
Abstract
The nuclear protein high-mobility group box 1 (HMGB-1) promotes inflammation in sepsis, but little is known about its role in brain ischemia-induced inflammation. We report that HMGB-1 and its receptors, receptor for advanced glycation end products (RAGE), Toll-like receptor 2 (TLR2), and TLR4, were expressed in normal brain and in cultured neurons, endothelia, and glial cells. During middle cerebral artery occlusion (MCAO), in mice, HMGB-1 immunostaining rapidly disappeared from all cells within the striatal ischemic core from 1 h after onset of occlusion. High-mobility group box 1 translocation from nucleus to cytoplasm was observed within the cortical periinfarct regions 2 h after ischemic reperfusion (2 h MCAO). High-mobility group box 1 predominantly translocated to the cytoplasm or disappeared in cells that colabeled with the neuronal marker NeuN. Furthermore, RAGE was robustly expressed in the periinfarct region after MCAO. Cellular release of HMGB-1 was detected by immunoblotting of cerebrospinal fluid as early as 2 h after ischemic reperfusion (2 h MCAO). High-mobility group box 1 released from neurons, in vitro, after glutamate excitotoxicity, maintained biologic activity and induced glial expression of tumor necrosis factor α (TNFα). Anti-HMGB-1 antibody suppressed TNFα upregulation in astrocytes exposed to conditioned media from glutamate-treated neurons. Moreover, TNFα and the cytokine intercellular adhesion molecule-1 increased in cultured glia and endothelial cells, respectively, after adding recombinant HMGB-1. In conclusion, HMGB-1 is released early after ischemic injury from neurons and may contribute to the initial stages of the inflammatory response.
Introduction
Ischemia induces inflammatory cell recruitment and migration (neutrophils followed later by monocytes) and upregulates inflammatory mediators (cytokines, chemokines, and endothelial-leukocyte adhesion molecules) in brain hours to days after the onset of injury (Huang et al, 1999). The significance of the inflammatory response to brain ischemia is complex, but there is evidence that in the early phase after ischemia, inflammation contributes to tissue injury, whereas in later stages, inflammation might participate in brain repair (Friedlander et al, 1997; Garau et al, 2005; Hara et al, 1997; Kumai et al, 2004; Lovering and Zhang, 2005; Marchetti and Abbracchio, 2005; Wang et al, 1997). In this early phase, in vivo and in vitro evidence is consistent with the paradigm that the endothelium promotes inflammation and recruits circulating leukocytes through the upregulation of adhesion molecules such as intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1, E-selectin, and P-selectin (del Zoppo and Mabuchi, 2003). These recruited leukocytes then release metalloproteinases, which participate in the breakdown of the neurovascular matrix with consequent blood—brain barrier disruption, edema, and/or hemorrhage (Justicia et al, 2003; Kolev et al, 2003; Maier et al, 2004; Ponnampalam and Mayberg, 2004; Veldhuis et al, 2003; Wang and Lo, 2003). The importance of the inflammatory response in the pathophysiology of ischemic stroke is generally recognized. However, very early specific event(s) and mediator(s) have remained elusive and/or considered nonspecific (e.g., DNA and other cellular material released from damaged cells). Herein, we report the important participation of high-mobility group box 1 (HMGB-1) as a potential candidate in a specific upstream pathway promoting inflammation after brain ischemia.
High-mobility group box 1 (also known as amphoterin or HMG-1) is a 216 amino acids (29kDa) DNA-binding protein with a highly conserved structure in several species (Thomas, 2001). High-mobility group box 1 participates in nucleosome formation and regulation of gene transcription (Park et al, 2003; Stros et al, 2002). High-mobility group box 1 has recently been characterized as a key cytokine (Lotze and Tracey, 2005; Yang et al, 2005). It is secreted by activated macrophages (Bonaldi et al, 2003; Wang et al, 1999), natural killer cells (Semino et al, 2005), and myeloid dendritic cells (Lotze and Tracey, 2005) in response to inflammatory stimuli. Interestingly, administration of anti-HMGB-1 antibodies confers significant protection against the lethal effects of endotoxin (Wang et al, 2004; Yang et al, 2004). It is unknown whether HMGB-1, released by neurons after brain ischemia, participates in neuroinflammation. The present in vivo and in vitro studies were aimed to test the hypothesis that brain cells most susceptible to ischemic injury (i.e., neurons) release HMGB-1 and that early release from neurons or delayed release from glia activates downstream inflammatory mechanisms within astrocytes and endothelial cells.
Materials and methods
Cell Cultures
Mouse brain endothelial cell line (bEnd3) was purchased from American Type Culture Collection (ATCC) and cultured in Dulbecco's Modified Eagle's Medium with 10% fetal bovine serum. Primary cultured neurons were isolated from embryonic day (E)-16 mouse fetuses and cultured in NeuroBasal medium with 2% B27 (Invitrogen, Carlsbad, CA, USA) on polyethylenimine-coated plates as described previously (Qiu et al, 2002). Neurons were grown for 10 to 12 days before use. Primary mixed glial cell cultures were prepared from postnatal day 1 pups and cultured in Dulbecco's Modified Eagle's Medium with 10% fetal bovine serum.
Middle Cerebral Artery Occlusion Model
C57 black/6j male mice (weighing 20 to 25 g, 8 to 10 weeks old) were used in all in vivo experiments, except for detecting HMGB-1 in cerebrospinal fluid (CSF) (male Sprague-Dawley rat, weighing 250 to 300 g, 8 to 10 weeks old). All experiments were performed after institutionally approved protocols in accordance with The National Institutes of Health Guide for the Care and Use of Laboratory Animals under institutional guidelines. The filament middle cerebral artery occlusion (MCAO) model was used, as described previously (Endres et al, 1998). Animals were anesthetized with 2% isoflurane and maintained on 1.5% isoflurane in 70% nitrous oxide and 30% oxygen. A silicone-coated 8-0 monofilament was introduced in the internal carotid artery and advanced to occlude the middle cerebral artery for 0.5, 1, or 2h. After 2 h MCAO, the animals were briefly reanesthetized and the filament was withdrawn for reperfusion studies. Regional cerebral blood flow was measured by laser-Doppler (PF2B; Perimed, Stockholm, Sweden) using a flexible probe, placed over the temporal bone after removal of part of the temporalis muscle, to confirm occlusion and reperfusion. Rectal temperature was maintained between 36.5°C and 37.5°C with a homeothermic blanket (Frederick Haer and Co., Brunswick, ME, USA). To assess HMGB-1 in CSF, rats were anesthetized and underwent filament occlusion with an 4-0 suture for 2 h followed by reperfusion. Two hours after reperfusion, the skin was incised, and the occipital bone was cleared of muscle to expose the atlanto-occipital membrane. A 27-gauge needle was inserted into the cisterna magna and approximately 100 μL of CSF was withdrawn. Only clear CSF was used for analysis.
Immunostaining
Immunostaining was performed as previously described (Qiu et al, 2004; Teramoto et al, 2003). Briefly, mice were killed at the indicated time points and transcardially perfused with cold saline. Brains were fixed with 4% phosphate-buffered paraformaldehyde. Coronal brain sections (floating sections) were prepared (40 μm thick). The following antibodies and dilution conditions were used: polyclonal anti-HMGB-1 antibody (1:200; Pharmingen, San Jose, CA, USA) and monoclonal anti-receptor for advanced glycation end products (RAGE) antibody (1:500; R&D System, Minneapolis, MN, USA). Neurons were stained with NeuN (1:1,000; Pharmingen) or microtubule-asso-ciated protein 2 (MAP2) (1:200; Sigma, St Louis, MO, USA); astrocytes were stained with glial fibrillary acidic protein (GFAP) (1:1,000; Sigma). Fluorescent-stained sections were analyzed by confocal microscopy as described (Qiu et al, 2004). For standardization of terminology, we define regions of periinfarct as corresponding to dorsal cortical regions 2 ± 0.5 mm lateral to midline spanning 1 mm rostral to the bregma and 1 mm caudal to the bregma.
Immunoblot
Immunoblot was performed using routine techniques (Qiu et al, 2002). The primary antibodies included the following: anti-HMGB-1 antibody (1:200; Pharmingen) and β-actin (1:1,000; Sigma).
Oxygen-Glucose Deprivation and Glutamate Treatment
Primary cultured neurons were subjected to 2 h oxygen-glucose deprivation (OGD) with balanced salt solution (Goldberg and Choi, 1993) and reoxygenation by replacing with fresh Neurobasal medium immediately after termination of ODG. For the conditioned medium, which contains neuronal HMGB-1, neurons (the number of initial seeding cells was one million per well in a six-well plate) were cultured in six-well plates with 0.6 mL of medium and treated with 0.25 mmol/L of glutamate for 18 h and then the media were collected. A volume of 5 μg/mL (final concentration) of anti-HMGB-1 antibody was added into the conditioned medium 2 to 3 mins before the medium was added into glial cultures.
Quantitative Real-Time Reverse Transcription-PCR
RNA was isolated from either cultured cells or brain tissues and treated with DNase I to remove any contaminating genomic DNA. Complementary DNA was generated by using SuperScript III (Invitrogen). Samples were run on an ABI Prism 7000 (Applied Biosystems, Foster City, CA, USA), in triplicates, with negative controls (no cDNA). Primers and Taqman FAM-labeled probes (Applied Biosystems) were mixed with AmpliTaq Gold Universal PCR Master Mix (Applied Biosystems) and then diluted with distilled deionized H2O up to 50 μL. Gene-specific products were normalized to an endogenous control of 18S rRNA (Applied Biosystems).
Statistics
mRNA expression levels were quantified by real-time reverse transcription-PCR. mRNA amounts were then averaged by group/treatment and compared by analysis of variance, followed by Dunnet post hoc analysis. P ≤ 0.05 was considered statistically significant.
Results
High-Mobility Group Box 1 Expression in Normal Brain and Cultured Brain Cells
In mouse brain coronal sections, robust HMGB-1 expression was observed in nuclei throughout the cortex and subcortical structures, in either NeuN or GFAP-positive cells (Figures 1A to 1F). High-mobility group box 1 was also observed in endothelial cells (Figure 1I, endothelial cells noted by arrow). In primary cultures, both neurons and astrocytes expressed nuclear HMGB-1 (Figures 1G—H). Immunoblotting of normal brain extract also confirmed expression of a 29kDa protein that was HMGB-1 (data not shown).
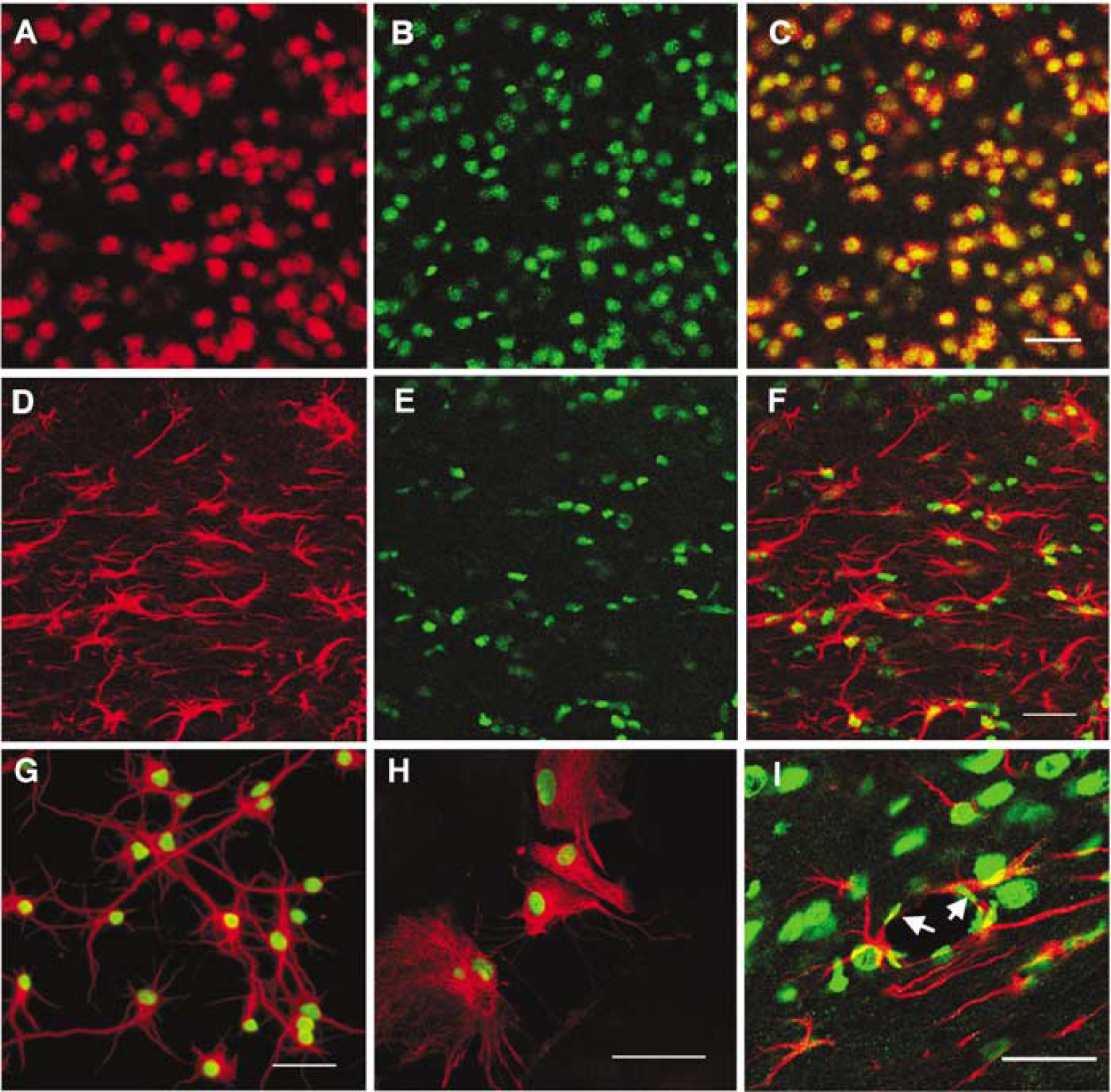
Nuclear localization of HMGB-1 expression in neurons and astrocytes from normal mouse brain and cultured neurons and glial cells. Within the brain, immunofluorescence staining shows the nuclear distribution of HMGB-1 (green,
Cerebral Ischemia Induces High-Mobility Group Box 1 Translocation and Release in Brain Cells
When cells become injured or undergo necrosis, HMGB-1, normally residing in nuclei, translocates to the cytoplasm and/or extracellular space (Scaffidi et al, 2002). Here, we examined several time points in a mouse MCAO model (Figure 2A) to determine whether HMGB-1 translocation and release occur after cerebral ischemia. There was no change in HMGB-1 staining in either the contralateral striatum (Figure 2D) or the contralateral cerebral cortex (Figure 2H) at any time point examined during ischemia or after reperfusion. During ischemia, HMGB-1 staining was slightly diminished in the ischemic core of the striatum as early as 30 mins (Figure 2B) with substantial and diffuse loss in the striatum 1 h after onset of MCAO (Figure 2C). Unlike the striatum with complete loss of HMGB-1 staining 1 h after onset of ischemia, the ipsilateral cortex had no observed change in HMGB-1 staining at 30 mins or 1 h during ischemia (Figure 2E). However, HMGB-1 loss/translocation was observed at 2 h after MCAO in cortex adjacent to the striatal ischemic core (data not shown).
To assess later time points, we analyzed tissue after reperfusion after 2 h of occlusion. High-mobility group box 1 staining was lost in ischemic striatum and cortex, but appeared localized to the cell cytoplasm at the margins of ischemic injury within the cortex (Figures 2F and 2G) as early as 2 h after reperfusion. The area of HMGB-1 translocation seemed to be maximum 2 h after reperfusion, as no significant change in staining pattern was noted at later time points (6 and 24 h) (data not shown). These results suggest rapid and region-specific temporal changes in HMGB-1 immunostaining after ischemia. Within the ischemic core, HMGB-1 appears to be released from the nucleus and cytoplasm into the extracellular space, whereas periinfarct regions appear to maintain translocation of HMGB-1 from the nucleus into the cytoplasm.
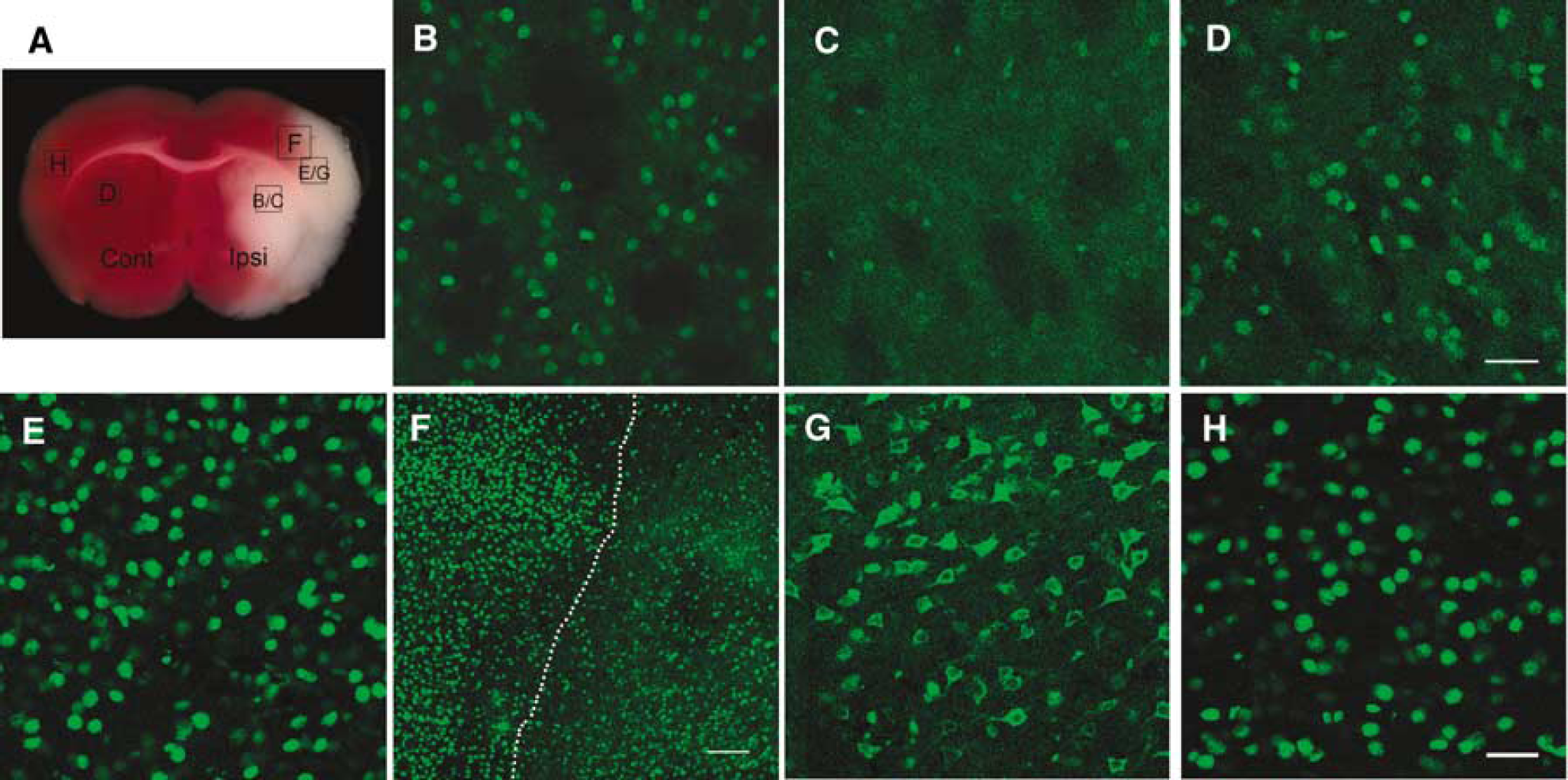
Cerebral ischemia induces HMGB-1 translocation and release. (
To assess the extracellular release of HMGB-1 after MCAO, we examined CSF for the presence of HMGB-1. Rats were used in this experiment because the volume of CSF was more readily available. High-mobility group box 1 was observed 2 h after reperfusion after 2 h MCAO (Figure 3A). No HMGB-1 was detectable in CSF from sham-operated animals. We ran immunoblots with lysates from contralateral nonischemic and ispsilateral ischemic mouse brain tissues using the same antibody as in immunostaining. As shown in Figures 3B and 3C, total HMGB-1 did not significantly differ between ischemic and nonischemic tissues from homologous brain regions after MCAO, despite the loss of nuclear and cytoplasmic staining. These data suggest that the loss of both nuclear and cytosolic immunostaining in the ischemic core was not due to degradation or modification of the HMGB-1 epitope, but was rather due to its release into the extracellular space. Furthermore, they suggest that the loss of HMGB-1 into the CSF is small relative to the total pool of HMGB-1. In contrast to CSF, HMGB-1 was not detected in plasma samples from either sham-operated mice or mice subjected to MCAO, for up to 24 h by Western blot.
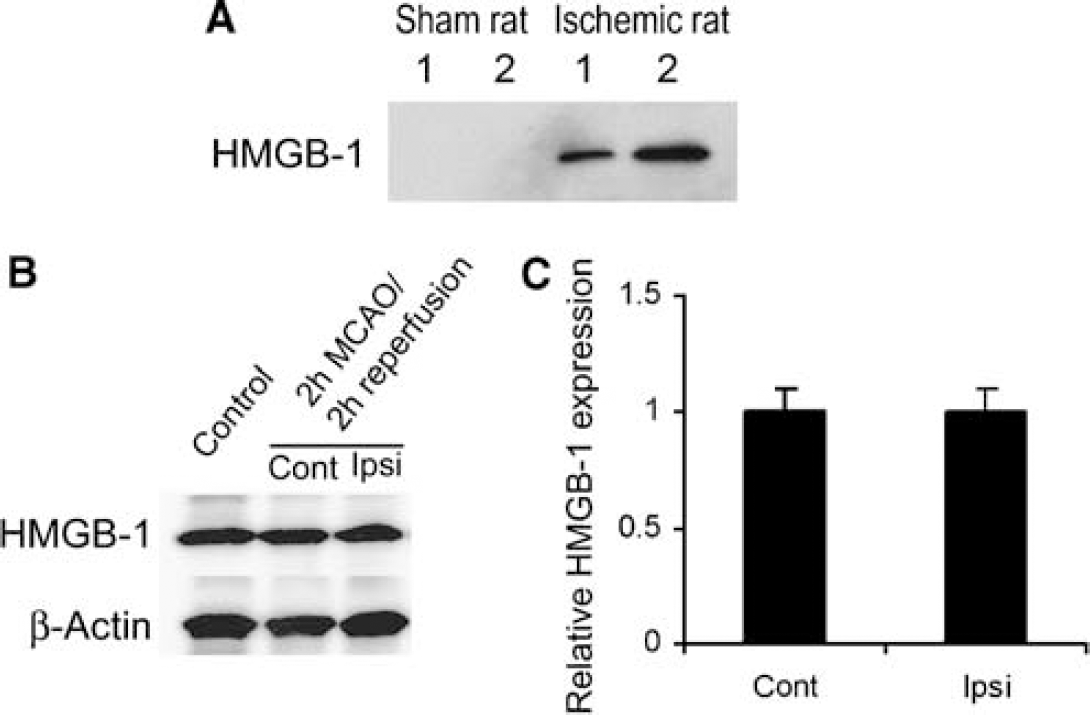
High-mobility group box 1 expression in CSF and brain parenchyma after MCAO. (
Early High-Mobility Group Box 1 Translocation and Release Induced by Middle Cerebral Artery Occlusion Occur Mostly in Neurons
To identify the cell types in which HMGB-1 translocation/release occurred after focal ischemia, coronal sections were costained with anti-HMGB-1 and anti-NeuN antibodies. Within the ischemic striatal core, HMGB-1 staining was negative or diminished in most NeuN-positive cells 1 h after MCAO (Figures 4A to 4C), suggesting that HMGB-1 had already been released from neurons by 1 h after MCAO. Two hours after reperfusion (2 h MCAO), HMGB-1 was observed in the cytoplasm of NeuN-positive cells, mainly within the outer margins of the cortical ischemic territory (Figures 4D to 4F), indicative of translocation from nucleus to cytoplasm. However, nuclear HMGB-1 staining was found in some of GFAP-positive astrocytes within the regions where HMGB-1 cytoplasmic translocation occurred in almost all neurons (Figures 4G to 41). This finding may in part reflect the selective vulnerability of neurons to ischemic injury and suggests that neurons could be one of the main sources of released HMGB-1, at least at this early time point.
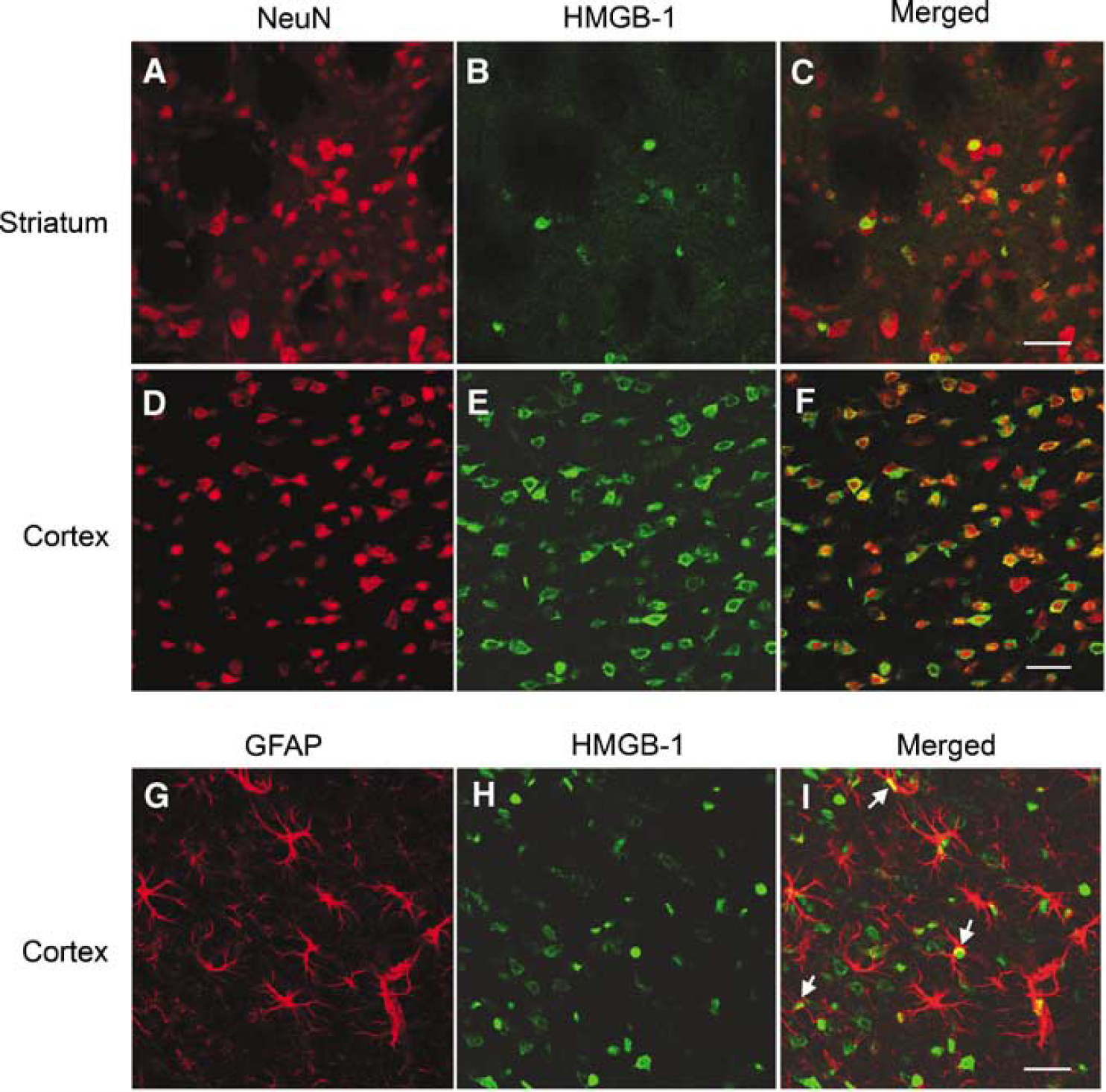
Early HMGB-1 translocation and release is induced by MCAO in neurons and some astrocytes. Immunofluorescence staining shows loss of HMGB-1 (green) in striatal neurons (NeuN, red,
To confirm the susceptibility of neurons to early HMGB-1 translocation/release, we subjected primary cultures to glutamate or 2 h of OGD and assayed time-dependent changes in HMGB-1 immunoreactivity in conditioned media. As shown in Figure 5A, HMGB-1 was undetectable in medium from untreated cells, but it was detected by immunoblot starting as early as 1 to 2 h after glutamate (0.25 mmol/L) or reoxygenation from OGD; moreover, there was no further substantial increase in the release of HMGB-1 out to 24 h after reoxygenation. Assuming no protein degradation in conditioned medium, this time course suggests that most of HMGB-1 release occurred in the first 2 h under these conditions. Despite HMGB-1 release at this time point, the gross morphology of neurons was still preserved and very few cells were lysed (Figure 5B). Thus, HMGB-1 release in vitro occurs as an early event after injury in cultured neurons, consistent with in vivo results. Moreover, HMGB-1 translocation was observed in some neurons exposed to OGD or glutamate by immunofluorescence (data not shown). A prolonged exposure to OGD (18 h, followed by 6 h reoxygenation) led to a loss of intracellular staining for HMGB-1 in astrocytes also, but such a loss did not occur after 2 h OGD and 6 h reoxygenation (data not shown).
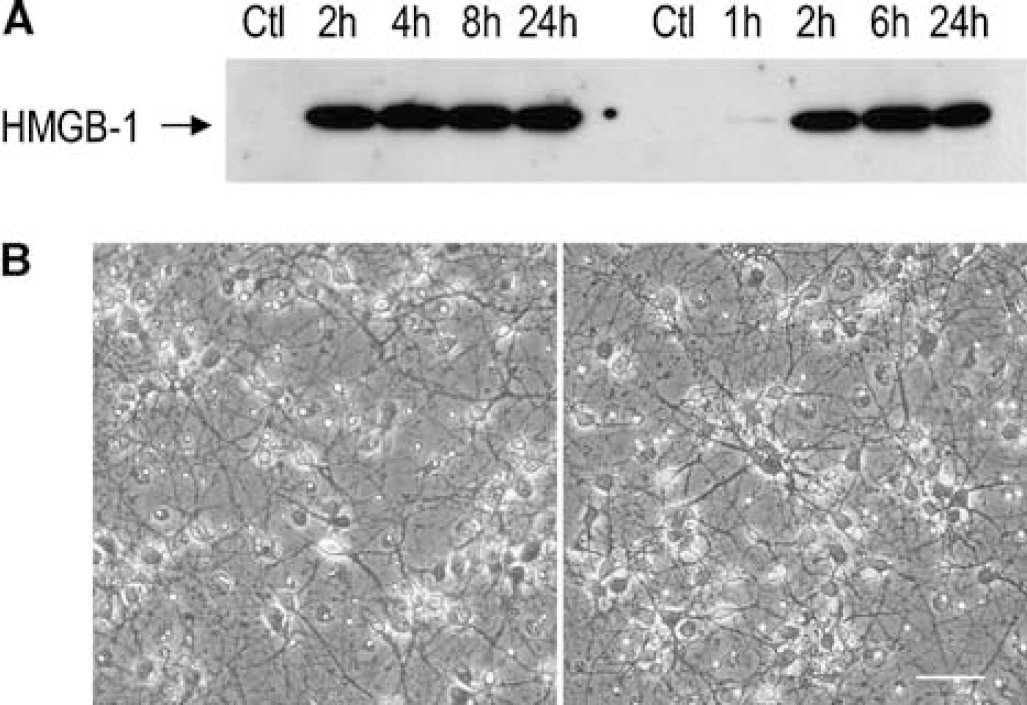
High-mobility group box 1 is released from injured cultured neurons. Primary cultured neurons were subjected to 0.25m mol/L glutamate or oxygen-glucose deprivation (OGD) and assayed for time-dependent changes in HMGB-1 (
Putative High-Mobility Group Box 1 Receptors (RAGE, TLR2, and TLR4) in Brain and Cultured Brain Cells
High-mobility group box 1 acts as a proinflammatory cytokine after interaction with RAGE (Hori et al, 1995), Toll-like receptor 2 (TLR2), and TLR4 (Park et al, 2004). We detected RAGE, TLR2, and TLR4 mRNA expression in bEnd3 cell line, cultured primary neurons, and astrocytes as well as in brain tissue (Figure 6A). This suggested that cells in the neurovascular unit might be susceptible to regulation by HMGB-1 signaling. Very low levels of RAGE expression were detected by immunohistochemistry in the normal brain (Figure 6B), but there was robust RAGE expression only in the cortical ischemic periinfarct region—the area between translocation and nontranslocation of HMGB-1 corresponding to Figure 2E 6 h after reperfusion (2 h MCAO) (Figure 6C). Robust RAGE immunoreactivity was mainly seen in cells exhibiting a neuronal morphology in layers 4 to 5. These results suggest that RAGE, a putative HMGB-1 receptor, may be upregulated after ischemic injury.
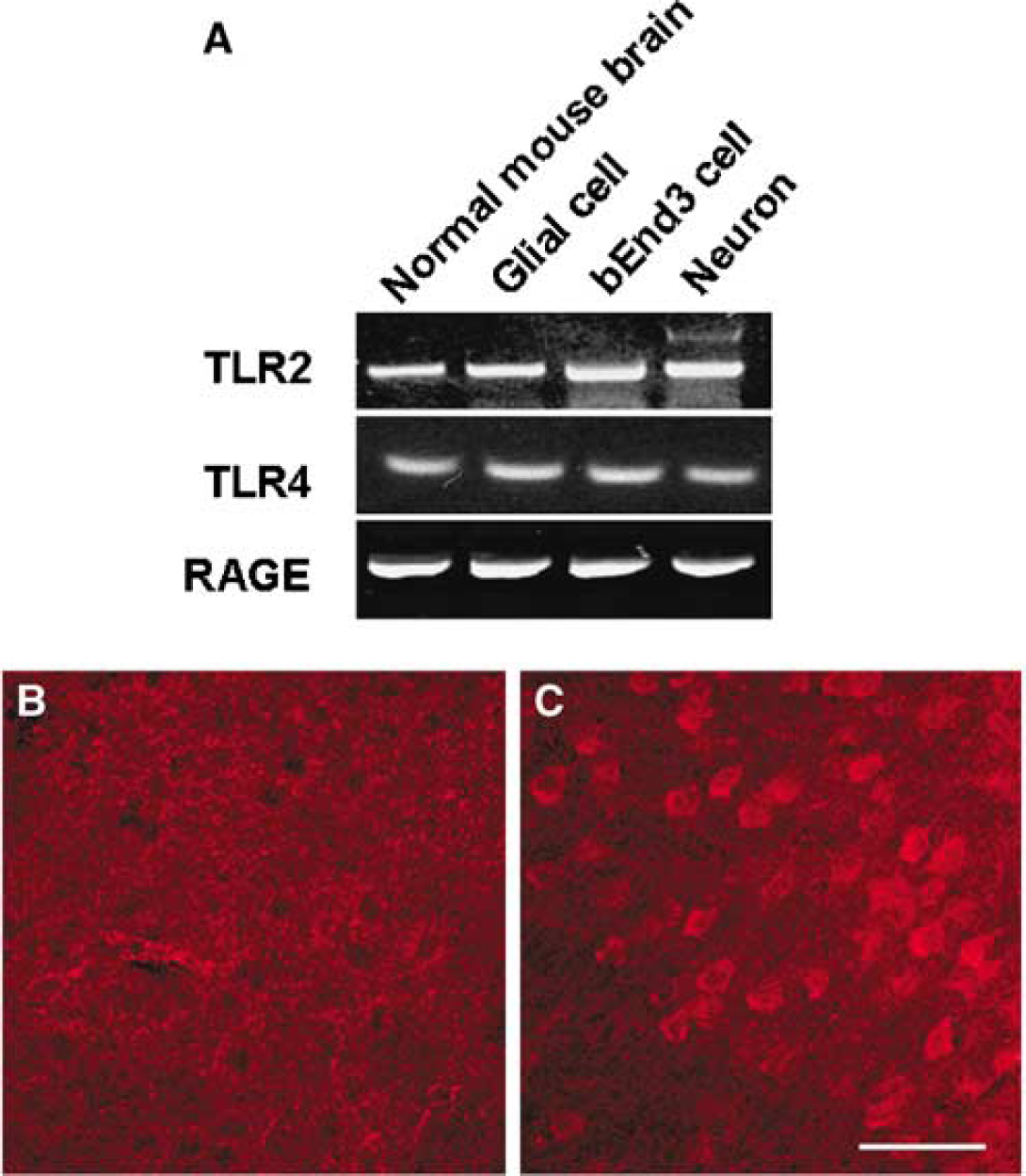
Putative HMGB-1 receptors (RAGE, TLR2, and TLR4) in brain and cultured brain cells. (
Extracellular High-Mobility Group Box 1 Induces Inflammatory Cytokines in Cultured Neurons, Astrocytes, and Endothelial Cells
Tumor necrosis factor α (TNFα), inducible nitric oxide synthase, and ICAM-1 expression are known to increase in ischemic brain and play a significant role in neuroinflammation. To determine whether extracellular HMGB-1 could induce these cytokines, we exposed cultured neurons to different concentrations of recombinant HMGB-1. In a concentration-dependent manner, HMGB-1 increased TNFα mRNA expression in astroglia (Figure 7A). Tumor necrosis factor α upregulation was noted as early as 1 h after HMGB-1 exposure and peaked at 3 to 6 h (Figure 7B). Tumor necrosis factor α expression was also found in the other cell types of the neurovascular unit such as in neurons and endothelial cells (data not shown). However, in neurons, TNFα upregulation was not as robust as in glia and endothelium. The expression of other inflammatory molecules was assessed after HMGB-1 treatment. Inducible nitric oxide synthase was increased in glia, and ICAM-1 was increased in endothelium at 3 h after HMGB-1 treatment (Figure 7C to TD).
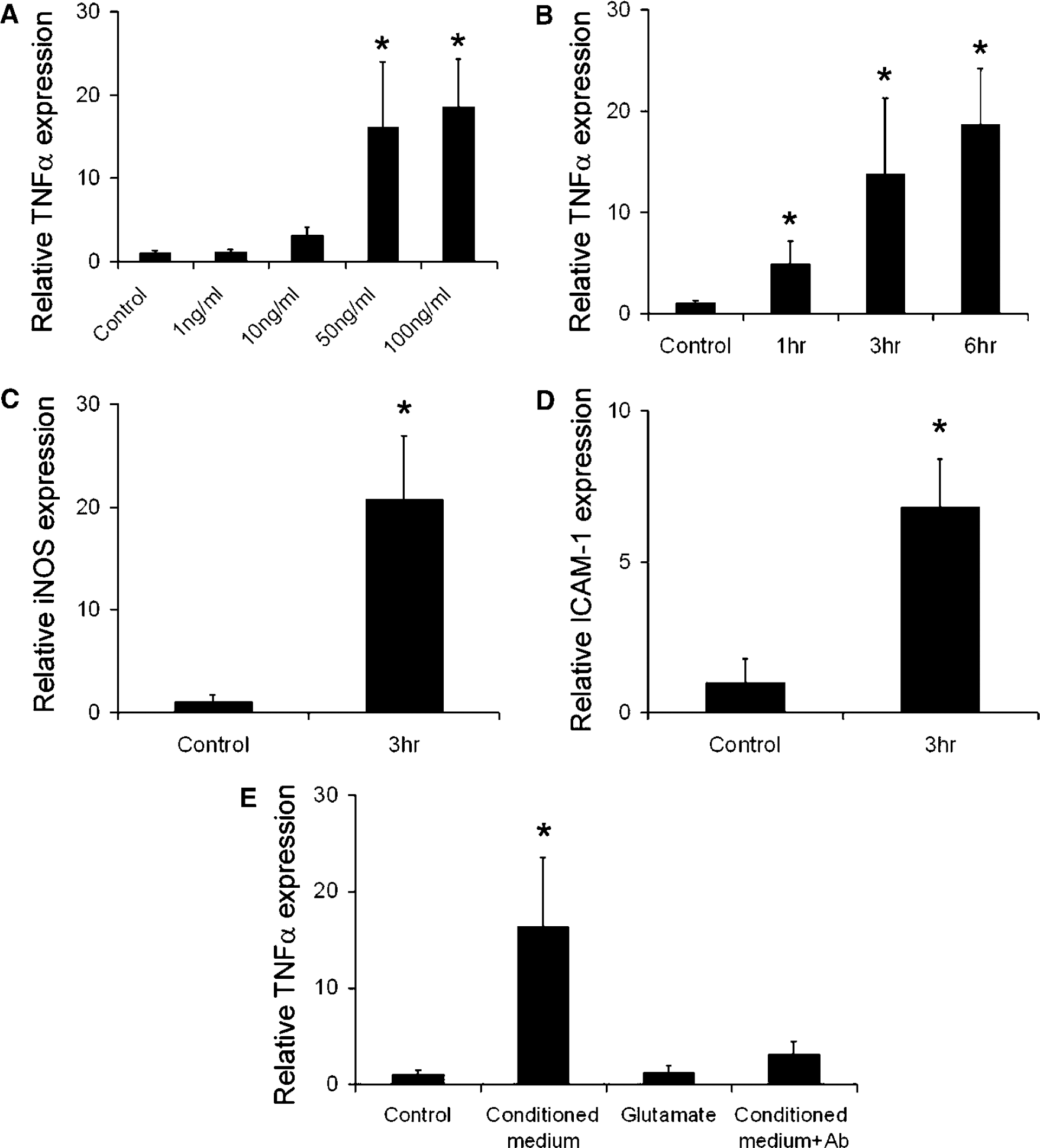
High-mobility group box 1 upregulates the expression of inflammatory mediators in cultured glia and endothelial cells. Tumor necrosis factor α, inducible nitric oxide synthase, and ICAM-1 mRNA expression was detected by real-time reverse transcription-PCR. (
Conditioned Medium from Glutamate-Treated Neurons Induces Tumor Necrosis Factor α in Astrocytes, an Effect Inhibited by Anti-HMGB-1 Blocking Antibody
To determine whether HMGB-1 released from injured cultured neurons was biologically active as a pro-inflammatory mediator, we treated primary astrocytic cultures with conditioned medium from glutamate-treated neurons. As shown in Figure 7E, conditioned medium robustly induced TNFα mRNA expression in astrocytes. Glutamate did not modify TNFα expression when added directly to the astrocyte culture medium. Tumor necrosis factor α expression was inhibited by 90% after pretreatment with anti-HMGB-1 blocking antibody, indicating that this response was mediated by soluble HMGB-1.
Discussion
HMBG-l is a key mediator of inflammation that we and others hypothesized might be released from damaged cells after focal cerebral ischemia. In this study, we showed that (1) HMGB-1 was translocated from nucleus to cytoplasm and release occurred in ischemic neurons as early as 1 to 2 h after MCAO; (2) HMGB-1 release also occurred from cultured neurons as early as 1 to 2 h after glutamate treatment or OGD followed by reoxygenation (astrocytes appeared more resistant); (3) putative HMGB-1 receptors were expressed in neurons, glia, and endothelial cells, and one of these receptors, RAGE, appeared upregulated in the cortical periinfarct region; and (4) recombinant HMGB-1 or HMGB-1 released by injured neurons in culture induced proinflammatory cytokine expression in neurons, astrocytes, and endothelial cells.
Previous studies have shown that HMGB-1 can be released into the extracellular space either actively, after HMGB-1 is hyperacetylated in the nucleus (Bonaldi et al, 2003), or passively, when HMGB-1 diffuses out of cells that are undergoing necrosis and cell membrane is damaged (Scaffidi et al, 2002). Active secretion typically occurs not only in macrophages and monocytes (Chen et al, 2004; Kalinina et al, 2004), but also in nonimmune cells such as hepatocytes where it is overexpressed after hypoxia (Tsung et al, 2005). Here, we suggest that HMGB-1 is released from the ischemic brain parenchyma within 1 h after MCAO, based on early loss of HMGB-1 immunoreactivity in the striatum. Kim et al (2006) showed that HMGB-1 was not hyperacetylated, suggesting that its release is passive. Our demonstration that HMGB-1 is present in rat CSF is also in agreement with Kim et al (2006), who detected this protein 3 h after transient MCAO. We further show that neurons are one of the principal sources of HMGB-1 release in the early stages of ischemic injury. In fact, HMGB-1 staining was lost in NeuN-positive cells in the ischemic core and cytosolic HMGB-1 staining colocalized with NeuN in the ischemic cortex. The presence of HMGB-1 in rat cisterna magna fluid probably reflects its release and diffusion from ischemic tissue. We did not detect any HMGB-1 in mouse plasma in contrast to a study in the rat (Kim et al, 2006).
High-mobility group box 1 may act as a proinflammatory signal and contribute to the delayed death of brain cells in the ischemic periinfarct region. Extracellular HMGB-1 binds to its receptors such as RAGE, TLR2, and TLR4. We detected mRNAs for these receptors in cell types of the neurovascular unit in vitro and in brain extracts. Toll-like receptor 2 and TLR4 expression has already been documented by others in endothelium (Faure et al, 2000) and in astrocytes (Bowman et al, 2003). We found upregulation of RAGE in the periinfarct region 6 h after reperfusion in the 2 h MCAO model. Because HMGB-1 is an endogenous ligand with high affinity for RAGE (Hori et al, 1995) and RAGE expression is reportedly induced by HMGB-1 (Li et al, 1998), the upregulation of RAGE in ischemic brain further supports the central hypothesis of a key proinflammatory role for HMGB-1 in ischemic stroke. Conceivably, increased expression of HMGB-1 receptors after ischemia may enhance the sensitivity of brain cells to HMGB-1. To our knowledge, RAGE overexpression in ischemic brain has not yet been reported, although its role in β-amyloid metabolism and Alzheimer's disease has been studied (Deane et al, 2003; Mackic et al, 1998). Receptor for advanced glycation end product signals through pathways that involve ERKl (extracellular signal-regulated kinase) and/or ERK2 as well as the mitogen-activated protein kinase p38, and it promotes the activation of nuclear factor-KB (Taguchi et al, 2000), which itself activates the transcription of proinflammatory genes (such as interleukin (IL)-1β, IL-6, and TNFα). Interaction of HMGB-1 with TLR2 and TLR4 promotes inflammatory responses that ultimately also lead to nuclear factor-kB activation. The expression of these other putative HMGB-1 receptors has been shown to increase during inflammation (Cipollone et al, 2003; Koedel et al, 2003). Both TLR2 and TLR4 are robustly upregulated in brain microvasculature of young rats injured by neonatal hypoxia-ischemia (Maslinska et al, 2004). Whether they are upregulated in experimental brain ischemia, however, remains to be determined.
If HMGB-1 acts as an early upstream inflammatory signal in cerebral ischemia, we expect its translocation/release to precede the upregulation of other cytokines and endothelial adhesion molecules and the subsequent leukocyte recruitment into the ischemic territory. We and others have shown that ERK activity increases as early as 0.5 to 1 h after ischemia in the middle cerebral artery territory (Alessandrini et al, 1999; Wang et al, 2003; Wu et al, 2000). This signaling is potentially linked to TLR2, TLR4, and/or RAGE activation (Ishihara et al, 2003; Park et al, 2003; Taguchi et al, 2000) and is known to promote transcription of cytokines such as TNFα (Fiuza et al, 2003). Tumor necrosis factor α mRNA reportedly increases within 1 h after induction of ischemia in the periinfarct and infarct area, peaks at 12 h, and remains elevated for up to 24 h (Gregersen et al, 2000; Lambertsen et al, 2002, 2005; Schroeter et al, 2003; Yang et al, 1999).
The data reported herein suggest that HMGB-1 may participate as an early upstream initiator of inflammation. In vitro, we observed a 3- to 4-fold TNFα mRNA induction in glial cultures already after 1 h incubation with exogenous HMGB-1; TNFα mRNA levels increased more than 10-fold after 3h. Noteworthy, TNFα mRNA induction in cultured glia was induced with 100 ng/mL HMGB-1; this concentration seems consistent with values that we can roughly estimate in CSF after MCAO, and this corresponds to approximately 3 nmol/L, which is close to the kDa value of HMGB-1 for RAGE (Hori et al, 1995). High-mobility group box 1 proinflammatory signaling might begin early, possibly in the first hour after focal ischemia, but may continue for hours thereafter (Faraco et al, 2007). Consistent with a more protracted HMGB-1 signaling, we have previously reported neuroprotection through ERK inhibition, even when the treatment had been initiated 3 h after MCAO (Namura et al, 2001).
Given its release from neurons, HMGB-1 may play multiple, proinflammatory roles in the neurovascular unit by upregulating cytokines and adhesion molecules in astrocytes and endothelium. Astrocytes directly contact and translate signals to neurons and blood vessels. The demonstration that cultured primary astrocytes upregulate TNFα as soon as 1 h after exposure to low concentrations of HMGB-1 seems particularly relevant. After brain ischemia, the astrocyte might represent an early target for released HMGB-1, as might endothelial cells. In this study, we found that HMGB-1 upregulated mRNA ICAM-1 in bEnd3 cell line. Similar studies showing an increase in protein levels of ICAM-1 at 4, 6, and 16 h in two other human cell lines are consistent with our data (Fiuza et al, 2003; Treutiger et al, 2003). Although bEnd3 cell lines constitutively express ICAM-1 and have been widely used for studying adhesion molecules, they may have a different response to inflammation compared to primary brain endothelial cells. Our unpublished data suggest that the baseline of ICAM-1 expression in bEnd3 cell line is lower than that in rat primary brain endothelial cells. Upregulation of ICAM-1 expression in endothelial cells would recruit immune cells into the ischemic area, leading to a more severe inflammatory reaction. Furthermore, it remains to be determined whether such responses by the endothelium are beneficial or deleterious, as their timing may be critical and have counterbalancing effects. Although the results of this study suggest that HMGB-1 may contribute to the early stages of inflammation in brain ischemia, Kim et al (2006) showed that HMGB-1 may also contribute to later stages of inflammation by activating microglia. Therefore, HMGB-1 may exert multiple proinflammatory effects in the ischemic brain.
The importance of neuroinflammation after focal brain ischemia has led to the development of drugs that target the mediators responsible for leukocyte recruitment. Unfortunately, clinical trials using either anti-ICAM-1 antibodies or a recombinant glycoprotein with selective binding to the CD11b integrin did not show any significant beneficial effect (Krams et al, 2003; Enlimomab Acute Stroke Trial Investigators, 2001). The complexity of inflammatory signaling raises the question whether targeting a single molecule or receptor would be an effective therapeutic strategy. A more important task is to identify central molecular mechanisms in the ischemic brain that may generate key, early mediators of the inflammatory response in the vasculature (endothelium) and in the circulating cell compartments. To date, very early event(s) and mediator(s) have not yet been identified. Our study points to a potential novel and pivotal role for HMGB-1 in a receptor-specific signaling pathway linking very early stages of cerebral ischemic injury with the activation of local neuroinflammatory responses within the neurovascular unit. Additional research will be required to clarify this point.
In conclusion, HMGB-1 is released by injured neurons early after ischemia and may represent a major upstream paracrine inflammatory mediator within the neurovascular unit in ischemic stroke. Targeting HMGB-1 signaling may be an important novel therapeutic strategy in acute ischemic stroke.
Footnotes
Acknowledgements
We thank Christian Waeber for preparation of figures and Igor Bagayev for his excellent technical assistance.