
Abstract
Recent advances in stroke reperfusion therapies have led to remarkable improvement in clinical outcomes, but many patients remain severely disabled, due in part to the lack of effective neuroprotective strategies. In this review, we show that 95% of published preclinical studies on “neuroprotectants” (1990–2018) reported positive outcomes in animal models of ischemic stroke, while none translated to successful Phase III trials. There are many complex reasons for this failure in translational research, including that the majority of clinical trials did not test early delivery of neuroprotectants in combination with successful reperfusion. In contrast to the clinical trials, >80% of recent preclinical studies examined the neuroprotectant in animal models of transient ischemia with complete reperfusion. Furthermore, only a small fraction of preclinical studies included long-term functional assessments, aged animals of both genders, and models with stroke comorbidities. Recent clinical trials demonstrate that 70%–80% of patients treated with endovascular thrombectomy achieve successful reperfusion. These successes revive the opportunity to retest previously failed approaches, including cocktail drugs that target multiple injury phases and different cell types. It is our hope that neurovascular protectants can be retested in future stroke research studies with specific criteria outlined in this review to increase translational successes.
Introduction
Acute ischemic stroke remains a leading cause of disability and mortality worldwide. 1 Following initially modest progress after the introduction of intravenous thrombolysis in 1995,2–5 dramatic gains in reperfusion therapies have recently been achieved with endovascular thrombectomy for the most severe strokes with large vessel occlusion (LVO). 6 On the other hand, many stroke patients remain severely disabled. The rates of functional independence achieved even after highly effective reperfusion treatments are just under 50%,7–9 underscoring an unmet clinical need for adjunctive neuroprotective treatments. There has been a long history of failed clinical trials of neuroprotection in stroke. 10 However, most clinical trials did not test the neuroprotective candidate in combination with successful reperfusion. Timely and successful restoration of blood flow may be essential to deliver therapeutic concentrations of neuroprotectants to the ischemic brain. Recognition of this key distinction unveils a new path for more fruitful partnerships between preclinical and clinical scientists. The primary goal of this review is to discuss the evidence-based rationale for retesting neuroprotectants in conjunction with reperfusion therapy in ischemic stroke patients. In this review, we hope to accomplish the following: (1) Highlight recent exciting advances in stroke treatments, (2) identify the remaining areas of paramount clinical need for neuroprotection in stroke, (3) synopsize our analysis of the preclinical literature on neuroprotective strategies, (4) identify mismatches between preclinical and clinical research that may have contributed to the failures of translation to date, (5) discuss the primary goals of stroke treatments and the biological mechanisms they must target, and (6) recommend future approaches for preclinical and clinical research, including combinatorial approaches to decelerate ischemic injury and extend the temporal window during which injured tissue can be salvaged.
Recent progress in stroke reperfusion therapies
Recent progress in stroke reperfusion therapies is largely attributed to the successful translation of key concepts about the pathophysiology of cerebral artery occlusion and ensuing tissue ischemia. Early laboratory and clinical studies demonstrated that acute LVO leads to an ischemic “core” consisting of irreversibly damaged, infarcted tissue, surrounded by a “penumbra” of functionally impaired, but potentially salvageable tissue.11–13 Following acute LVO, the ischemic core may expand into the penumbral area over time, but this process may be aborted if tissue reperfusion is achieved sufficiently early.14–16 The growth of the ischemic core progresses at varying rates across individuals, depending on collateral blood flow capacity and parenchymal ischemic tolerance.17–19 Thus, some individuals with poor collateral flow and low ischemic tolerance after LVO display a larger ischemic core soon after symptom onset. These individuals are known as “fast progressors,” who tend to suffer worse clinical outcomes, with the most severe symptoms, larger infarcts, and increased risk of malignant cerebral edema and hemorrhagic transformation. 19 Further studies are needed to better understand the molecular and clinical predictors of this devastating stroke phenotype. Conversely, patients with superior collateral flow and ischemic tolerance tend to exhibit a smaller ischemic core up to several days after LVO stroke onset. These may be the “slow progressors,” who typically experience superior clinical outcomes in response to reperfusion therapy.13,19,20
Pivotal clinical trials in the development of modern stroke reperfusion therapies.
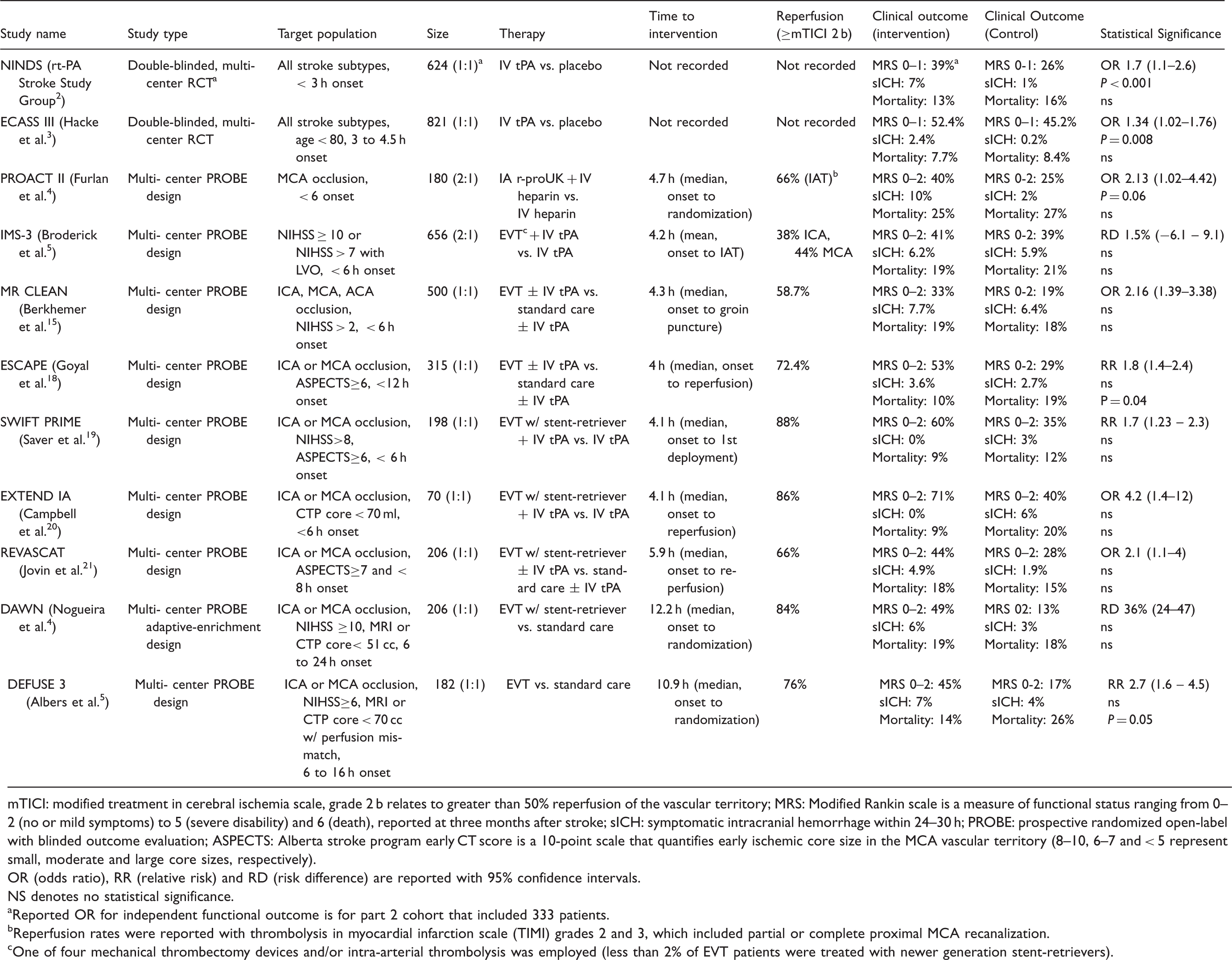
mTICI: modified treatment in cerebral ischemia scale, grade 2 b relates to greater than 50% reperfusion of the vascular territory; MRS: Modified Rankin scale is a measure of functional status ranging from 0–2 (no or mild symptoms) to 5 (severe disability) and 6 (death), reported at three months after stroke; sICH: symptomatic intracranial hemorrhage within 24–30 h; PROBE: prospective randomized open-label with blinded outcome evaluation; ASPECTS: Alberta stroke program early CT score is a 10-point scale that quantifies early ischemic core size in the MCA vascular territory (8–10, 6–7 and < 5 represent small, moderate and large core sizes, respectively). OR (odds ratio), RR (relative risk) and RD (risk difference) are reported with 95% confidence intervals. NS denotes no statistical significance.
Reported OR for independent functional outcome is for part 2 cohort that included 333 patients.
Reperfusion rates were reported with thrombolysis in myocardial infarction scale (TIMI) grades 2 and 3, which included partial or complete proximal MCA recanalization.
One of four mechanical thrombectomy devices and/or intra-arterial thrombolysis was employed (less than 2% of EVT patients were treated with newer generation stent-retrievers).
Why does reperfusion therapy not improve stroke outcome in all patients?
Despite the successful modernization of reperfusion therapies as outlined in Table 1 and recognition of the importance of selecting patients with salvageable brain tissue rather than solely by fixed time-windows, there remains a large need to improve acute stroke treatments in multiple areas. Although overall rates of partial or complete arterial recanalization after early administration of intravenous tissue plasminogen activator (iv tPA) reach 33%,26,27 complete recanalization rates with iv tPA are much lower for occlusions of the intracranial internal carotid artery (ICA; 4%), proximal middle cerebral artery (MCA; 21%), and basilar artery (4%). 27 Moreover, a significant number of LVO patients remain severely disabled despite achieving successful reperfusion with the gold standard intervention of intravenous tPA combined with endovascular thrombectomy. In a 2015 meta-analysis of the first five successful trials of stroke endovascular therapy, 18.5 to 32.5% of those patients who achieved successful reperfusion within 3 to 8 h after symptom onset still experienced severe disability or death at 90 days. 28 In the DAWN and DEFUSE-3 trials of delayed reperfusion, 84% and 76% of respective endovascular patients achieved successful reperfusion, but only 49% and 45% of these patients experienced long-term functional independence. These data indicate that some patients are less responsive to initially successful reperfusion. In some cases, early re-stenosis or re-occlusion of the recanalized vessel may account for unfavorable long-term outcomes. In the DAWN and DEFUSE-3 trials, the rates of artery recanalization at 24 h were 77% and 78%, respectively, which were only slightly lower or similar to the rates of initial angiographic recanalization. Therefore, in the majority of patients, failure to achieve good outcomes despite successful reperfusion is more readily attributed to a large pre-intervention infarct and to a lesser extent, the eloquence of the affected brain area. 29 Another possible explanation is that some LVO patients experience relatively fast infarct growth due to collaterals failing between the initial time of brain imaging and subsequent vessel recanalization. 19 Other factors, most notably age but also other co-morbidities, in-hospital complications, and rehabilitation quality may also influence the long-term efficacy of reperfusion therapies. Further studies are needed to characterize the mechanisms of ineffective arterial recanalization and suboptimal response to adequate reperfusion.
Among current stroke therapies, another need for improvement relates to hemorrhagic transformation and other deleterious effects of reperfusion after intravenous thrombolysis with or without endovascular thrombectomy. 30 Intravenous thrombolysis has been associated with an increased risk of symptomatic intracranial hemorrhage (sICH) ranging between 2 and 7% within the first 24 to 30 h after ischemic stroke onset.31,32 The risk of sICH or large parenchymal hematoma ranged from 4 to 5% in both the intervention and control groups in the HERMES meta-analysis, which included more than 1200 patients pooled from the landmark endovascular therapy trials in 2015. 7 Furthermore, approximately 60 to 80% of LVO patients who developed early ICH after endovascular thrombectomy experienced severe disabilities based on the modified Rankin score (mRS 4 or 5) or mortality at 90 days in a large single-center retrospective study. 33 To maximize the benefits of reperfusion therapies, future neuroprotective therapies must protect the blood–brain barrier, reduce the risk of hemorrhagic transformation, and minimize other types of reperfusion injury after ischemic strokes.
How can stroke reperfusion therapies be further improved?
Although stroke reperfusion therapy is effective under ideal circumstances, its effectiveness in the real world remains limited because the majority of stroke patients lack timely access to intravenous thrombolysis, with or without endovascular therapy. This is due in large part to failure of early recognition, unknown duration of symptoms, or late arrival of stroke patients to the hospital in remote geographical locations. Early epidemiological studies demonstrated that less than 10% of patients diagnosed with acute ischemic stroke in US hospitals are treated with reperfusion therapies, mainly because of delayed presentation. 34 This number is likely to increase with the modernization of stroke healthcare systems, and as more patients with LVO are now eligible for endovascular therapy up to 24 h after symptom onset, based on the DAWN and DEFUSE-3 trials. 35 On the other hand, no more than 25% of all acute ischemic strokes in the general population are thought to be due to LVO. 36 Therefore, the majority of non-LVO patients with distal arterial branch occlusions or small vessel occlusion who could be eligible for intravenous thrombolysis remain excluded from reperfusion, strictly due to delayed clinical presentation beyond 4.5 h of symptom onset. This problem underscores the unmet demand for neuroprotective therapies to preserve penumbral brain tissue (with consequent delay in tissue infarction), prevent hemorrhagic transformation, and extend the therapeutic time window for reperfusion.
Why did earlier clinical trials of neuroprotection in stroke fail despite previous success in preclinical models?
The last two decades have witnessed a remarkable increase in the number, breadth, and depth of preclinical research studies on acute ischemic stroke. 37 Recent animal studies have identified a significant number of neuroprotectants that reduce acute ischemic brain injury in preclinical models. 38 Between 1990 and 1994, there were ∼25 publications per year assessing the effects of neuroprotectants in animal models of acute ischemic stroke. This number increased three-fold (77 per year) between 2000 and 2004 and almost six-fold after 2010 (144 per year). Between 1990 and 1994, approximately 500 clinical trials had commenced on >200 neuroprotective candidates for ischemic stroke, and ∼350 of these trials had been completed as of 28 February 2018 (data from www.clinicaltrials.gov ). However, among the ∼200 neuroprotective candidates tested in patients, only nine successfully transitioned to Phase III trials with the support of promising preclinical studies. An additional candidate, DP-B99, transitioned to Phase III trials without the support of published preclinical work.39–41 However, none of these completed Phase III trials demonstrated significantly positive treatment effects to support translation of neuroprotectants into standard clinical practice.11,13,19,20
The neuroprotectant uric acid accounts for as much as two-thirds of total antioxidant capacity in plasma 42 and is among the top 10 candidates from the abovementioned Phase III stroke trials. Preclinical studies have demonstrated that uric acid can prevent glutamate-induced cell death in vitro 43 and improve functional outcomes in animal models of ischemic stroke by neutralizing reactive oxygen species. 44 Consistent with these preclinical data, the administration of uric acid decreased lipid peroxidation in a Phase II pilot study without eliciting significant adverse reactions. 45 However, the randomized, double-blinded, placebo-controlled Phase IIb/III multicenter URICO-ICTUS trial demonstrated that co-administration of uric acid with tissue plasminogen activator (tPA) was not superior to reperfusion (intravenous or intra-arterial) alone; the addition of uric acid failed to change primary outcomes, or the percentage of patients with modified Rankin scores of 0–1 during follow-up at 90 days. 46 However, a post hoc analysis conducted on patients undergoing thrombectomy in URICO-ICTUS revealed statistically significant differences in clinical outcomes favoring the uric acid arm. 47 A plethora of other preclinically validated neuroprotective candidates, including cerebrolysin, 48 magnesium sulfate, 49 citicoline, 50 human albumin, 51 ginsenoside Rb1, 52 and recombinant human erythropoietin (rhEPO) 53 all failed to provide meaningful clinical benefits, raising important questions about the low rate of translation of drug efficacy from experimental models to clinical trials. 54 Why has it been so difficult to achieve meaningful neuroprotective effects in clinical trials of stroke? This failure may be attributed to multiple factors, including a series of key mismatches between preclinical and clinical stroke research that are identified below.
Analysis of pre-clinical and clinical research mismatches
In order to identify the discrepancies between basic and clinical stroke research, we analyzed all the original preclinical studies on neuroprotectants in ischemic stroke that were indexed in PubMed and Web of Science databases between January 1, 1990 and February 28, 2018 and published in English. The MeSH search terms entered into PubMed were: “Neuroprotective Agents”[Majr] AND “Brain Ischemia”[Majr]. The search terms for the Web of Science database were: (TS = (((Brain OR cerebral OR intracranial OR cranial OR intracerebral) AND (ischemic OR ischemia OR infarct OR hypoxic OR hypoxia OR anoxic OR anoxia)) AND (neuroprotective OR neuroprotectant OR neuroprotect))) AND LANGUAGE: (English) AND DOCUMENT TYPES: (Article). These search strategies identified a total of 3943 publications in PubMed and 6749 publications in Web of Science. We excluded studies duplicated in both databases (3182 in number) and all records that did not directly report the testing of neuroprotectants (2928 in number). The remaining 4582 studies were screened to ensure compliance with remaining inclusion/exclusion criteria. For example, review articles, clinical studies, in vitro studies, studies focused on physiotherapy, hemorrhagic stroke, or global ischemic brain injuries were excluded. In total, 2489 publications were closely examined in the final analyses (Figure 1). All of the included studies evaluated biological, chemical, and cell-based therapies in preclinical settings, and we identified a number of important inconsistencies between preclinical models and clinical protocols that we believe may have influenced the general failure to translate animal work. Here we list five major characteristics of the clinical population that are not faithfully recapitulated in most preclinical research design:
Flowchart of study selection. A total of 7510 studies on Pubmed and Web of Science were screened by evaluators. After exclusion for lack of relevance and various other criteria, 2489 studies were eligible for the qualitative synthesis.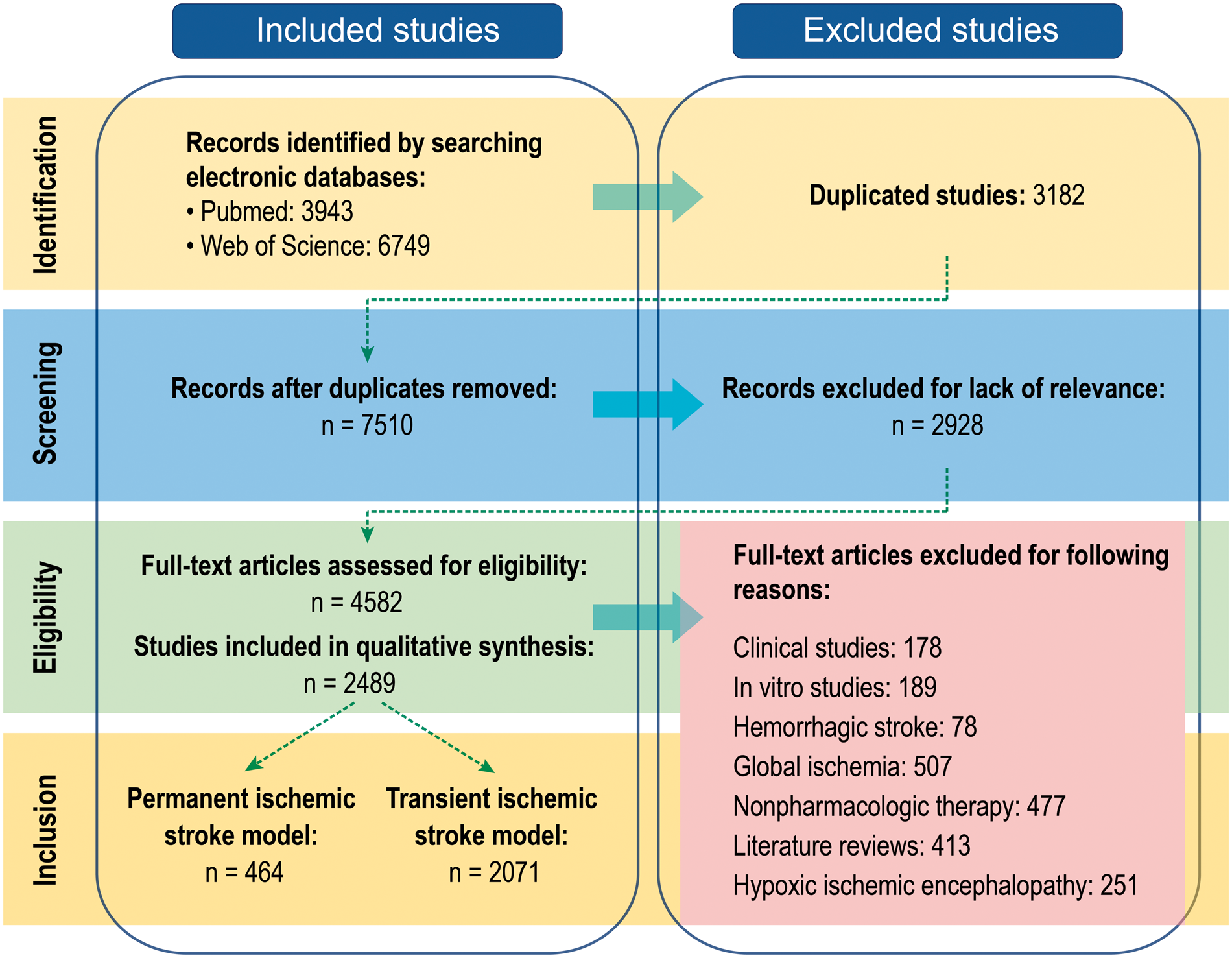
Mismatch I: Neuroprotectants have been tested in preclinical models of transient ischemia but not tested in combination with adequate reperfusion in stroke clinical trials
Our attention was first drawn to 10 therapies that had entered Phase III clinical trials and were initially deemed neuroprotective based on preclinical studies. Further scrutiny of the preclinical work on these neuroprotectants revealed that most were tested in models of transient ischemia (Figure 2(a)). As experimental models of transient ischemia typically last for 1 h in mice or 1.5–2 h in rats, the likelihood of successful translation of the preclinical neuroprotectant might be greater in patients who are recanalized with tPa and EVT within these time frames. Preclinical models of permanent cerebral ischemia are much less frequently used, although the vast majority of stroke patients had not experienced significant reperfusion before the recent successes of EVT.
55
For example, uric acid was only tested and proven efficacious in transient injury models—all four preclinical studies adhering to our inclusion criteria consistently found a reduction in brain damage after transient focal ischemia.43,44,56,57 As mentioned earlier, the Phase III clinical trial, URICO-ICTUS, revealed that uric acid combined with intravenous tPA within 4.5 h of stroke onset had no additional salutary effects on functional recovery at 90 days post-injury compared to tPA alone.
46
However, further analyses of the URICO-ICTUS trial suggested that uric acid was more effective than the placebo in reducing infarct volume in patients with early onset recanalization (P = 0.015), but not in patients with delayed recanalization (P = 0.889) or permanent occlusion (P = 0.233).
60
These important observations highlight the importance of accounting for the temporal kinetics of ischemia in both preclinical and clinical work.
Neuroprotectants in preclinical models of transient and permanent ischemia. (a) Preclinical studies of ten neuroprotectants that entered Phase III clinical trials are divided into those employing permanent (pink) or transient (blue) ischemic insults. (b) Neuroprotectants with positive effects in transient models but neutral or negative effects in permanent models are listed.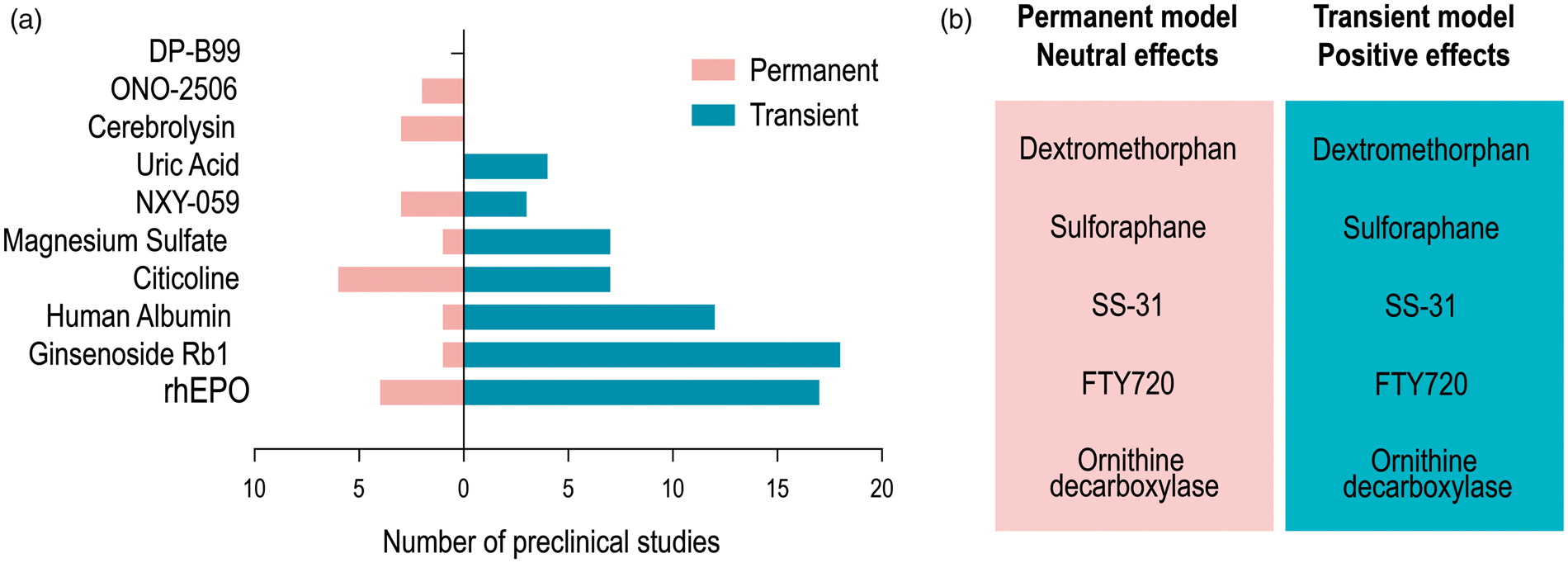
In addition to uric acid, magnesium sulfate,49,59,60 human albumin,51,61,62 and Ginsenoside Rb163–65 were also tested in preclinical models of transient ischemia (Figure 2(a)), and also failed in Phase III trials. Indeed, more than 80% of preclinical studies involved transient ischemic stroke, in contrast to the clinical studies, in which only about one-third of patients exhibited revascularization. 27 This mismatch between preclinical experimental models and human subjects offers one likely explanation for the high rate of failure of neuroprotectants in clinical trials.
A series of preclinical rodent studies employing transient middle cerebral artery occlusion (MCAO) reported that human albumin reduced infarct volumes, ameliorated brain edema, and improved neurobehavioral function.57,61,66,68 Among numerous potential mechanisms, an improvement in collateral perfusion to the ischemic penumbra and regulation of the microcirculatory environment were identified.62,69 These preclinical results led to initiation of a Phase III randomized, double-blinded, placebo-controlled multicenter clinical trial, ALIAS, to examine the potential neuroprotective effects of human albumin on acute ischemic stroke patients. 70 The ALIAS clinical trial included 841 patients across 89 sites in four countries and failed to report any significant difference between patients administered high-dose human albumin treatment versus placebo. Although 89% of the patients enrolled in ALIAS received either intravenous thrombolytic therapy with recombinant tissue plasminogen activator or underwent an endovascular procedure, the clinical report did not include recanalization rates. Administration of thrombolytic therapy is no guarantee of sufficient recanalization. Only 33% of patients with proximal LVO receiving intravenous thrombolysis typically undergo partial or complete early recanalization according to a meta-analysis of 26 studies encompassing 2063 stroke patients. 27 In other words, the majority of patients enrolled in clinical trials involving neuroprotectants do not undergo recanalization. This conclusion is further substantiated by the SAINT-II trial testing NXY-059 as a potential treatment for acute ischemic stroke, as only 44% of 3306 subjects received tPA in this trial. 71 Given that only 33% of patients with proximal LVO who receive tPA achieve successful recanalization, 27 one might estimate that ∼85% of subjects in the SAINT-II trial received NXY-059 without significant reperfusion. In contrast, almost all animals undergo complete recanalization once the suture is removed in experimental models of transient ischemia, revealing a mismatch in the degree of reperfusion achieved in preclinical versus clinical studies of neuroprotection in stroke.
Preclinical studies also support the notion that neuroprotection is reperfusion-dependent because the treatment effect size is typically smaller after permanent ischemia than transient ischemia. 72 For example, dextromethorphan,73,74 sulforaphane,75,76 SS-31, 77 FTY720,78,79 and ornithine decarboxylase80,81 were significantly protective in transient stroke models but failed to elicit any protection against permanent cerebral ischemia (Figure 2(b)). Therefore, recent advances in EVT offer unprecedented opportunities to test neuroprotective therapies that were previously unsuccessful on non-reperfused ischemic tissue. 55 Restoration of blood flow with EVT is likely to improve the delivery of neuroprotectants into ischemic brain tissue. 82 In previous clinical work, the lack of reperfusion may have acted as a bottleneck preventing effective delivery of neuroprotectants to cells in the ischemic penumbra that would be the most dynamically responsive to therapeutic intervention. In light of these arguments, we believe that some of the neuroprotectants that are effective in transient MCAO models may be worth reevaluating clinically in combination with modern EVT.
The efficacy of reperfusion after ischemic stroke is determined not only by recanalization of the occluded vessel, but also by the inherent properties of the collateral circulation. The collateral circulation in the vicinity of the necrotic infarct core may be recruited after vessel occlusion to supply missing nutrients and oxygen to the endangered surrounding tissue and exerts a profound influence over the evolution of the penumbral infarct zone.83,84 If there is insufficient collateral blood flow in stroke patients, the infarct tends to grow in size. 84 It is important to bear in mind that specific features of the collateral circulation in humans, such as number of collaterals, arterial diameter, the anatomical structure of the circle of Willis, and the presence of anterior communicating arteries are not always readily modeled in rodents. 85 Although collateral blood flow may be useful as a prognostic factor, various types of collateral blood flow measurements have been underemployed in preclinical work. A recent comprehensive analysis has demonstrated that 80% of the genetic variation of leptomeningeal collateral anatomy has been linked to a single nucleotide polymorphism of the Rabep2 gene across numerous mouse strains. 86 Therefore, researchers should stratify animals according to natural variations in collateral perfusion to control against intragroup and strain variability. 83
Mismatch II: Treatment effect sizes are determined by infarct volumes in preclinical models but by long-term functional outcomes in clinical trials
Aside from the mismatch between transient ischemia models and the lack of reperfusion in clinical trials of neuroprotection in stroke, another concern is that 70% of the preclinical studies relied on “infarct volume” as the single endpoint for the evaluation of therapeutic effects. In contrast, the effectiveness of a clinical treatment is mainly based on the 90-day modified Rankin score for long-term neurological function.41,87,88 A significant discrepancy between the severity of the clinical deficits and the infarct volumes is not uncommon in human subjects, 89 suggesting that overreliance on infarct volumes in preclinical studies should be avoided. The recent DAWN trial on thrombectomy revealed that acute stroke patients with a mismatch between clinical deficits and infarct volumes, or “clinical-core mismatch,” were excellent candidates for successful EVT irrespective of time of presentation within 24 h after stroke onset. 8 These clinical findings highlight the critical necessity of functional evaluations in preclinical studies, at both acute injury and chronic recovery/repair phases. Furthermore, the preclinical measurements of infarct volume are typically made at the histological level on postmortem tissue, which may be difficult to relate to the neuroimaging that is typically performed in clinical subjects.
Among the 2489 preclinical studies included in our analyses, only 819 evaluated neurological functional outcomes in stroke animals (Figure 3(a)). Even among those that reported neurological function, fewer than 30% of studies monitored functional recovery over extended periods (defined here as >14 days) (Figure 3(a)). Among those neuroprotectants that improved long-term functional recovery in animal studies, only 20 agents transitioned to Phase II or III trials. These agents include 17 beta-estradiol,
90
atorvastatin,
91
cilostazol,
92
erythropoietin,
93
ginsenoside Rd,
94
filgrastim,
95
hydrogen (Recruiting), melatonin (registered), minocycline,
96
NXY-059,
97
human albumin,
98
cerebrolysin,
99
citicoline,
100
propofol (Recruiting), JPI-289 (Recruiting), memantine (Recruiting), 3K3A-APC (NCT02222714, completed), FTY720 (Recruiting), YM872 (NCT00044057, NCT00044070, completed), and edaravone.
101
Thus, more than 80% of the neuroprotectants that elicit improvements in long-term functional outcomes in preclinical studies have never been tested clinically. For example, preclinical evidence support the neuroprotective role of progesterone following acute CNS injuries, including traumatic brain injury and ischemic stroke.102–104 Progesterone significantly reduces infarct volumes and improves long-term functional outcomes in both permanent and transient models of ischemia.105,106 Although the therapeutic potential of progesterone following traumatic brain injury has been tested in two multi-center Phase III clinical trials,107,108 no clinical trials have tested the efficacy of progesterone as a treatment for ischemic stroke.
Neurological functional assessments in preclinical studies of neuroprotectants. (a) Only 32% of the eligible preclinical studies included measurements of neurological functional outcomes in stroke animals. Even among those that reported neurological function, fewer than 30% of studies monitored functional recovery over extended periods (defined here as >14 days). (b) Among the 85 preclinical studies on the ten neuroprotectants that entered Phase III clinical trials, only 39 measured neurological function as an endpoint, and only 12 evaluated neurological functions longer than 14 days after stroke.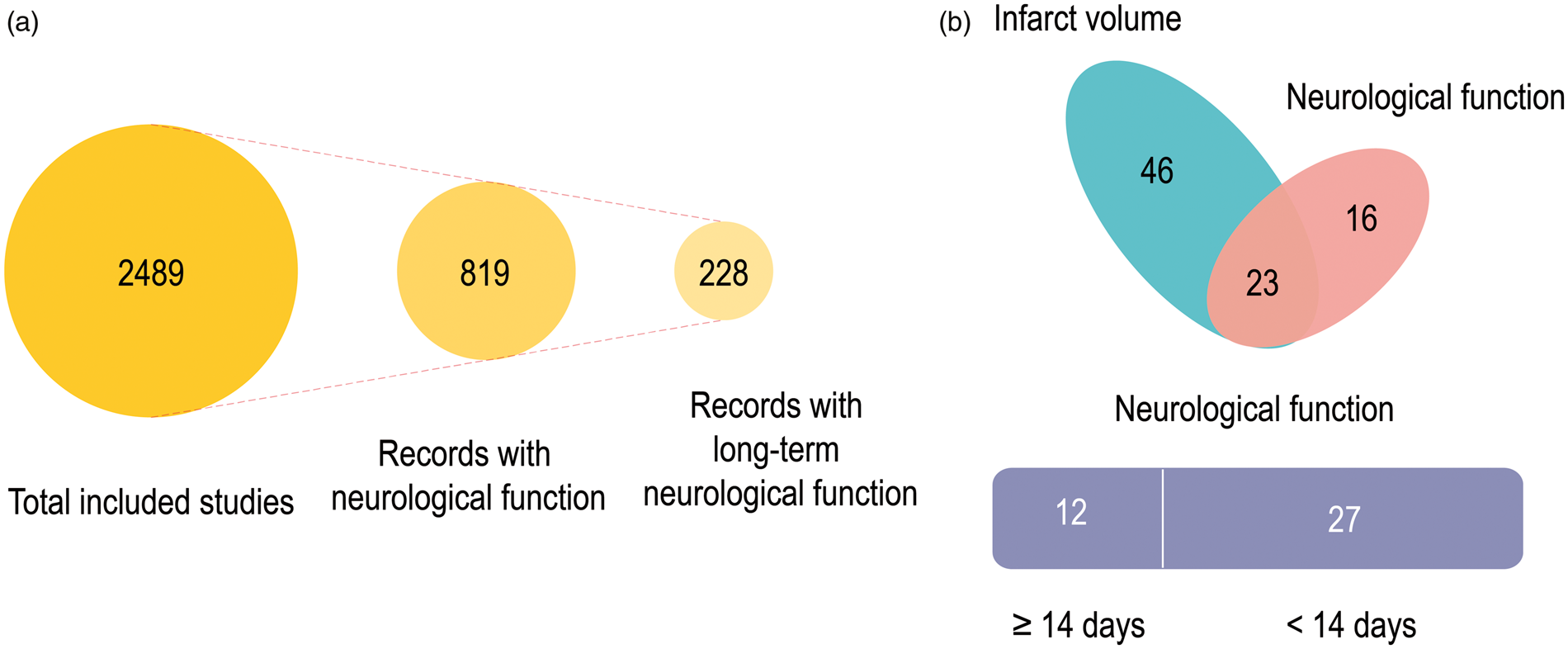
Of the 85 preclinical studies on the 10 neuroprotectants that entered Phase III clinical trials, only 39 measured neurological function as an endpoint, and only 12 evaluated neurological functions longer than 14 days after stroke (Figure 3(b)). For example, DP-B99 is a lipophilic chelator with a moderate affinity for zinc and was observed to exert beneficial effects against ischemic stroke in double-blinded placebo-controlled Phase II trials. 40 However, the Phase III Membrane Activator Chelator Stroke Intervention trial on 446 subjects with acute ischemic stroke reported that only 20.6% patients receiving DP-B99 displayed modified Rankin scores ≤ 1, which was lower than placebo (28.8%). 39 To the best of our knowledge, there are no preclinical studies on the impact of DP-B99 in experimental models of acute ischemic stroke. In addition to DP-B99, the therapeutic candidates ONO-2506, cerebrolysin, and uric acid have also not received sufficient preclinical support. Citicoline has been tested in various ischemic models involving different species, but only one of these studies measured neurological functions longer than 14 days after stroke. 109 Similar concerns apply to some of the other neuroprotectants, including human albumin, magnesium sulfate, and NXY-059. Therefore, it seems worthwhile to focus future clinical tests on those neuroprotectants that achieve significant improvements in long-term functional recovery in preclinical studies conducted by multiple independent laboratories.
Mismatch III: Stroke is most prevalent in elderly men and women, whereas preclinical models mostly test young animals
The third concern raised by our analysis is that the majority of preclinical studies employ young adult animals, although it is well established that the risk for human stroke rises dramatically with age.
110
As shown in Figure 4(a), out of more than 2000 treatments, only two (3K3A-activated protein C
111
and magnesium chloride
112
) have been evaluated in an aged rodent model of permanent ischemia, and nine have been assessed in aged rodent models of transient ischemia. The remarkable differences in stroke pathophysiology and outcomes between young and old animals have been known for many years.
113
Aging is associated with a notable decline in the capacity of the brain to restore cellular and biochemical functions after ischemic stroke.57,113 Preclinical studies indicate that the mortality rate for animals subjected to transient middle cerebral artery occlusion is 6.3% for young adult rats and 43.5% for aged rats.
114
Aging can result in greater oxidative DNA damage and impair neuroprotective responses in the peri-infarct cortex in the early stages after ischemic stroke.
115
Our group previously observed that aged mice exhibited worse long-term learning and memory deficits after experimental stroke than young mice, and this age-related exacerbation of functional outcomes was associated with impaired cerebral perfusion, greater infarct sizes, white matter injury, and dysregulation of pro versus anti-inflammatory microglia.
116
Similar age-related declines in endogenous defenses are also observed in clinical settings. Almost 72% of stroke occurrences are recorded in patients above the age of 65.
117
A recent report from the American Heart Association (2018) indicated that the 30-day mortality rate of patients with ischemic stroke was 9.0% between 65 and 74 years of age, 13.1% between 74 and 84 years of age, and 23.0% in those 85 years of age or older.
118
Aged animals can respond quite differently to treatment regimens than young animals.
119
For example, melatonin prevents neuronal apoptosis by protecting nuclear and mitochondrial DNA damage when administered chronically to aged rats, but increases neuronal apoptosis when administered to younger rats,
120
perhaps because melatonin levels drop with age.
121
Thus, future experimental stroke studies need to account for age-related changes in the brain and test the effects of neuroprotective agents in appropriately aged animals. In preclinical studies, researchers may also need to focus on distal branch occlusion for better translation to elderly humans, whereas in preclinical studies, middle cerebral artery occlusion is commonly employed.
Aging imbalance, therapeutic targets, and publication bias in preclinical studies of ischemic stroke. (a) Out of more than 2,000 treatments, only two have been evaluated in an aged rodent model of permanent ischemia, and nine neuroprotectants have been assessed in aged rodent models of transient ischemia. (b) The potential mechanisms underlying the therapeutic properties of neuroprotectants in both transient and permanent stroke models. More than 80% of the neuroprotectants in the current analyses target inflammation, apoptosis, oxidative stress, neurotrophy, and excitotoxicity. (c) 95% of studies fitting all of our inclusion criteria reported positive outcomes in preclinical models. PISM: permanent ischemic stroke model; TISM: transient ischemic stroke model.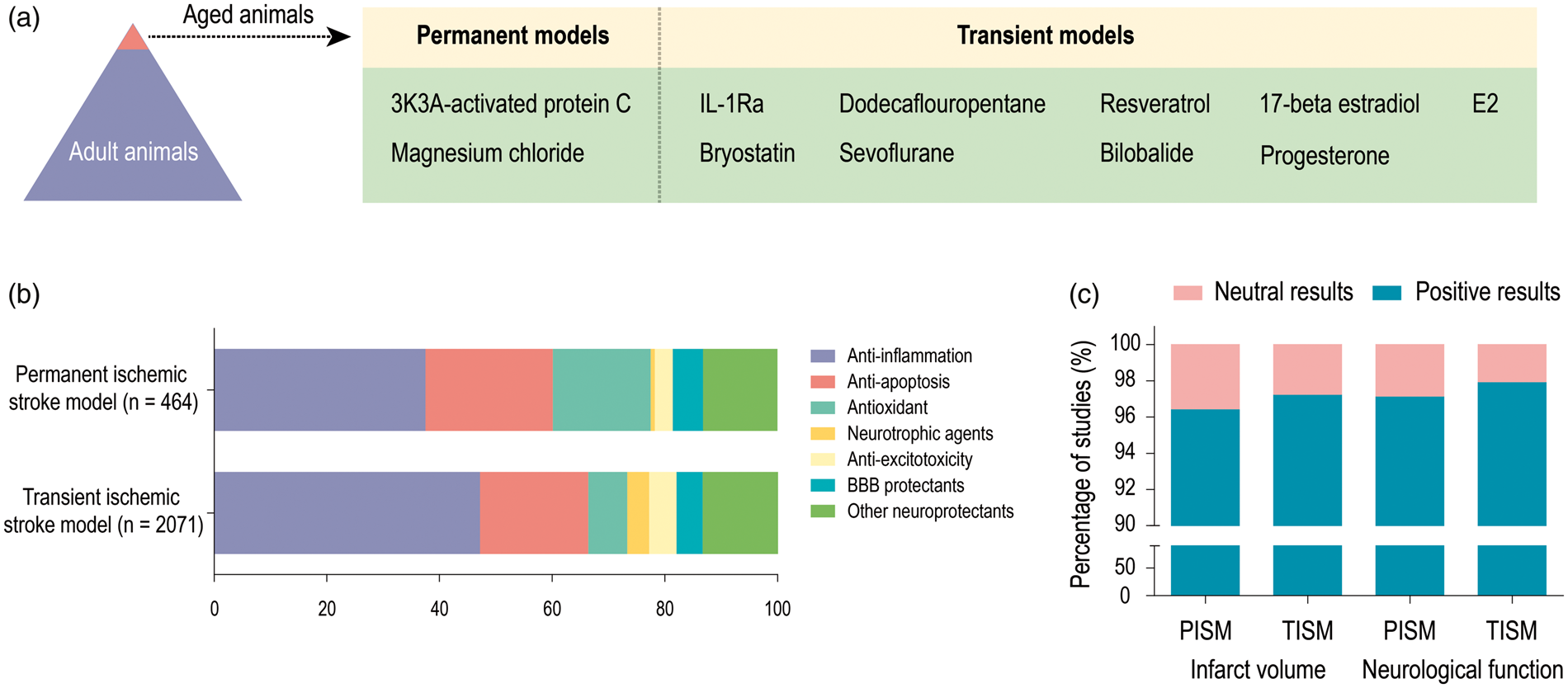
The National Institutes of Neurological Disorders and Stroke reported that ∼55,000 more women are afflicted annually by stroke than men. 122 Women have a lower incidence of ischemic stroke than men when they are younger, but this trend is reversed after 85 years of age. 123 In addition, women have higher post-stroke rates of mortality, disability, depression, and dementia compared to men. 124 Although this has been partially attributed to higher life expectancies in women, this gender disparity remains present even after adjusting for age differences in medical history and presentation. 125 In preclinical studies, young female rodents exhibit less brain injury than males after ischemic stroke. 126 This relative resilience can be abolished by ovariectomy or after reproductive senescence, 127 perhaps because the circulating sex steroid hormones, estrogen and progesterone, are potential neuroprotective factors after ischemic stroke.128,129
In our analysis, only 3% of the preclinical ischemic stroke studies reported the effects of potential neuroprotectants in female animals. Given the gender dimorphisms presented above, it seems necessary to assess neuroprotective strategies in both males and females prior to clinical translation. For example, uric acid failed to achieve any overall improvement in patients with acute ischemic stroke, but it exerted a positive effect on functional recovery in female patients. 46 In the URICO-ICTUS trial, women displayed significantly lower serum uric acid concentrations than men, but their levels rose by 13% after uric acid therapy. 46 Therefore, it seems plausible that uric acid may be more valuable as a therapeutic agent against oxidative stress after ischemic stroke in female patients. Nevertheless, our extensive analysis did not reveal any preclinical studies that assess the effects and potential mechanisms of uric acid in female animals.
Mismatch IV: Stroke is more devastating in patients with multiple comorbidities not captured in preclinical models
As mentioned above, almost 72% of ischemic strokes occur in patients above the age of 65. 117 Serious comorbidities are present in this elderly cohort, including hypertension and diabetes, which are likely to influence the efficacy of neuroprotectants. 130 The INTERSTROKE study estimated that 90% of ischemic strokes arise due to 10 established risk factors, including hypertension, diabetes, high alcohol intake, current smoking habits, poor dietary choices, obesity, physical inactivity, psychosocial stress, and cardiac and lipid abnormalities. 131 Despite these observations, the vast majority of experimental stroke studies have been performed on young and healthy rodents housed under optimal pathogen-free conditions. 132 Of course, no single model can simulate all the comorbidities known to influence stroke outcomes in humans. However, several attempts have been made to devise animal models displaying the most frequent comorbidities, including diabetes133,134 and hypertension.135,136 Approximately 50 preclinical stroke studies have employed hypertensive animals and reported larger infarct volumes compared to normotensive animals. 137 Fewer preclinical investigations have assessed the effect of diabetes on the response to therapeutic interventions for ischemic stroke.138,139 For example, the genetically altered db/db mouse is used as a model of adult onset type-2 diabetes. 140 The db/db mice display larger infarct volumes, increased cerebral edema, and impaired functional recovery following ischemic stroke than db/+ mice. 140 In addition, db/db mice display higher expression of matrix metalloprotease(MMP)-9 than db/+ mice, resulting in increased blood–brain barrier (BBB) permeability, greater infiltration of macrophages and neutrophils into the infarct area, and higher inflammation. 141
A number of issues need to be addressed in order to properly evaluate therapeutic interventions in animals with comorbid diseases, including selection of appropriate animal models. For example, there are important post-stroke sequelae not commonly assessed in animals, such as depression and other psychiatric consequences, 142 obstructive sleep apnea, 143 and immunodepression, which frequently results in fever. 144 Given these complexities, it has been argued by some authors that “experimental models of stroke can cover only individual, specific aspects of this multifaceted disease.” 145 Therefore, it is recommended that the effects of a therapeutic intervention on stroke injury and recovery should first be examined in adult male control and diseased (diabetic, hypertensive, or hyperlipidemic) animals, followed by studies in female and aged animals. 146 Ensuring that therapies are effective in multiple independent models that encompass specific comorbidities may increase the likelihood of clinical translation.
Mismatch V: Therapeutic targets and publication bias in preclinical versus clinical studies
The primary goal of neuroprotectants is to salvage the ischemic penumbra surrounding the infarcted core (Figure 4(b)). More than 80% of the neuroprotectants in the current analyses aid in neurotrophy and target inflammation, apoptosis, oxidative stress, and excitotoxicity (Figure 4(b)). The actions of these neuroprotective agents range from pharmacological blockade of neurotransmitter receptors to interference with cell death pathways. It is well established that one of the first molecular mechanisms underlying ischemic brain damage involves excitotoxicity. 147 Aside from massive and rapid release of the excitatory amino acid glutamate after ischemic stroke, the reuptake of this transmitter is also inhibited, 148 and the subsequent extracellular accumulation of glutamate activates downstream signaling pathways that flood cells with intracellular calcium, leading to activation of proteases and failure of mitochondrial bioenergetics. 148 Numerous pharmacological inhibitors of glutamate receptors have been examined in models of ischemic stroke and in stroke patients, but all failed to show efficacy in clinical trials, including N-methyl-D-aspartate (NMDA) receptor antagonists that reduce glutamate excitotoxicity, such as Selfotel, Aptiganel, and Gavestinel.149,150
NMDA antagonists have been shown to reduce neurogenesis in the recovery phase of ischemic stroke, apart from inhibiting proteasomal function, which can lead to selective neuronal apoptosis. 151 Although NMDA antagonists may alleviate early neuronal injury by suppressing glutamate excitotoxicity in the acute phase, the inhibition of long-term neurogenesis during the chronic recovery/repair phase may counteract the former benefits and contribute to failure of clinical translation. Thus, investigators have begun to target the signaling pathways downstream of glutamate receptor binding. For example, NA-1 is an inhibitor of the postsynaptic density-95 protein (PSD95), which couples NMDA receptor activation to nitric oxide neurotoxicity.152 NA-1 has been shown to halt glutamate excitotoxicity, reduce infarct volume, and improve long-term functional outcomes in rodents and non-human primates after ischemic stroke. 153 Although clinical trials confirmed the safety of NA-1, no significant effects on functional recovery have been achieved. 154 However, the clinical study was geared towards testing neuroprotective effects in patients with stroke following endovascular aneurysmal repair and were therefore unlikely to harbor a salvageable penumbra anyway. Previous studies testing NA-1 in ischemic stroke primarily focused on its ability to protect neurons from excitotoxic damage. 155 Few studies were designed to characterize the influence of NA-1 on neurogenesis. A randomized double-blinded clinical trial, ESCAPE-NA-1 (NCT02930018), is currently ongoing to test the safety and efficacy of NA-1 administration in LVO patients with salvageable penumbral tissue and sufficient collateral circulation who have been treated with EVT.
In addition to the aforementioned issues contributing to the failure to translate preclinically effective treatments into clinical interventions (e.g. the absence of recanalization or functional evaluations, overreliance on young male animals, and a neglect of comorbidities), there have been many other impediments to meaningful progress. For example, neutral or negative results are difficult to publish in major peer-reviewed journals, which may introduce biases into preclinical and clinical research and may therefore influence the conclusions drawn in this report. Surprisingly, 95% of studies fitting all of our inclusion criteria reported positive outcomes in preclinical models (Figure 4(c)). As we do not believe that our search terms or inclusion criteria would have skewed the data dramatically, this degree of bias towards only reporting positive outcomes may be serious. There are too few rewards for publishing neutral or negative data and the resulting publication bias needs to be addressed at the levels of reviewers, editors, publishers, and funding bodies.
Perspectives
The primary function of stroke treatments should be to improve recanalization rates in the early stages after ischemic stroke. Recently, a number of novel thrombolytic drugs (e.g. tenecteplase, desmoteplase) with superior pharmacological profiles compared to alteplase (tPA) have been tested in Phase III trials. 156 Our previous meta-analysis of 819 patients from five clinical trials indicated that desmoteplase administered over an extended time window (3 to 9 h after stroke onset) exerted no significant effects upon functional recovery, but had a favorable safety profile in patients with acute ischemic stroke. 157 Furthermore, greater early-stage (4 to 8 h) reperfusion was observed in the desmoteplase-treated group (47.4%) compared to the placebo-treated group (37.5%). Although tenecteplase was not superior to alteplase in improving recanalization rate in ischemic stroke patients, it was associated with a reduced risk of bleeding. 158 Recent data indicate that tenecteplase is superior to alteplase as a bridging agent for patients with LVO stroke undergoing intravenous thrombolysis in the 0–4.5 h time window prior to intended thrombectomy. 159
Reperfusion therapies with thrombolysis and mechanical thrombectomy are aimed at achieving tissue reperfusion and not simply recanalization. Clinical trials have shown that 70–80% of patients receiving thrombectomy can be successfully reperfused. 7 This area of clinical intervention has great potential for growth. Stroke researchers will need to identify more effective and safer thrombolytic agents and improve the mechanical thrombectomy technique to not only achieve LV recanalization in the largest number of ischemic stroke patients, but also obtain maximum small vessel reperfusion in ischemic brains.
Effective neuroprotectants are needed to alleviate ischemia–reperfusion injury in multiple cell types. Ischemic stroke causes a failure in cellular bioenergetics in the ischemic core, including in neurons, astrocytes and endothelial cells of the BBB. 160 Impairments in the function of the ischemic BBB facilitate the entry of water and pro-inflammatory cytokines into the brain, leading to further activation of inflammatory responses and production of excess free radicals after reperfusion, culminating in the phenomenon of “reperfusion injury.” 160 Furthermore, ischemia and reperfusion injury can both induce hemorrhagic transformation. 160 Hence, drugs that protect the BBB and maintain its integrity might alleviate reperfusion injury and prevent hemorrhagic transformation after complete recanalization.
Neuroprotective candidates should be tested in both transient and permanent ischemia stroke models, and in young and aged animals of both genders before the initiation of a clinical trial. Permanent ischemia models may be superior for the testing of neuroprotectants that can maintain salvageable ischemic penumbra until reperfusion (i.e. bridging therapy), whereas transient ischemia models may be most suited to test reperfusion injury. In addition, preclinical studies are essential for establishing a comprehensive toxicity profile prior to any human exposures, and for identifying and then characterizing the precise molecular mechanism of action, which can accelerate the discovery of additional drug targets. Hypertensive and diabetic animals are particularly valuable in revealing the therapeutic potential of the neuroprotective candidate in more realistic settings of age-related diseases. Furthermore, experimental studies need to focus not only on short-term neuroprotection, which may be transient and only delay injury, but also on long-term repair and recovery processes, including neurogenesis and other structural changes in the brain. Finally, all stroke studies should incorporate long-term assessments of multiple functional outcomes into their study design, particularly those that encompass higher order cognitive functions as well as anxiety and depression. In order to properly compare clinical testing conditions such as Rankin scores to animal research, assessments at 90 days post-injury may also be necessary. Furthermore, live animal imaging may be easier to relate to the functional imaging on live stroke patients rather than relying only on postmortem histological assessments.
Based on our extensive literature analyses, we recommend that combinations of neuroprotectants with thrombolytic drugs or endovascular thrombectomy should be studied as new therapeutic strategies for acute ischemic stroke. Rather than targeting one biological system/pathway or cell type with monotherapies, a multidimensional cocktail approach that includes thrombolytic agents, neuroprotectants, glioprotectants, vasoprotectants, anti-inflammatory drugs, anti-excitatory agents, and antioxidants delivered at both the optimal dose and time after onset of symptoms might improve success rates. The combination drug cocktail approach may also extend the duration of the ischemic penumbra. 161 Other important points to consider are whether the temporal window of tPA or other intravenous thrombolytic agents such as tenecteplase can really be extended without increasing the risk for hemorrhagic transformation or blunting thrombolytic efficacy, but these risks may be mitigated by the concurrent use of neuroprotective interventions such as hypothermia, which is thought to mitigate reperfusion injury.162,163
Conclusions
To summarize, the lack of success of preclinically validated neuroprotectants in clinical trials of stroke may be attributed to: (1) Failure to test the candidate molecule in combination with rapid, substantial reperfusion in patients, as in the experimental models, which suffer from overreliance on transient rather than permanent brain ischemia models; (2) paucity of preclinical studies designed to analyze treatment effects on clinically relevant long-term outcomes; (3) use of healthy young adult male animals instead of aged males and females with diabetes and other conditions; and (4) a publication bias toward reporting statistically significant, positive outcomes in preclinical studies versus large clinical trials.
Previous efforts in facilitating the success of clinical translation have prompted STAIR, STEPS, and RIGOR research guidelines, which have also proposed the use of females, aged animals, long-term functional outcomes, and increased rigor with blinding, power analyses, and appropriate statistical tests. 164 Based on our literature analyses, we also encourage technical development of superior preclinical models that recapitulate permanent ischemia or removal of a thrombus followed by reperfusion. We recognize that these recommendations might involve completing fewer studies due to higher costs and greater animal numbers, but these higher demands are likely to reward biomedical researchers in the long run by accelerating the translation of their benchwork to the bedside and clinic.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: MR is supported by University of Pittsburgh Physician Foundation Research grant and NIH/NINDS grant R01NS097876 (Co-I/Site PI). RKL is supported by NIH/NINDS grants 1R21NS107960-01 (RKL PI), 1R15NS093539-01 (RKL PI), 1R01NS095029-01 (RKL Co-I), and 2R01NS036736-15 (RKL Co-I). JC is supported by NIH/NINDS grants and VA Merit Review Grants. Jun Chen is a recipient of the VA Senior Research Career Scientist Award and the Richard King Mellon Endowed Chair.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: TGJ: Consultant: Cerenovus (Steering Committee – modest), Stryker Neurovascular (PI DAWN – unpaid), Fundacio Ictus (PI REVASCAT/Steering committee RACECAT – unpaid). Stock: FreeOx Biotech, Anaconda, Silk Road, Blockade Medical (modest). RGN: Stryker-Neurovascular (Trevo-2 & DAWN Trial PI), Covidien (SWIFT & SWIFT PRIME/Steering Committee, STAR Trial/ Core Lab), Penumbra (3-D Separator Trial/Executive Committee). The other authors have no conflicts of interest to declare.