate inside to counteract intracellular acidification.4,5 In several reports, mostly using rodent models, a rapid pH reduction occurs within minutes of ischemia, to the range of 6.5–6.0.17,21–23 At about 30 min after occlusion, brain pH is typically down to 6.2 or even below 6 in some cases. Hyperglycemia further worsens the degree of acidification while hypoglycemia alleviates it.17,19,24–26 When there is no reperfusion, this level of pH reduction is maintained for up to 6 hr. In cats, acidosis persists to the second day, though the degree of acidosis becomes milder. 27 In human, at an average of 6 days after ischemic stroke, the brain exhibits a slight alkaline shift. 28
pH changes following reperfusion
In transient ischemia, reperfusion quickly normalizes the metabolites and brings pH back to the normal range. 15 In another study, Maruiki et al performed 12 min complete ischemia in dogs and found that brain pH returned to pre-ischemia level within 30–60 min of reperfusion. 23 However, if the ischemic event lasts longer, the rise in oxidative stress accompanies disrupted metabolism at the reperfusion stage can lead to another phase of prolonged reduction in brain pH.18,29,30 In one study, following 60 min transient middle cerebral artery occlusion (tMCAO), a transient rebound into the alkaline range occurs at 2-hr after reperfusion, then brain pH reaches ∼6.5 at the 4-hr time point. 18 It is worth noting that while the extracellular brain pH typically maintains in acidic phase for hours following reperfusion, intracellular pH can become slightly alkaline within an hour. 15 Figure 1(a) illustrates qualitatively the dynamics of pH changes in ischemia, either with or without reperfusion.
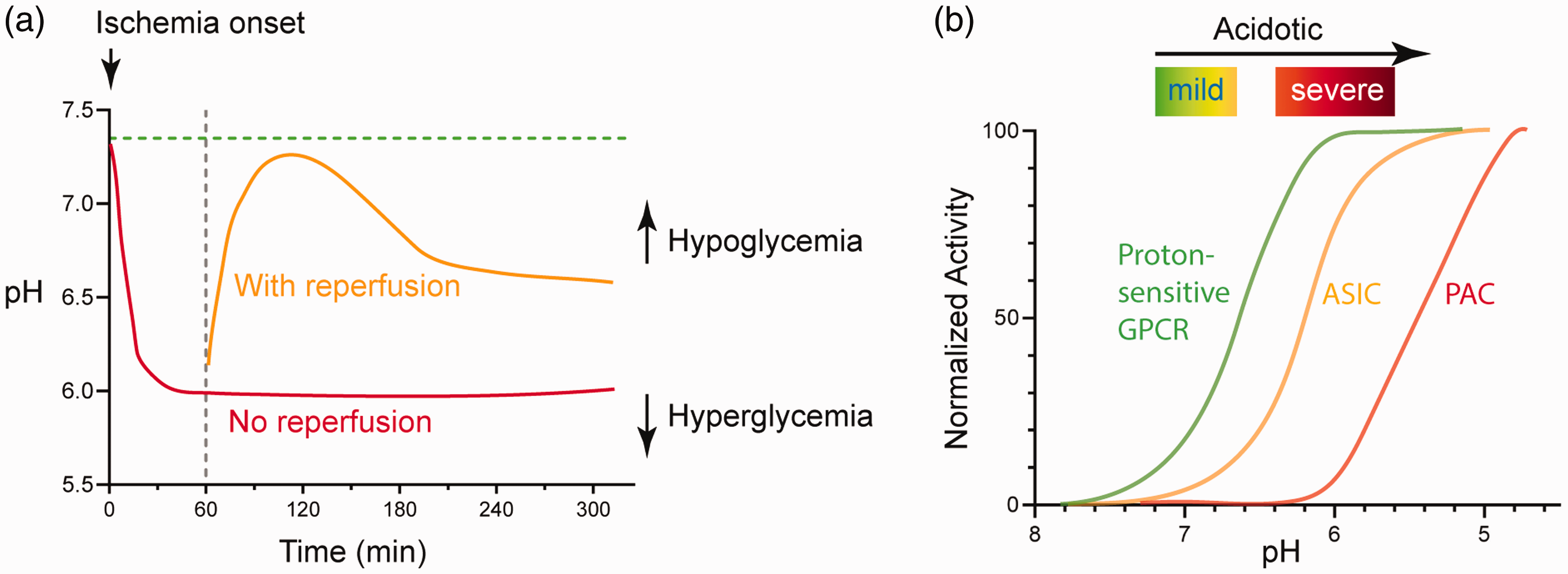
pH dynamics and the three classes of acid-sensitive receptors in the brain. (a) Illustration showing brain pH changes during brain ischemia and following reperfusion. The red line illustrates the change in permanent occlusion. The orange line illustrates the approximate change following reperfusion. These curves may shift upward or downward in hypoglycemic or hyperglycemic conditions, respectively. See text for details. (b) Diagram illustrating the pH sensitivity of the three classes of proton receptors. The curves are qualitative representation of the approximate/average pH response curves of a group of receptors within that family. See text for more explanation.
The proton-sensitive receptors
The first receptor identified to functionally mediate acid-sensing in the brain is the acid-sensing ion channel 1a (ASIC1a) . 31 It is now evident that the brain expresses most members in the three families of proton-sensitive receptors, which include the ASICs, the PAC/ASOR, and the proton-sensitive GPCRs. However, these receptors exhibit distinct patterns of expression and downstream signaling. In Table 1, we summarized the genomic location, main downstream signaling of these receptors, and some of the reagents which are relatively specific for the subtypes of the receptors. It is important to note that, especially for ASIC channels, there are additional reagents which are less selective among the subtypes. For more extensive reviews on ASIC modulation, including additional pharmacological reagents, see references.32,33
Summary of signaling and pharmacological reagents for proton-sensitive channels and GPCRs.
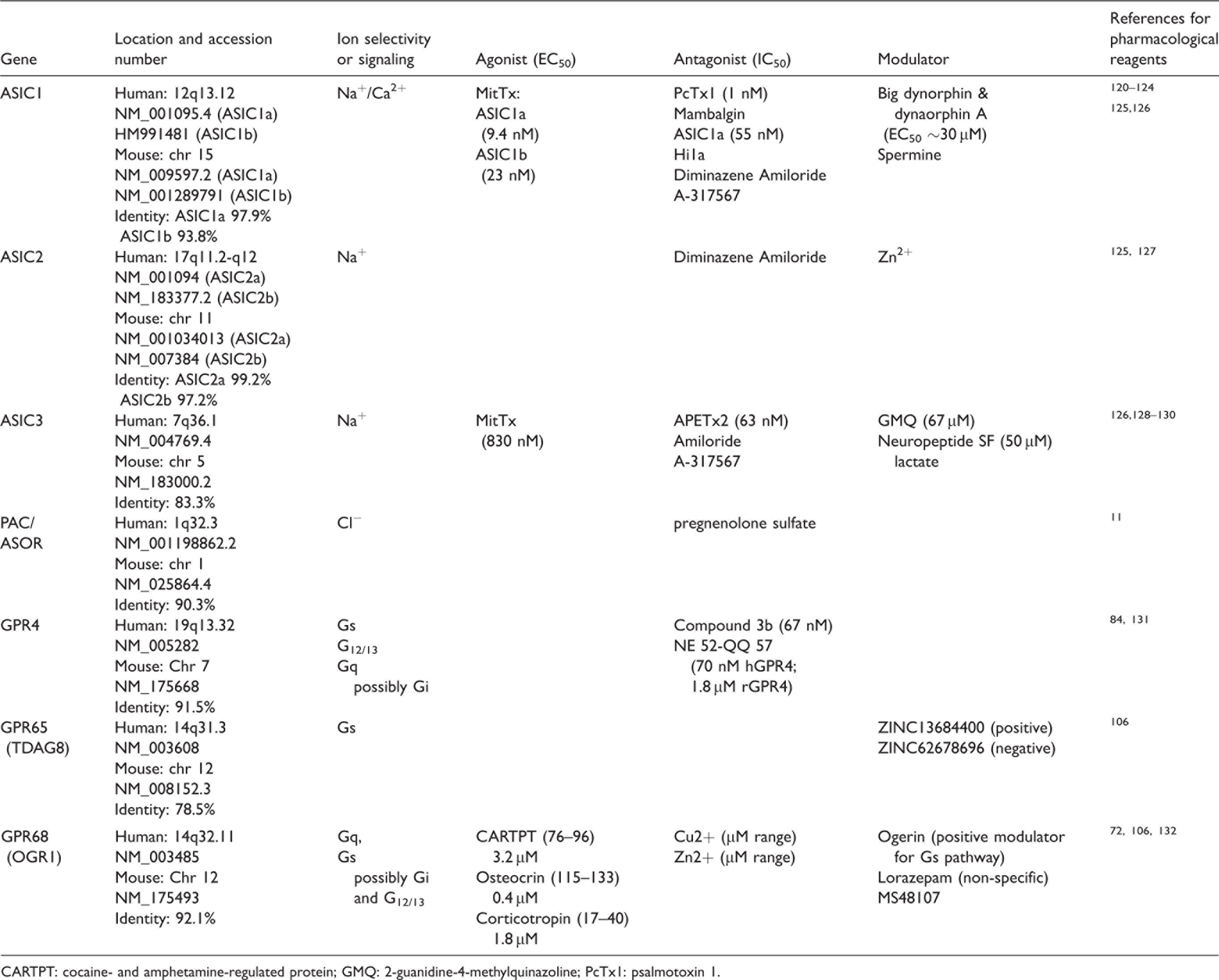
CARTPT: cocaine- and amphetamine-regulated protein; GMQ: 2-guanidine-4-methylquinazoline; PcTx1: psalmotoxin 1.
Expression of proton-sensitive receptors in the brain and genetic tools available.
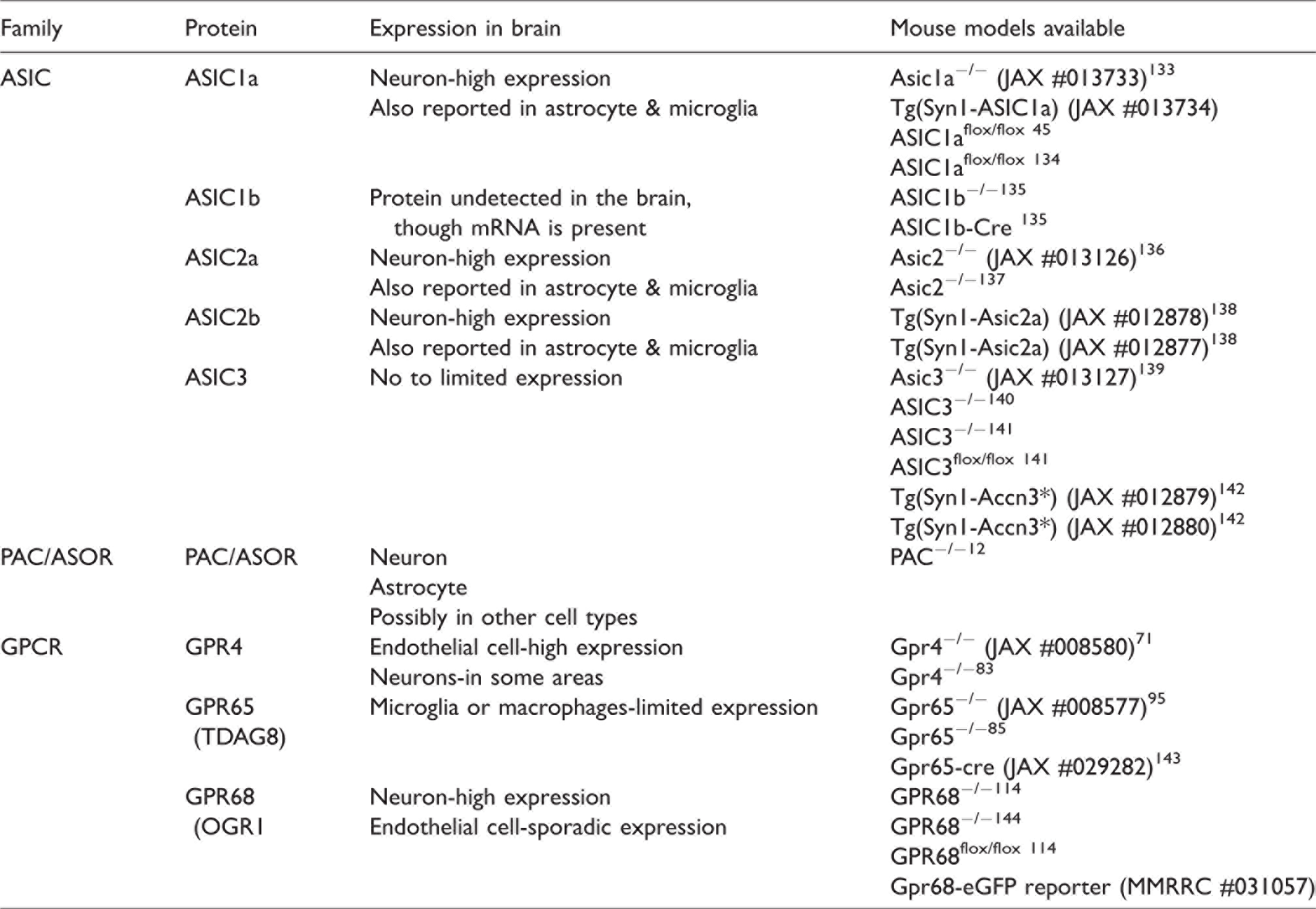
In previous studies, multiple groups have generated various kinds of mouse models for these receptors, including global knockout, conditional knockout, and reporter lines. Table 2 summarizes the mice which have been reported in the literature. Table 2 also presents a summary of the overall expression of these receptors in the central nervous system. In the brain, ASICs are mostly present in neurons.32,34 ASIC1a, 2a, and 2 b are the major ASIC subunits expressed in brain neurons. The PAC/ASOR channel are more ubiquitously expressed in both neurons and non-neuronal cells.11,12,35,36 For the GPCRs, GPR4, GPR65, and GPR68 exhibit preferential expression in endothelium, neuron, and microglia, respectively (see text below for more detailed discussion).
Besides differential expression, another important difference exist among these receptors is their pH sensitivity and kinetics. The ASICs, depending on the subunit composition, have a pH50 around 6.8–5. 37 The PAC requires much lower pH to get activated, with pH50 around 5.11,12,35 Compared to the ASICs and PAC, the GPCRs exhibit higher pH sensitivity (Figure 1(b)). All three receptors start to get activated at about pH 7.4 or even higher, and typically reach maximal activation at pH 6.8–6.238–40. One caveat of these studies, however, is that the majority of these studies on pH responses in GPCRs used ectopic expression systems. Nevertheless, it is apparent that the proton-sensitive GPCRs do not exhibit fast desensitization, making them an attractive mediator of acid signaling during persistent acidosis.
Acid-sensing ion channels (ASICs)
The ASICs are a family of proton-gated cation channels. There are four genes encoding ASICs: ASIC1-4.10,32 ASIC1 and ASIC2 have two splice variants a and b. The ASIC subunits have two transmembrane domains with a huge extracellular domain. Three subunits form one functional ASIC channel. 41 ASIC1a, 1 b, 2a, and 3 can conduct acid-activated cation currents. ASIC1 and 3 respond to pH drop with a threshold range of 7.0-7.2 while ASIC2a are much less pH sensitive and starts to get activated at pH close to 5.5.33,37 ASIC channels mostly conduct Na+. However, homomeric ASIC1a, human ASIC1b, as well as ASIC1a/2b heteromers, exhibit a permeability to Ca2+.31,42 Though proton is the only known ligand, various cations, neuropeptides, and oxidants can have important modulatory effects on ASIC function.34,37 As a neuronal and synaptic acid receptor, ASICs contribute to synaptic function, plasticity, and learning.10,32,34 Detailed review of ASIC biophysical properties and regulation can be found in multiple review articles.33,43
ASIC and hypercapnia-induced cerebral vasodilation
Besides its function in neuron physiology, ASIC also contributes to cerebral blood flow regulation. In one study, ASIC1a deletion or local infusion of psalmotoxin (PcTx1), a specific inhibitor of homomeric ASIC1a and heteromeric ASIC1a/2b channels, attenuated hypercapnia-induced vasodilation. 44 The authors further generated a synapsin-cre driven syn-ASIC1a knockout mouse, which had reduced ASIC current in interneurons and principal neurons. 45 The syn-ASIC1a knockout mice had attenuated response to CO2. This result suggests that ASIC1a activation in neurons contributes to hypercapnia-induced vasodilation. While the finding is interesting, it is worth noting that GPR4 in endothelium may play a more direct role in CO2-induced vasodilation (see below).
ASICs in acidotoxicity and ischemic injury
In disease conditions which induce large pH reduction, ASICs are one important mediator of acid-induced neuronal injury. ASIC1a−/− exhibits significant reduction in ischemia-induced brain injury.18,46 Inhibiting ASIC activity by either amiloride, a non-specific ASIC inhibitor, or either PcTx1 or Hi1a, two disulfide-rich spider venom peptides, all have protective effect following tMCAO.46,47 Deleting the ASIC2 gene, through reducing ASIC1a trafficking and possibly biogenesis as well, also reduces tMCAO-induced brain injury.48,49 The protective effect of ASIC inhibition is additive to that of NMDA receptor inhibition.18,50 These data suggest that ASIC targeting can be combined with other approaches to offer enhanced protection against ischemia-induced brain injury. In most of the above studies, the pro-injury function of ASICs correlates with its channel activity. However, there are data showing that ASIC1a can elicit necroptosis independent of its channel activity. 51 This new mechanism requires the ASIC1a C-terminal tail, which elicits cell death through an interaction with serine/threonine kinase receptor interaction protein 1.
Proton-activated chloride channel
Discovery
It has been known for some years that protons induce an acid-sensitive outward rectifying anion channel (ASOR), which passes chloride.52–54 With a cell-based RNA interference screening, two groups recently identified the molecular constituent of this anion current.11,12 The ASOR channel turns out to be a previously uncharacterized protein, TMEM206 (transmembrane protein 206 or C1orf75). One group named it based on its function as “proton-activated chloride channel (PAC)”. 12 PAC/ASOR/TMEM206 is present in multiple organisms, including zebrafish, chicken, rodents, and human. Ectopic expression of PAC in heterologous cells reconstitutes the typical proton-activated ASOR current.11,12
General properties
PAC/ASOR has two transmembrane domains, with the N- and C-termini inside the cell and a large extracellular domain.11,12 A recent Cryo-EM study shows that PAC also forms a trimer. 55 Thus, the overall topology and stoichiometry resemble that of the ASICs, even though the two types of channels do not share any close sequence homology. At room temperature, PAC has an activation threshold of about pH 5.5 and a pH50 of close to pH 5. At 37 °C, PAC starts to get activated at about pH 6.2 with its pH50 shifted to around ∼5.7. These data indicate that PAC is less pH sensitive than ASICs (also see Figure 1(b)). Consequently, PAC activation requires relatively more severe acidosis, i.e., when pH is reduced to 6 or lower.
Cells expressing PAC are present in multiple anatomical places, with high expression levels in brain, bone marrow, kidney, lung, lymph node, spleen, and bladder. In the brain, neurons possess robust PAC current.12,35 In addition, PAC is also present in non-neuronal cells in the brain, including astrocytes and microglia. 36
PAC/ASOR and intracellular trafficking
Besides its presence at cell membrane, PAC/ASOR can traffic to endosomes and forms a functional chloride channel there. 56 The trafficking to endosome apparently requires the YXXL motif of PAC/ASOR. PAC regulates endosomal pH and chloride concentration. Deleting PAC abolishes endosomal chloride leakage, which consequently raises chloride concentration and reduces endosomal pH. 56 In PAC deficient HEK293 cells, transferrin uptake exhibits a 30% increase. 56 This finding suggests that, through modulating endosomal pH and ion gradient across endosomal membrane, PAC/ASOR serves as one regulator of vesicle recycling and/or receptor trafficking.
Role in acidotoxicity and ischemic injury
PAC/ASOR activation in acidotic conditions leads to cell swelling, which suggests that its activation contributes to acidosis-induced cellular injury. In addition, its pH sensitivity indicates that the PAC/ASOR pathways contribute to injuries when the acidosis is more severe (e.g. when pH is lower than 6). In HEK293 cells, deleting PAC/ASOR protects the cells from pH 4.5-induced swelling and cell death. 11 In cultured cortical neurons, PAC deletion or shRNA knockdown offers protection against pH 6 and 5.6-induced cell death.12,35 In a permanent model of brain ischemia, PAC null mice exhibit a reduction in MCAO-induced brain infarction. 12
Proton-sensitive GPCRs
Introduction
Around mid-1990s, homologous cloning led to the identification of three proton-sensitive GPCRs: GPR4, GPR65, and GPR68.57–59 These three GPCRs belong to the orphan family of GPCRs. Protons are the only well-established ligand which activates these receptors. 60 Phylogenic analysis shows that GPR4, 65, 68 evolved from a common ancestor, GPR132 (or G2A, G2 accumulation protein). 38 Early studies showed that GPR132 also responds to proton.61,62 However, based on structural modeling and mutagenesis analysis, GPR132 does not contain the key proton-sensing residues which are conserved in the other three receptors. 38 In addition, GPR132 does not exhibit a robust pH-dependent signaling as compared to the other three GPCRs. In qPCR and RNA-Seq analysis, GPR132 had little expression in either mouse or human brain.63,64 For these reasons, we focus here on GPR4, GPR65, and GPR68.
For all three receptors, when ectopically expressed together with various G alpha subunits, they are capable of coupling to most G alpha subunits tested. 38 This result indicates that the exact signaling these receptors conduct will depend on the specific system/cell type and the availability of the G alpha subunits. In the discussion below, we will mainly cover the results obtained from the brain and/or mammalian cells without the overexpression of G alpha. Table 1 presents a summary of the signaling and main pharmacological reagents which are currently available for these receptors.
GPR4
GPR65
GPR68
A recent study examined the contribution of GPR68 in a sevoflurane-induced neurotoxicity. 117 In this model, 4.9% sevoflurane for 2 hrs induces neuronal loss in hippocampus in neonatal rats. This change correlates with a reduction in GPR68 expression. AAV-mediate GPR68 overexpression reverted sevoflurane-induced loss of neurons. This effect correlates with a reduced neuronal apoptosis with GPR68 overexpression. Functionally, GPR68 overexpression improves the behavioral performance in the Morris water maze test. Compared to the group injected with control AAV, the group receiving AAV-GPR68 had reduced escape latency, increased time in target quadrant, and increased number of platform crossings. 117 The study further shows that GPR68 overexpressed group exhibits an increase in oligodendrocyte proliferation and myelination. Another interesting observation here is that sevoflurane treatment reduces BDNF expression while GPR68 overexpression partially rescues the reduction in BDNF. Though the mechanism is unclear, the finding is consistent with a neuroprotective role of GPR68 in the brain, and suggests a potential link to neurotrophic signaling pathways.
In organotypic cortical slices, inhibiting PKC activities with Go6983 worsened acidosis-induced neuronal injury in WT slices, but had no significant effect in GPR68−/− slices. 63 In our RNA-Seq analysis, GPR68 deletion reduced the expression of Hsp70 and Grp78, 64 suggesting a link to protein misfolding/ER stress function. In previous studies, ischemia leads to upregulation of neuronal hemoglobin or neuroglobin, which is linked to increased antioxidant activities.118,119 Interestingly, we found that deleting GPR68 abolished this increase. 64 These data suggest that GPR68 may contribute to neuroprotection through modulating chaperone and/or ER function, and PKC-mediated signaling likely mediates part of the effect in acidotic conditions. The exact downstream effector of GPR68 in ischemia or neuronal injury in vivo warrants further investigation.
Summary and speculations
We have gained much knowledge on brain acid signaling from the ASICs. With the emerging evidence regarding metabotropic proton receptor function in the brain, together with the discovery of PAC/ASOR anion channel, it is apparent that brain acid signaling is more complex than what was initially thought. As discussed above, the three classes of proton receptors differ in their pH sensitivity. The proton-sensitive GPCRs are most sensitive to acidification and are activated at ∼pH 7.438–40. The ASICs are in the middle and start to open at ∼7.34,37 The PAC is least pH sensitive and only activates when pH is at or below 6.11,12,35 Together, these receptors cover a wide range of pH changes, starting from the resting pH (∼7.3–7.4) down to ∼5. Besides their differences in pH sensitivity, these receptors differ in their expression and signaling (Figure 2). This combination suggests that these receptors work in concert to determine the outcome of cerebrovascular function, neuroinflammation, and brain injuries. Given that some receptors mediate protection while others elicit injury, discovering specific pharmacological reagents will be a key step and offer new opportunities for therapeutic targeting of these receptors in brain ischemia or other neurological diseases which involve acidotoxicity.
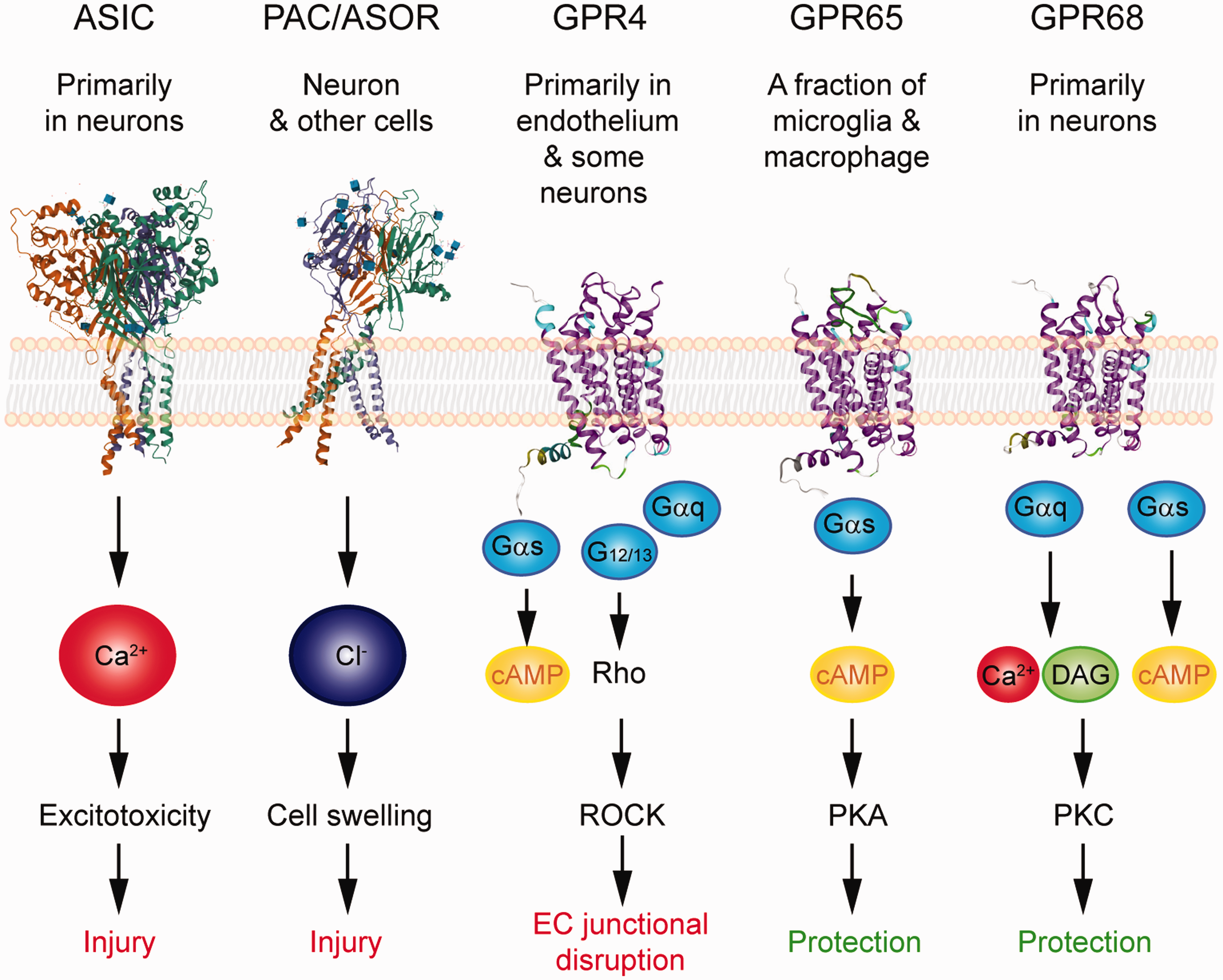
Summary of the expression, signaling, and impact on ischemic injury of the acid-sensitive receptors. 3D illustration of ASIC and PAC/ASOR was based on crystal structure deposited in NCBI PDB database (PBD ID 3S3W and 7JNA) and created with the NGL viewer.145–147 Note that these structures do not contain most of the intracellular tails. For GPCRs, the 3D illustrations were generated using GPCR homology modeling located on GPCRdb.148,149 The signaling illustrates the key pathways which have been either demonstrated or implicated in the receptor’s contribution to neuronal injury, vascular dysfunction, or neuroinflammation.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Xiangming Zha is supported by the NIH/NINDS R01NS102495 and startup funds from University of Missouri-Kansas City.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors' contributions
XMZ reviewed the literature and wrote the manuscript. ZGX and RPS provided important discussions. All authors reviewed the manuscript.