
Abstract
Most neurological diseases, including stroke, lead to some degree of blood–brain barrier (BBB) dysfunction. A significant portion of BBB injury is caused by inflammation, due to pro-inflammatory factors produced in the brain, and by leukocyte engagement of the brain endothelium. Recently, microRNAs (miRNAs) have appeared as major regulators of inflammation-induced changes to gene expression in the microvascular endothelial cells (BMVEC) that comprise the BBB. However, miRNAs’ role during cerebral ischemia/reperfusion is still underexplored. Endothelial levels of miR-98 were significantly altered following ischemia/reperfusion insults, both in vivo and in vitro, transient middle cerebral artery occlusion (tMCAO), and oxygen–glucose deprivation (OGD), respectively. Overexpression of miR-98 reduced the mouse’s infarct size after tMCAO. Further, miR-98 lessened infiltration of proinflammatory Ly6CHI leukocytes into the brain following stroke and diminished the prevalence of M1 (activated) microglia within the impacted area. miR-98 attenuated BBB permeability, as demonstrated by changes to fluorescently-labeled dextran penetration in vivo and improved transendothelial electrical resistance (TEER) in vitro. Treatment with miR-98 improved significantly the locomotor impairment. Our study provides identification and functional assessment of miRNAs in brain endothelium and lays the groundwork for improving therapeutic approaches for patients suffering from ischemic attacks.
Introduction
Stroke accounts for 55% of all neurological diseases and is considered the leading cause of permanent physical and mental weaknesses.1,2 While inflammation plays a critical role in the pathogenesis of ischemic stroke/reperfusion, the underlying mechanisms remain largely unclear.2–5 One possibility is that cerebral ischemia could disrupt the dynamic equilibrium between pro-inflammatory and anti-inflammatory responses, and aggravate the activation and recruitment of inflammatory cells into the brain. Hence, attenuation of inflammatory responses can diminish infarct size following stroke and prevent neurological deficits.1,4 Endothelial dysfunction represents the earliest event of inflammatory damage and vascular compromise following ischemia and reperfusion (IS/R). Quickly after ischemic injury, brain endothelium and astrocytes produce large numbers of chemokines and cytokines. Such molecules (interleukins and TNFα) stimulate the expression of adhesion molecules on the endothelium, leading to leukocyte adhesion and degradation of endothelial tight junction proteins (TJP) and of the extracellular matrix.6–8 IS/R induces a time-dependent recruitment and activation of inflammatory cells, such as neutrophils, T cells, and monocytes/macrophages; hence, inhibition of such inflammatory responses reduces the infarct size and attenuates neurological deficits. 4 Critically, the injured blood–brain barrier (BBB) promotes both the infiltration of peripheral inflammatory cells into the brain, and the secretion of deleterious mediators, thereby resulting in permanent barrier injury.
Despite stroke’s high incidence, morbidity, and mortality, treatment options remain limited. Plasminogen activators are the only thrombolytic agents approved by the FDA to treat the thrombosed vessel, 5 and are used in only a small percentage of patients. Beyond treating the clot, however, there are few current methods for treating inflammatory responses to stroke within the brain. Therefore, it is crucial to develop alternative or complementary treatment strategies. Brain microvascular endothelial cells at the site of blockage are both active participants in, and regulators of, extensive inflammatory processes, which involve numerous changes to gene expression. microRNA (miRNA) have emerged as a class of gene expression regulators. However, the relationship between inflammation and miRNA expression in brain endothelium remains largely unexplored. Based on previous studies by our laboratory 9 and others, we propose that BBB protection is most effectively achieved when the intervening agents possess both anti-inflammatory and TJP stability enhancing properties, which can reduce both leukocyte–endothelial interactions and leukocyte extravasation.10–13 Recently, we have identified miRNAs belonging to the let-7 family that were significantly down-regulated during inflammation. 9 We have demonstrated the anti-inflammatory effects of let-7 and miR-98 in in vitro and in vivo models of acute and systemic inflammation. 9 let-7 and miR-98 overexpression in activated endothelial cells decreased secretion of pro-inflammatory factors and expression of adhesion molecules and attenuated monocyte adhesion/migration across the BBB. 9 In the current study, we demonstrate in both an in vitro model of IS/R (oxygen–glucose deprivation (OGD) followed by reoxygenation (OGD/R)) and an in vivo model of IS/R (transient middle cerebral artery occlusion (tMCAO)), that expression of miR-98 was reduced in both primary brain microvascular endothelial cells (BMVECs) and in isolated cerebral microvessels (MVs). miR-98 has also been described to stabilize hypoxia/reperfusion injury in human umbilical vein endothelial cells (HUVEC). 14 We determined that overexpression of miR-98 improved barrier tightness in vitro, while mice treated with miR-98 exhibited faster neurological recovery after tMCAO. These mice also displayed attenuated BBB disruption and diminished infiltration of proinflammatory leukocytes into the brain.
Taking these results together, we propose that miR-98 confers significant neuroprotective effects following ischemia. Owing to direct effects on BBB integrity, treatment with miR-98 could represent an important new approach for stroke therapies, whether given alone or in conjunction with other therapeutics.
Methods and materials
Cells and oxygen-glucose deprivation followed by re-oxygenation
Primary BMVEC, isolated from vessels obtained from brain resection tissue (showing no abnormalities) of patients undergoing surgery for treatment of intractable epilepsy, were supplied by Michael Bernas and Dr. Marlys Witte (University of Arizona, Tucson, AZ, USA) and were maintained as described.9,15,16 Ischemic conditions were simulated using an OGD/R protocol. 17 Cell culture media was replaced with glucose-free DMEM medium (Invitrogen, Life Technologies, Carlsbad, CA, USA) and the plates were placed into a gas exchange chamber (MIC-101, Billups-Rothenberg, Inc., Del Mar, CA, USA). Anaerobic conditions were achieved with gas (1% O2, 5% CO2, balance N2) and the plates were left in OGD conditions for a further 2–4 h in the incubator; 17 medium was then exchanged for re-oxygenation with glucose-containing DMEM medium and left for 24–76 h in the incubator.
Animals
All animal experiments were approved by the Temple University Institutional Animal Care and Use Committee and were conducted in accordance with Temple University guidelines, which are based on the National Institutes of Health (NIH) guide for care and use of laboratory animals and with the ARRIVE (Animal Research: Reporting In Vivo Experiments) guidelines (study design, experimental procedures, housing and husbandry, statistical methods) (www.nc3rs.org.uk/arrive-guidelines). Eight-week-old male C57BL/6 mice purchased from the Jackson Laboratory (Bar Harbor, ME, USA).
Transient middle cerebral artery occlusion
Mice were subjected to 60 min focal cerebral ischemia produced by transient intraluminal occlusion with a monofilament made of 6–0 nylon with a rounded tip (Doccol Corp., Sharon, MA, USA, cat# 602312PK10) into the middle cerebral artery (MCAO) as described previously.1,18 To perform transient MCAO, the mice were re-anesthetized and the suture was withdrawn. Body temperature was monitored throughout surgery with a rectal probe and maintained at 37.0 ± 0.5℃ using a heating pad (Sunbeam, Neosho, MO, USA). Sham-operated mice were subjected to the same surgical procedure, but the filament was not advanced far enough to occlude the middle cerebral artery. At either 24, 48, 72, or 96 h after the induction of MCAO, mice were anesthetized with 5% isoflurane and euthanized by cervical dislocation, decapitated, and the brain collected. To ensure rigor in tMCAO experiments, each mouse was monitored for regional cerebral blood flow (rCBF) before ischemia, during MCAO, and after reperfusion using a Laser Speckle PeriCam PSI System (Perimed AB, Järfälla, Sweden). If rCBF was not reduced to at least 25% of initial level, the animal was excluded from the study and euthanized. 19
Neurological assessment
The neurological score assessment was performed 72 h after the onset of ischemia. An overall neurological improvement was observed in enhanced locomotor activity (measured by total ambulation counts).20–23 The corner test, which is sensitive to sensorimotor and postural symmetries, was used. “Stroked” mice usually turn toward the affected side (right), while nonaffected mice have almost 50–50% left to right turn distribution. All mice tested were allowed to enter a corner with an angle of 30°, which required them to turn either to the right or the left to exit the corner. This test was repeated and documented 10 times, with as a minimum of 30 s between trials, and the percentage of right turns out of total turns was calculated.23–25
In vitro and in vivo miRNA transfection
A successful BMVEC transfection protocol was recently developed in our laboratory.
9
In short, BMVEC were transfected at 2 × 106 cells/mL with miRNA oligos at 50 nM with the Neon
miRNA (similar to siRNA) can infiltrate cell membranes. However, miRNA can be degraded if injected in a nonencapsulated form. Hence, we used a protocol recently developed in our laboratory 9 using a liposome-based miRNA transport system. To generate miRNA-containing liposomes, 5 nmol synthetic miRNA (GE Healthcare Dharmacon, Inc., Lafayette, CO, USA) were mixed with Lipofectamine 2000 (Life Technologies) in RNase and DNase-free water (Life Technologies) per the manufacturer’s directions. Lipid-oligo complexes were incubated at room temperature for at least 1 h and, if required, stored at 4℃ overnight. The miRNA–liposome complex was diluted in PBS and 100 µL was injected per mouse. To verify efficiency of miRNA delivery into microvessels (MVs), the mouse brains were harvested at different time points (6, 24, or 48 h) following miRNA administration. MVs were isolated and the amount of transfected miRNA was estimated by qPCR as described below. While a single injection of liposome-miRNA complexes resulted in a 22-fold increase in miRNA expression in MVs, best results were obtained when mice were injected with miRNAs twice (with 24 h between injections). The maximum miRNA expression levels were obtained in MVs after 48 and 72 h with 40- to 60-fold increases respectively, vs. injection with liposomal solution only. Next, mice were injected (i.v.) once with miRNA-liposomes as described9,26,27 at different times after tMCAO (2, 4, or 24 h) with a second injection 24 h after the first injection. Alternatively, mice were injected 48 h and 24 h prior to tMCAO.
Mouse brain MV isolation
Mouse brain MVs were isolated using a protocol based on previously published studies.9,28,29 For each preparation, mice (n is described in figure legends) were overdosed with 5% isoflurane and their brains were harvested and placed in 4℃ HBSS. The cerebellum, meninges, choroid plexus, brain stem, and large superficial blood vessels were removed. The residual tissue was diced in HBSS (4 mL/g) and then homogenized using a Potter-Thomas homogenizer (0.25 mm clearance) (Thomas Scientific, Swedesboro, NJ, USA). The homogenate was centrifuged (1000 g for 10 min at 4℃) to remove HBSS, resuspended in 17.5% dextran (Sigma/Aldrich, St. Louis, MO, USA) and centrifuged again to separate the MVs. The MV pellet was resuspended in 1% BSA in HBSS and the supernatant was centrifuged (4000 g for 10 min at 4℃). The MVs from each centrifugation were combined. The MV suspension was passed through a 100-μm nylon mesh filter and then through a 40-μm nylon mesh filter (Corning Life Sciences, Tewksbury, MA, USA). The material retained on the 40-μm nylon mesh filter contained the MVs.
Flow cytometry (FACS) for brain infiltrating leukocytes and microglia
Brain infiltrating leukocytes (BIL) and microglia were isolated from infarcted and noninfarcted hemispheres by Percoll/Ficoll centrifugation30,31 followed by surface staining with antibodies against mouse CD45 (PE, clone 30-F11), CD11b (PE-CY7, clone M1/70), Ly6g (APC-eFluor780, clone HK1.4), Ly6c (APC, clone 1A8-Ly6g), all purchased from eBiosciences (Thermo Fisher Scientific, Waltham, MA, USA), at 4℃ for 30 min. Cells were then fixed using IC fixation buffer (eBiosciences, San Diego, CA, USA). For intracellular markers, cells were permeabilized after fixation using permeabilization buffer (eBiosciences) following the manufacturer’s instructions and incubated at room temperature for 30 min in antibodies against mouse IBA (FITC, clone 1022-5, Abcam, Cambridge, UK). Cytometric acquisition was performed using a BD FACS Canto II flow cytometer and analyzed with FlowJo software (Tree Star, Inc., Ashland, OR, USA).
In vivo BBB permeability assay
Mice were injected retroorbitally with 100 µL of 40 kDa dextran-fluorescein in phosphate-buffered saline. At 15 minutes post-injection, mice were perfused with cold PBS. Following perfusion, the cortex was removed, homogenized, and centrifuged at 10,000 r/min for 10 min at room temperature. The supernatant was then isolated and relative fluorescence was measured as described 9 using a Synergy 2 plate reader (BioTek, Winooski, VT, USA). Fluorescent dye content was calculated using external standards, including collected blood plasma. All data are expressed as amount of tracer per mg of tissue. 9 Additionally, to better visualize the modifications in BBB permeability, the small molecular weight fixable fluorescent tracer, sodium fluorescein (Na-F, Sigma/Aldrich), was used as described. 32 Meninges were removed and the brain was segmented through the impact site and imaged to visualize the underlying extent of BBB disruption under short-wave UV light using a G:Box gel and blot imaging system (Syngene, Frederick, MD, USA).
Immunohistochemistry (IHC)
Mice were anesthetized with 5% isoflurane and transcardially perfused with 20 mL cold PBS, delivered over a period of 10 min. Following perfusion, brains were removed and fixed in 4% formaldehyde solution for 24 h, then transferred to 30% sucrose solution until the brains no longer floated in the solution. Brains were then sectioned coronally on a Leica CM1860 cryostat (Leica Biosystems, Wetzlar, Germany) into 6 µm sections, which were then washed with PBS to remove cryoprotectant, permeabilized with 0.1% Triton X-100 in PBS and blocked for 2 h in 1% BSA, 5% normal donkey serum in PBS/0.1% Triton X-100. Primary antibody (anti-IBA-1 (1:500, Wako Chemicals, Richmond, VA, USA; Cat# 019-19741 Lot#WDK2121 RRID:AB_839504)) was diluted in blocking solution and incubated overnight at 4℃. Samples were then washed four times in PBS/0.1% Triton X-100 and incubated in secondary antibody diluted in blocking solution for 2 h at room temperature. Imaging histology was performed using a CoolSNAP EZ CCD camera (Photometrics, Tucson, AZ, USA) coupled to a Nikon i80 Eclipse (Nikon Instruments Inc., Melville, NY, USA). Images were taken at 20× and 100×.
Transendothelial electrical resistance (TEER)
BMVEC transfected with miRNAs (targeting or nontargeting sequences) were plated on collagen type I coated 96W20idf electrode arrays (Applied Biophysics, Troy, NY, USA) and were maintained for 72 h to form a monolayer; basal levels of TEER were 800–1500 Ω.10,15 A time “zero” was assigned for the experiment with or without TNFα treatment. TEER measurements were performed using the 1600R ECIS System (Applied Biophysics) as described.9,15,16 The results are presented as an average of the resistance values (Ohm, Ω) as well as the average percent change from baseline TEER (expressed as average ± SD) from at least three independent experiments consisting of four to six replicates each.
Quantitative RT-PCR
RNA was isolated utilizing the mirVana miRNA extraction kit (Life Technologies). Real-time RT-PCR was used to detect the differential expression of target genes. Real-time RT-PCR was performed using the mirVana qRT-PCR miRNA detection kit (Life Technologies) per the manufacturer’s protocol as described previously. 9 PCR primer pairs for reverse transcription and detection of mature miRs were purchased from Life Technologies (hsa-mir-98 and control U6). In general, quantitative real-time RT-PCR (qRT-PCR) on primary BMVEC was performed on three independent experiments using 25 ng of template using the Quantstudio S3 real-time PCR system (Life Technologies). For each sample, qRT-PCR was performed in triplicate. Amplification was analyzed using the ΔΔCt method, using a web-based data analysis tool (SABiosciences, Qiagen Inc., Valencia, CA, USA) by normalization to the corresponding values of housekeeping gene (U6) and fold-change calculated from the difference between experimental condition and untreated control.
Statistical analysis
Data are expressed as the mean ± SD of experiments conducted multiple times. Data were tested for normality using the Shapiro-Wilk test, and, if data were normally distributed, for multiple group comparisons. Multiple group comparisons were performed by one-way ANOVA with Tukey post hoc test with significance at p < 0.05 (TEER, FACS, permeability assays, animal experiments). A paired two-tailed Student’s test was used to compare before and after effects. Differences were considered to be significant at p < 0.05. Statistical analyses were performed utilizing Prism v6.0 h software (GraphPad Software Inc., San Diego, CA, USA).
Results
miR-98 levels are diminished in brain endothelial cells in in vitro and in vivo stroke models
Recently, our group has demonstrated that miR-98, belonging to the let-7 family, was drastically down-regulated during inflammation.
9
To test whether miR-98 expression levels would be affected during IS/R conditions, we first exposed primary human BMVEC to oxygen and glucose deprivation followed by reperfusion (OGD/R). The cells displayed a pronounced reaction to stroke-like conditions, showing a 2-fold reduction in miR-98 expression levels following OGD/R (Figure 1(a)). Next, to determine the bidirectionality of these effects, we transfected BMVEC with miR-98, utilizing a method established by us,
9
prior to assessing barrier integrity by TEER (see section “Materials and methods”). We determined that overexpression of miR-98 was effective in restoring the loss of electrical resistance in BMVEC to near baseline levels (p < 0.01), when compared with noncoding miRNA (scramble)-treated cells. miR-98-expressing cells showed faster recovery after OGD/R and a sustained increase in barrier tightness (Figure 1(b) and (c)).
OGD/R decreases miR-98 expression in BMVEC and its overexpression restores BBB tightness in vitro. (a) qPCR analysis of miR-98 expression in BMVEC after 3 h of OGD and 24 h reperfusion. Results are presented as an average from at least two independent experiments (**p < 0.01 vs. scramble control) consisting of at least four replicates. Mimic oligos of miR-98, or noncoding (scramble) were transfected into endothelial cells. (b) Absolute level of TEER (in ohms, Ω) after transfection of miR-98 or scramble miRNAs. (c) Quantification of the difference in percent change in TEER at 11, 16, 22, 33, and 42 h after OGD/R. Results are presented as an average from at least two independent experiments (*p < 0.05 or **p < 0.01 vs. scramble control) consisting of at least three replicates.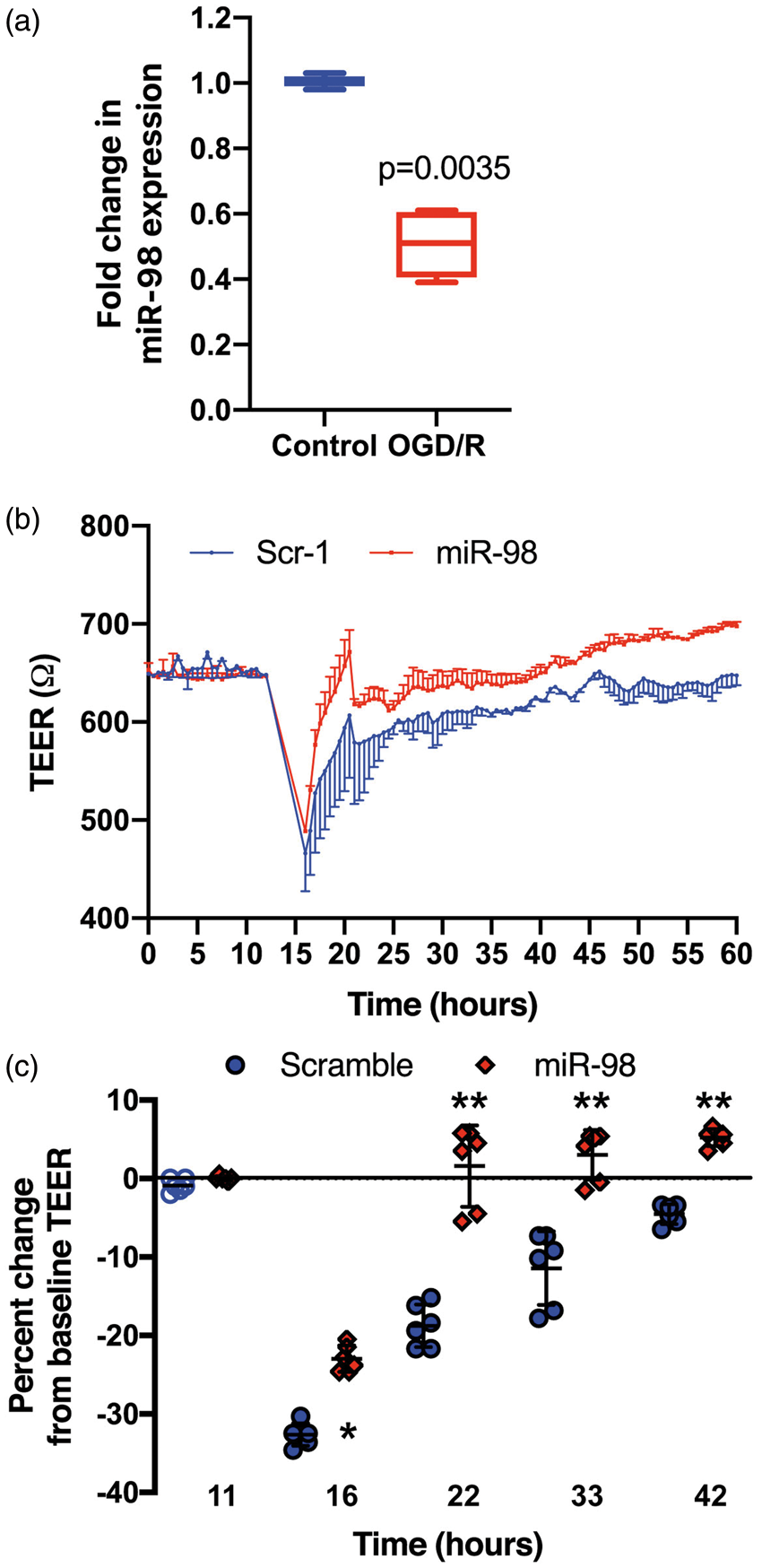
IS/R conditions also induced profound reductions in miR-98 expression in MVs in the in vivo model. Occlusion of the middle cerebral artery during tMCAO was measured as a function of regional cerebral blood flow via laser speckle imaging (Figure 2(a)). Upon blocking, the right cerebral hemisphere demonstrated a ∼75% reduction in blood perfusion (p < 0.01), relative to the control hemisphere, until the filament was removed (Figure 2(b)). Such arterial blockage (tMCAO) produced a ∼3.3-fold reduction in miR-98 expression in brain MVs (p = 0.0292) (Figure 2(c)).
Ischemia-reperfusion reduces miR-98 gene expression in brain microvessels. 60 min ischemia and 24 h reperfusion were performed as described in the “Materials and methods” section. Blood perfusion was confirmed by laser speckle analysis. Representative images (a) and quantitative analysis of blood perfusion (b). Results are presented in laser speckle perfusion units (LSPU) as ± SD (**p < 0.01 vs. scramble control) comprising > 4 replicates. Microvessels from the ischemic part of the brain were isolated, miRNA extracted and analyzed in triplicate by qPCR (c). Results are presented as mean ± SD from at least two independent experiments (n = 4).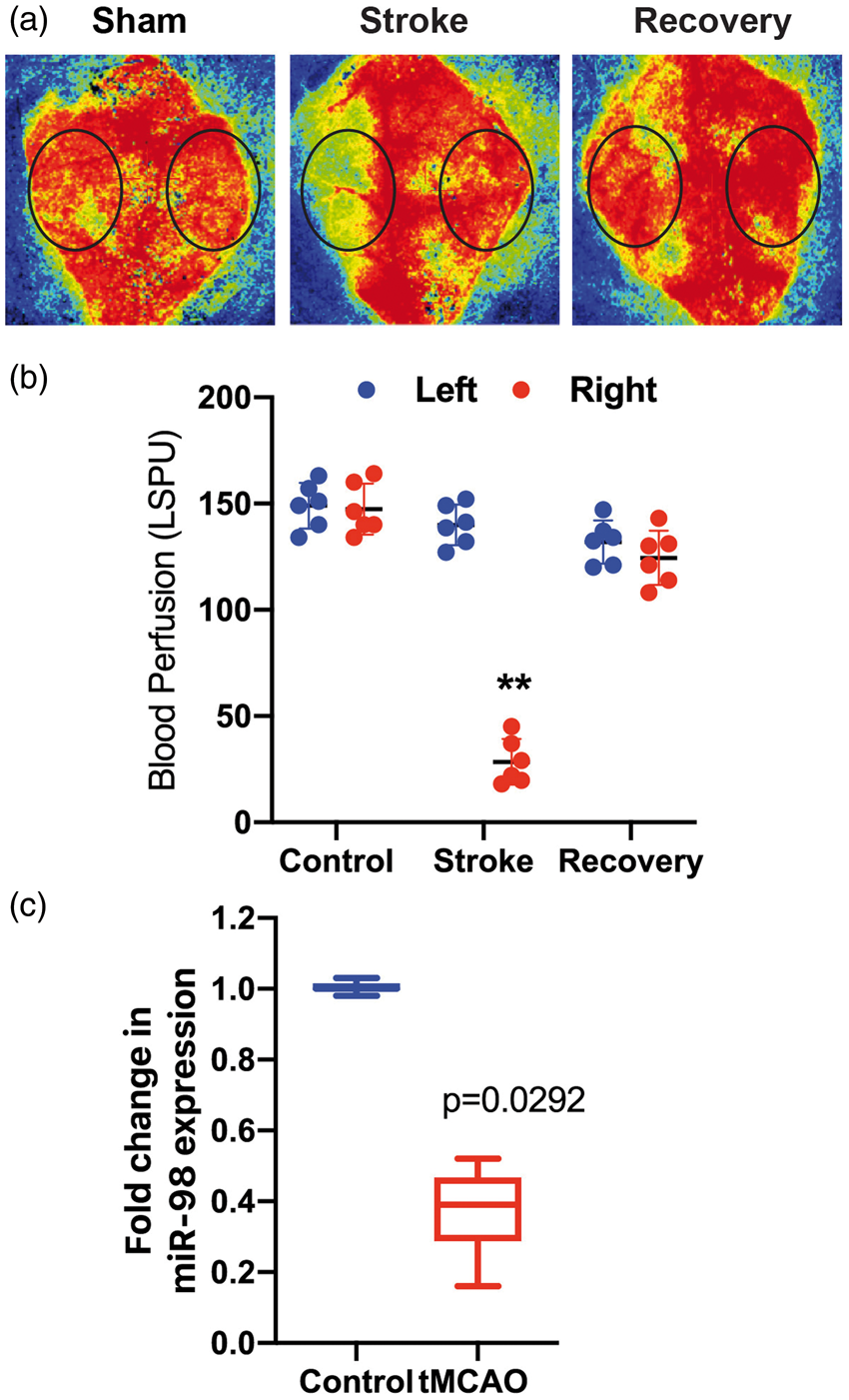
miR-98 overexpression alleviates neurological outcomes of tMCAO
Next, we tested whether restoration of miR-98 levels in MVs would result in the improvement of neurological outcome. We have previously shown that mice injected i.v. with liposome-miRNA complexes demonstrated a 22-fold increase of miR-98 in MVs 2 h after injection and the overexpression lasted several days (40- to 60-fold increases after 48 and 72 h, respectively). 9 In this study, we performed tMCAO and i.v. injected mice twice with miR-98 or noncoding (scramble, control) liposomal complex at different time points (2 and 24 h, 4 and 24 h, and 24 and 48 h after tMCAO), and assessed neurological outcome during the three days following the stroke.
The overall neurological improvement was observed in enhanced locomotor activity20–23 (measured by the total ambulation counts) (Figure 3(a)). tMCAO caused ∼69-fold reduction in locomotor activity. Mice injected with miR-98-liposomal complex showed the best recovery from stroke. miR-98 treatments at 2 and 24 h or 4 and 24 h after tMCAO improved locomotor activity 24.3- and 4.1-fold, respectively, vs. vehicle only or noncoding miR complex (p < 0.05).
miR-98 expression improves neurological scores in tMCAO. miR-98 was injected at specified times (2 and 24 h, 4 and 24 h, and 24 and 48 h) following tMCAO. Sham vehicle only or scramble (NC) were injected as negative controls. Total ambulation activity test (a) and Corner test (b) were acquired prior to surgery and after reperfusion following ischemia. Results are presented as an average from 8 to 12 mice per condition. #p < 0.05 vs. nonspecific miRNA control.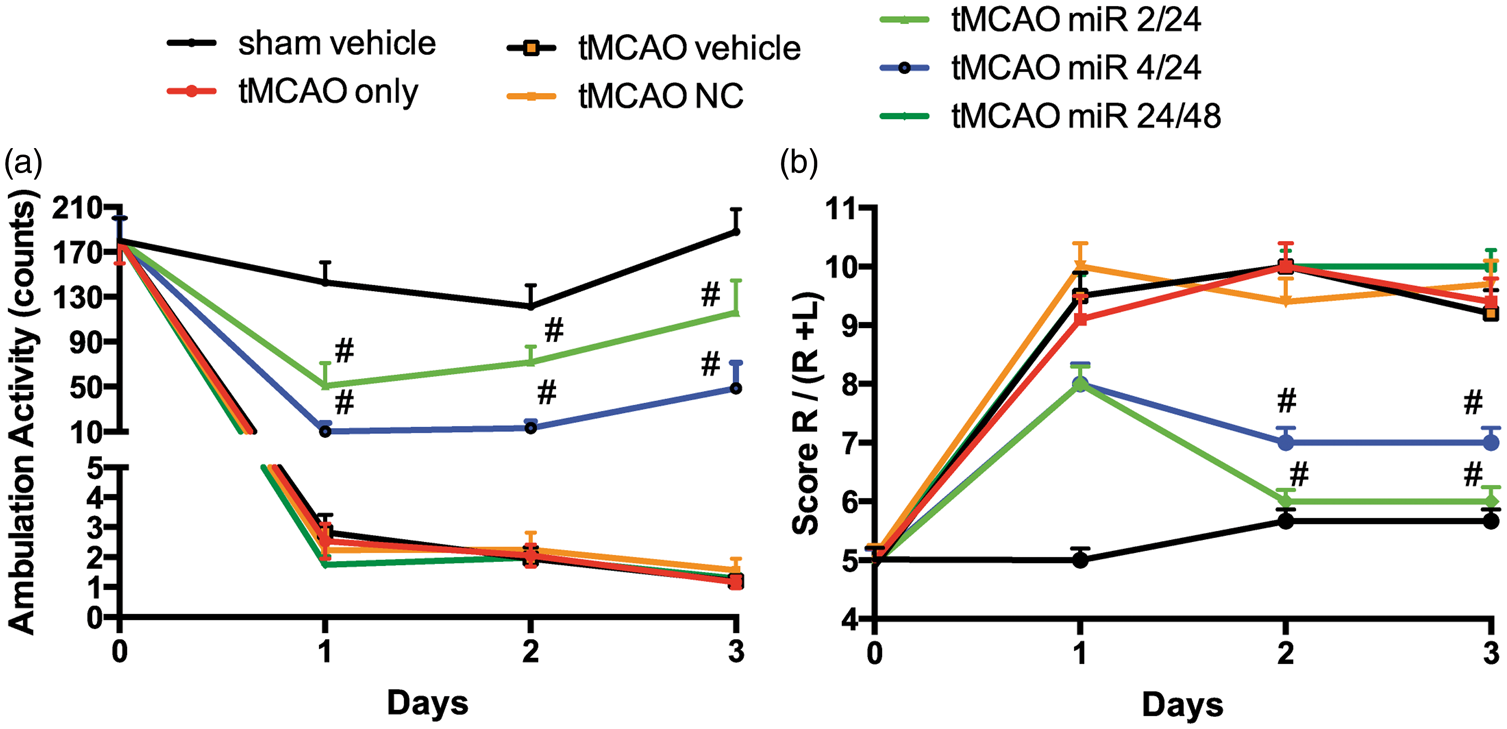
Next, we used the corner test, which is sensitive to sensorimotor and postural symmetries. “Stroked” mice usually turn toward the affected side (right), while nonaffected mice have almost a 50–50% left-to-right distribution.23,24 After tMCAO, mice showed a 9 ± 0.6 out of 10 right-to-left turn distribution (a feature of high impairment), whereas sham vehicle presented a normal 5 ± 1 out of 10 right-to-left turn allocation. miR-98-liposome-treated mice showed significant improvement in a time-dependent manner (Figure 3(b)). Mice that were treated at 2 h and 4 h after tMCAO displayed corner results similar to the sham animals on the second and third days after the procedure. Treatment with miR-98 that was applied 4 h and 24 h after stroke resulted in 55% ± 3% improvement in corner score compared to tMCAO only (p < 0.05), while treatment at 24 h and 48 h did not produce any effect (Figure 3(b)). These results suggest that anti-inflammatory treatment (in our case miR-98) should be given as soon as possible after the stroke in order to prevent neurological impairments.
miR-98 diminishes inflammatory responses upon IS/R conditions in vivo
IS/R induces a time-dependent recruitment and activation of inflammatory cells, such as neutrophils, T cells, and monocytes/macrophages; inhibition of inflammatory responses reduces infarct size and improves neurological deficits.1,4 The impaired, activated BBB promotes the infiltration of peripheral inflammatory cells into the brain, which secrete deleterious mediators resulting in long-lasting barrier injury. Our working hypothesis was that miR-98 is reduced during inflammation and IS/R in endothelial cells, which leads to BBB dysfunction; miR-98 overexpression would restore the BBB and reduce inflammatory responses. First, we assessed the profile of brain infiltrating leukocytes (BIL) after tMCAO. Monocytes (CD45hiCD11b+Ly6g-Ly6chi (further Ly6chi)) recently have been shown to display a proinflammatory phenotype
33
and to infiltrate the ischemic brain three days after stroke in the mouse tMCAO model. Conversely, Ly6clow is considered to have an anti-inflammatory phenotype.
33
We isolated BILs from mice three days after tMCAO and found a 5-fold increased presence of Ly6chi monocytes in the infarcted hemisphere (Figure 4). In agreement with our neurological assessments, injection with miR-98, that was applied within shortest time window after the tMCAO, led to a 1.79-fold (p < 0.05) diminution in Ly6chi proinflammatory monocytes three days after induction of tMCAO (Figure 4), while even a 4 h delayed treatment had no significant effect on the amount of infiltrated Ly6chi monocytes, as a noncoding miR treatment.
miR-98 expression reduces CNS infiltration of proinflammatory Ly6Chi monocytes. Mice were subjected to tMCAO and treated with liposome/exogenous miRNA complexes of mimic oligos of miR-98, noncoding (NC = scramble), or vehicle only. Flow cytometry analysis of BILs isolated from infarcted hemisphere (n = 5). 10,000 events were recorded per tube. MFI for Ly6C was analyzed in CD45hiCD11b+monocytes. Data are presented as mean ± SD. *p < 0.05 miR-98-treated vs. noncoding miRNA, #p < 0.01 tMCAO vs. sham-operated animals.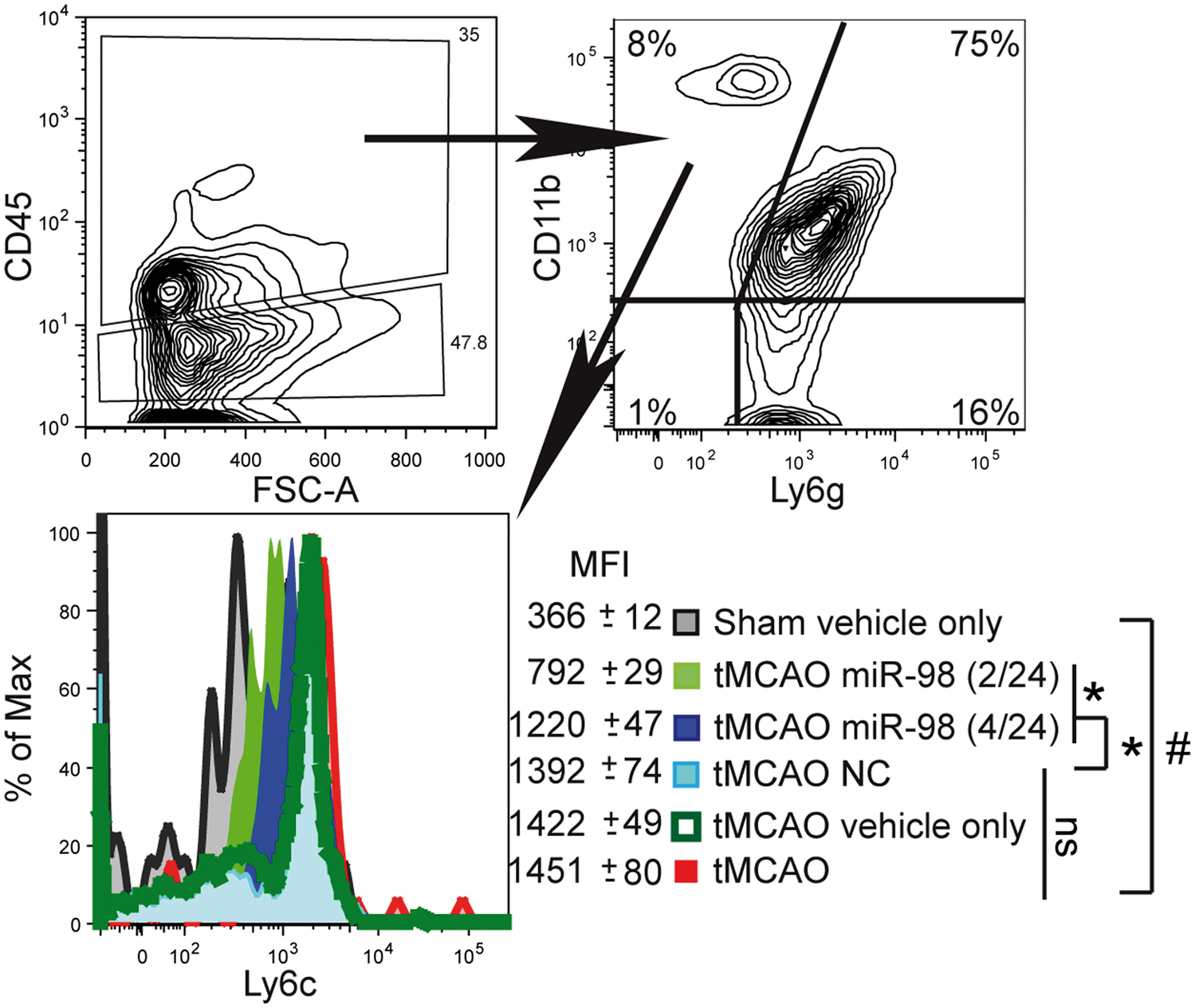
Next, we tested whether miR-98 treatment would affect microglia activation following tMCAO. tMCAO mice injected with noncoding miR (scramble) presented ∼3-fold increase in microglia activation (as measured by IBA expression within CD45lowCD11b+ cells) vs. sham-operated animals (p < 0.05). Mice treated with miR-98 liposomal complex displayed a 2-fold (p < 0.05) reduction in activated microglia within the ischemic brain at three days (Figure 5(a) and (b)). Taken together, these results demonstrate that restoring miR-98 levels in MVs protects the BBB from crossing by proinflammatory Ly6chi monocytes and therefore prevents further microglial activation.
miR-98 overexpression lowers microglia activation after tMCAO. Mice were subjected to tMCAO and treated with liposome/exogenous miRNA complexes of mimic oligos of miR-98, noncoding (NC = scramble), or vehicle only. Microglial activation was measured by FACS in isolated microglia from the affected hemisphere. Data are presented as (a) MFI for IBA analyzed in CD45lowCD11b+cells. (b) Immunohistochemistry of IBA in brain sections, shown at 20× and 100×. Data are presented as mean ± SD. *p < 0.05 miR-98-treated vs. noncoding miRNA, #p < 0.01 tMCAO vs. sham-operated animals.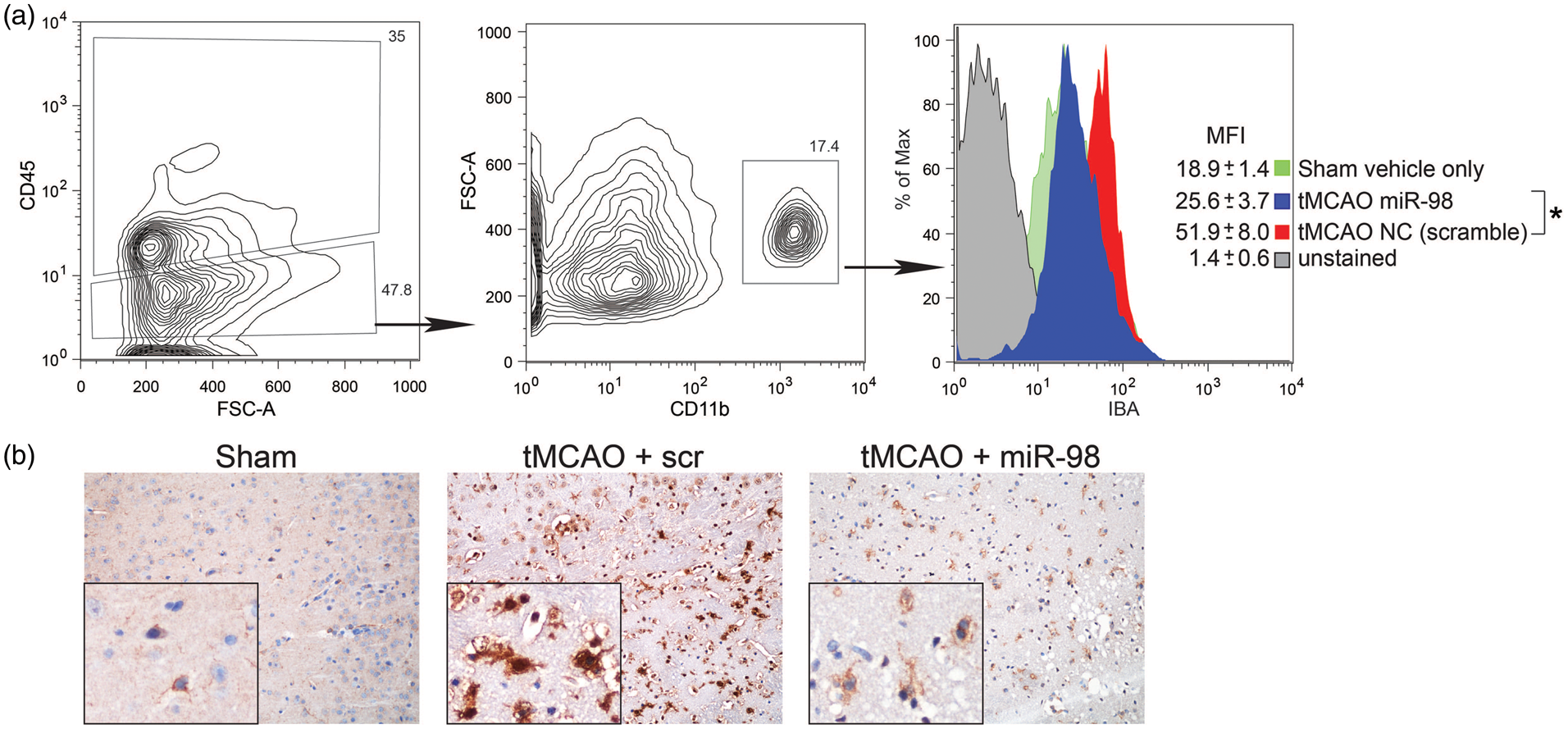
miR-98 overexpression mitigates BBB disruption upon IS/R conditions in vivo
The miR-98-induced improvements in BBB tightness in vitro (Figure 1(b) and (c)), and concurrent reduction of monocyte infiltration (Figure 4) and microglial activation (Figure 5) prompted us to assess whether miR-98 would affect BBB stability in vivo. Barrier integrity was explored using a modified fluorescein permeability assay.
32
At 72 h after tMCAO, FITC-labeled dextran was introduced by i.v. injection and allowed to circulate for 15 min prior to perfusion and tissue harvest. Images of sectioned brain showed a significant 1.39-fold increase in fluorescence in tMCAO animals treated either with scramble or vehicle alone vs. sham condition (Figure 6). Post-stroke treatment with miR-98 reduced brain permeability to 40 kD dextran by 21% (p < 0.05) when compared to scramble-treated mice (Figure 6).
Overexpression of miR-98 attenuates BBB disruption. Mice were subjected to tMCAO and injected (i.v.) with liposome/exogenous miRNA complexes of mimic oligos of miR-98, noncoding (NC = scramble), or vehicle only, with sham serving as the control. Quantification of FITC-labeled dextran (40 kDa) accumulation and representative images of each section of the brain are shown for each treatment group (sham, vehicle, scramble, miR-98). Experiments were performed in triplicate with four mice in each group. Permeability is represented as mean fluorescence ± SD.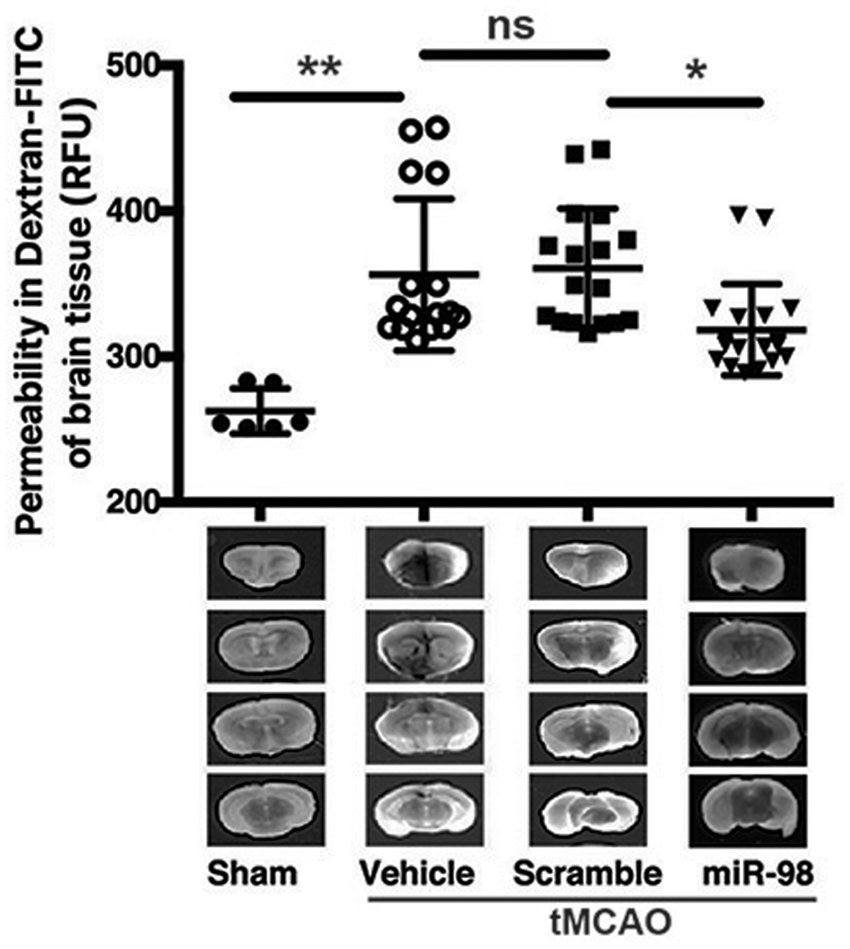
Next, we tested the effects of miR-98 treatment prior to IS/R conditions. We injected mice 24 h prior to tMCAO with miR-98 liposomal complex or with noncoding scramble and demonstrated that miR-98 attenuated the permeability of ischemic brain tissue by 2-fold (p < 0.0057) (Figure 7(a)). miR-98 treatment shrank the overall area of infarct by more than 50% (Figure 7(b) and (c)). Further, the substantial reduction in overall infarct size was associated with significantly improved clinical outcomes. Animals treated with miR-98 demonstrated significantly better ambulation activity in the days following surgery (p < 0.005) (Figure 7(d)), as well as improved performance in the corner test (p < 0.001) (Figure 7(e)). Taken together, our results indicate that overexpression of miR-98 effectively suppresses leukocyte infiltration and microglia activation, leading to diminution in the infarcted area and improved neurological outcomes.
Pre-tMCAO overexpression of miR-98 reduces infarct area and improves neurological scores. Mice were injected retroorbitally at two time points (as previously described
9
) with liposome/exogenous miRNA complexes of mimic oligos of miR-98, noncoding (NC = scramble), or vehicle only 24 h prior to tMCAO. (a) Quantification of FITC-labeled dextran (40 kDa) accumulation in the stroke brain hemisphere. Experiments were performed in triplicate with four mice in each group. Total ambulation activity test (d) and corner test (e) were acquired prior to surgery and after reperfusion following ischemia. Results are presented as an average from 7 to 9 mice per condition. The results are shown as mean fluorescence ± SD. Representative images (b) and quantification of the infarct volume (c) by TTC staining of brains 24 h after ischemic onset.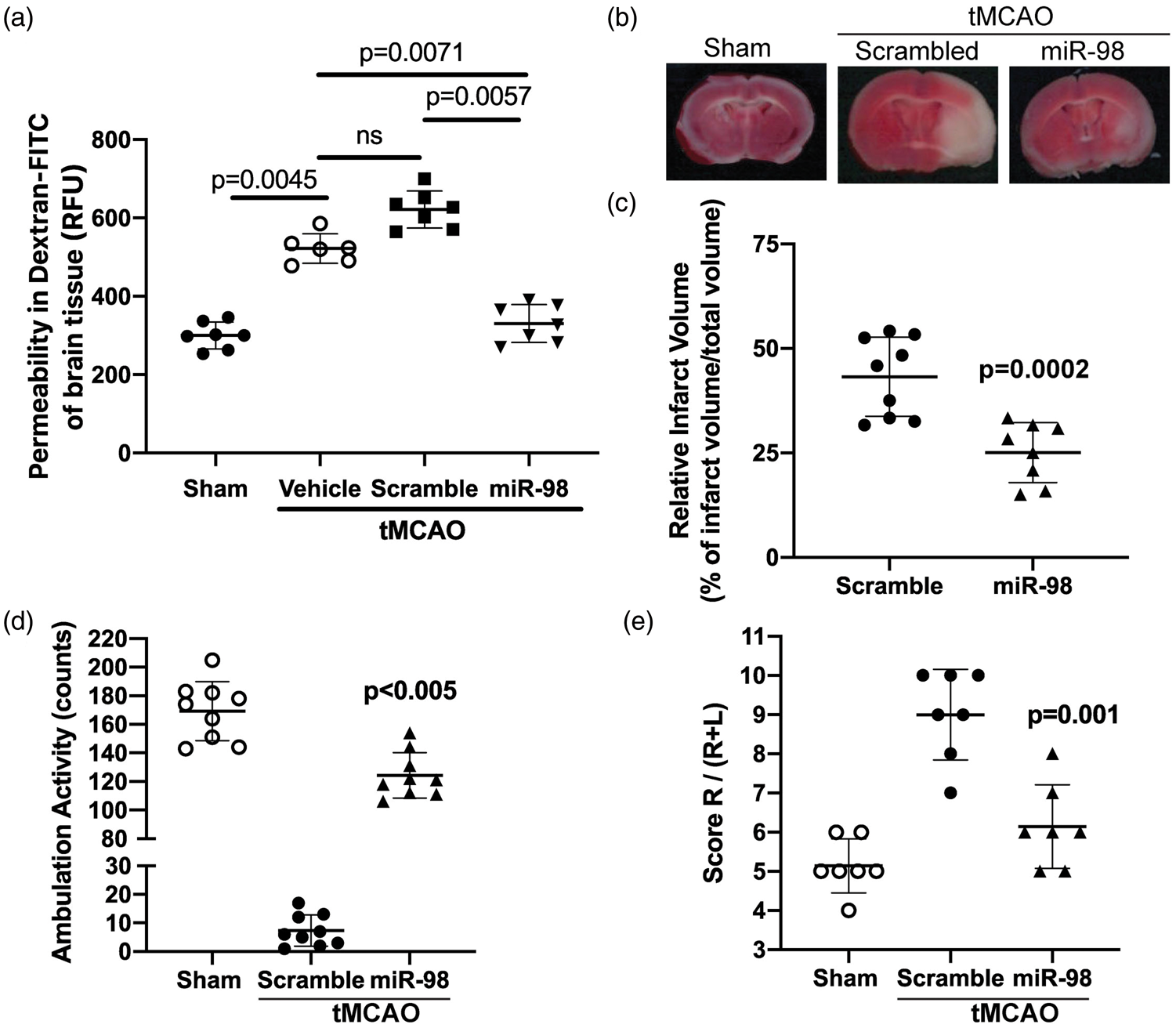
Discussion
In the current study, we demonstrate a critical role for miR-98 as a key regulator of the extent of CNS damage following ischemia. First, we demonstrate that miR-98 is significantly downregulated following stroke-like conditions in in vitro and in vivo stroke models. These data are consistent with the growing body of literature,9,34,35 which points to downregulation of miR-98 and other let7 miRNAs as a key cause of damage following inflammatory events. By reversing this downregulation, indeed by augmenting the expression of mir-98 following stroke, we were able to ameliorate many of the neuroinflammatory and behavioral features of stroke, preserve more tissue within the region of infarct, and contribute to a greater post-stroke prognosis. Our findings denote the importance of expression of miR-98, a member of let-7 miRNA family, as a critical element of the ischemic cascade, which may have strong significance for potential future therapies for stroke treatment or prevention.
One of the main factors underlying the importance of let-7 miRs, such as miR-98 and let-7 g, 9 is the ability to improve and restore the integrity of the BBB. BBB compromise is one of the earliest and most critical measures of neuroinflammation; indeed, vascular endothelial cells are often the first area of compromise.36,37 It is also well known that endothelial dysfunction contributes to and amplifies the effects of further events such as cytokine release, 38 leukocyte adhesion, 39 and microglial activation. 40 Hence, treatments which preserve the functionality of endothelial cells have the potential to drastically improve clinical outcomes. In our in vitro model, we determined that treatment with miR-98 preserved the integrity of BMVEC following stroke-like conditions and maintained BBB integrity for an extended period of time (Figure 1). Such improvements in BBB tightness have been shown to slow the release of cytokines 41 and limit the recruitment of other inflammatory cells, 41 ultimately reducing the damage caused by both types of cells. Dramatic behavioral performance improvements as well as a decrease in BBB permeability and infarct size in mice treated prior to tMCAO, open the possibility of potential therapeutic applications for miR-98. Such applications include the prevention of perioperative stroke, which occurs in small incidence rate ∼1–1.9% during noncardiac, nonneurologic, or nonmajor surgery, whereas it increases by 5- to 9-fold in people who undergo high-risk cardiac or brain surgery procedures. 42 Perioperative stroke occurrence rates might go even higher in patients with a thrombosis or other comorbidities.43,44
Recently, we demonstrated that overexpression of miR-98 and let-7 g resulted in direct targeting and decreased expression of MCP-1 and RANTES cytokines. 9 MCP-1/CCL2 and RANTES/CCL5 are enablers of acute inflammatory responses involved in attracting leukocytes to adhere to and migrate across BBB. 45 MCP-1 has been indicated to modify BBB rigidity by stimulating small Rho GTPases, triggering actin cytoskeleton rearrangement and redistribution of TJ proteins, such as ZO-1, ZO-2, occludin, and claudin-5. 46 Many studies have shown that maintaining barrier integrity is essential for limiting the damage caused by inflammation. The level of vascular compromise is often proportional to the concentration of toxic products. 47 Indeed, the current study denoted reduced markers of inflammation, smaller infarct sizes, and greater clinical outcomes, in animals treated with miR-98. While such impacts would be useful in treating stroke, a more intact BBB is also strongly associated with greater clinical outcomes in other forms of neuroinflammation, including traumatic brain injury, 48 HIV-1 encephalitis, 49 and diabetes. 50 The results of the current study suggest that increased or restored miR-98 levels can be an effective means of treatment for many inflammatory brain disorders, alone, or in conjunction with other pharmacotherapies.
Another critical advantage of compounds that target the endothelium, such as miR-98, is the high bioavailability and ease in reaching the target areas, which lay on the proximal vasculature. Without the considerable obstacle of crossing the BBB, compounds such as miR-98 can begin working immediately, and can have high bioavailability when delivered intravenously. Indeed, we observe significant effects when administering miR-98 within 2 h of stroke. At the same time, the barrier-tightening effects on endothelial cells can persist for many hours following an ischemic insult as demonstrated in Figure 1.
Such improvements may not necessarily be confined to the brain; miR-98 is present in most tissues and cell types in the body.51–54 Therefore, it is possible that some of the miR-98 extracted from the microvessels in Figure 2 could have been produced by mural cells such as pericytes, in addition to BMVECs. Endothelial to pericyte ratios in normal tissues vary between 1:1 and 10:1, while pericyte coverage of the endothelial abluminal surface ranges between 70% and 10%, with highest ratio in retina vessels.55–58 In Figure 1, ischemia/reperfusion conditions reproduced by OGD/R resulting changes to miR-98 expression, were attributable only to expression occurring within BMVECs themselves. Hence, it is possible that mural cells contribute to the overall expression of miR-98, but endothelial cells appear to be responsible for a significant proportion of its expression within the microvasculature. Generally, miR-98 is downregulated during inflammatory insults, and restoration of expression can lead to preservation of cellular function across a variety of conditions, including resistance to oxidative stress within hepatocytes. 59 Due to the systemic application of the liposomal-miR-98 complex, it is possible that other organs/tissues were also targeted. We identified that miR-98 expression was robust within the brain microvasculature, to a lesser extent within the brain itself, and much less in other organs. Based on our current data and results from our previous work, 9 we believe that the ameliorative effects of miR-98 on the stroked brain are almost exclusively due to direct impact on the microvasculature. Recently we demonstrated that miR-98 directly targets production of proinflammatory cytokines, CCL2 and CCL5, 9 which are well-known to facilitate acute inflammatory responses and are associated in drawing leukocytes to adhere to and migrate across the BBB, 45 as well as aggravating BBB tightness by modifying small Rho GTPases activation and causing rearrangements in actin cytoskeleton and inciting TJ proteins, ZO-1, ZO-2, occludin, and claudin-5 redistribution. 46 Rather, we think that most of the stroke protective effects are coming from endothelial cells transfected with miR-98.
Finally, miR-98 offers the potential for studying and treating stroke because of a relatively long window of clinical utility. Following ischemic or traumatic insults, the brain vasculature undergoes significant damage, and this damage can persist for days or weeks following insult.60,61 Compounds that impact on the integrity of the BBB specifically, such as miR-98, may have a long therapeutic window. While early interventions are associated unequivocally with better clinical outcomes, miR-98 is also effective at reducing infarct size and improving behavioral outcomes when administered prior to stroke. These attributes offer great promise for future clinical utility. While research on miR-98 itself is still sparse, studies such as this one lay the groundwork for more effective clinical and preclinical treatments for neuroinflammatory diseases such as stroke, by protecting the BBB and slowing the ischemic cascade.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by NIH research grants R01NS101135 (SR), R01AA015913 (YP) and R01MH115786 (YP).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
David L Bernstein – data acquisition and analysis, drafting, revising, and final approval article; Viviana Zuluaga-Ramirez – data acquisition and analysis, revising, and final approval article; Sachin Gajghate – data acquisition and analysis, revising, and final approval article; Nancy L Reichenbach – data acquisition and analysis, revising, and final approval article; Boris Polyak – data interpretation, revising, and final approval article; Yuri Persidsky – data interpretation, revising article, and final approval; Slava Rom – conception and design, data acquisition, analysis and interpretation, drafting and revising article, and final approval.