
Abstract
The calcium-permeable transient receptor potential M2 (TRPM2) ion channel is activated following oxidative stress and has been implicated in ischemic damage; however, little experimental evidence exists linking TRPM2 channel activation to damage following cerebral ischemia. We directly assessed the involvement of TRPM2 channels in ischemic brain injury using pharmacological inhibitors and short-hairpin RNA (shRNA)-mediated knockdown of TRPM2 expression. Each of the four TRPM2 inhibitors tested provided significant protection to male neurons following in vitro ischemia (oxygen–glucose deprivation, OGD), while having no effect in female neurons. Similarly, TRPM2 knockdown by TRPM2 shRNA resulted in significantly reduced neuronal cell death following OGD only in male neurons. The TRPM2 inhibitor clotrimazole reduced infarct volume in male mice, while having no effect on female infarct volume. Finally, intrastriatal injection of lentivirus expressing shRNA against TRPM2 resulted in significantly smaller striatal infarcts only in male mice following middle cerebral artery occlusion, having no significant effect in female mice. Data presented in the current study demonstrate that TRPM2 inhibition and knockdown preferentially protects male neurons and brain against ischemia in vitro and in vivo, indicating that TRPM2 inhibitors may provide a new therapeutic approach to the treatment of stroke in men.
Introduction
Stroke is a leading cause of death and disability in the United States and a great deal of experimental data have revealed several interrelated mechanisms of ischemic damage, including excitotoxicity, oxidative stress, apoptosis, and necrosis. Excessive rise in intracellular calcium appears to be a common trigger shared by these distinct cell death pathways. Therefore, research has focused on calcium permeation pathways engaged following ischemia, most notably N-methyl-
The calcium-permeable TRPM2 ion channel was first described and characterized, before cloning of the gene, as an NAD+-activated nonselective cation (NSNAD) channel activated by hydrogen peroxide (H2O2)-induced oxidative stress (Herson and Ashford, 1997; Herson et al, 1999; Smith et al, 2003). Identification of the TRPM2 gene led to confirmation that TRPM2 is activated by H2O2-induced oxidative stress and gated directly by intracellular adenine dinucleotide phosphate ribose (ADPr). Indeed, the most well-characterized role for TRPM2 is as an executioner of cell death following oxidative stress (for review see McNulty and Fonfria, 2005; Eisfeld and Luckhoff, 2007). Expression in sensitive neuronal populations, coupled with sensitivity to oxidative stress, provides a strong rationale to hypothesize that TRPM2 channels have a significant role in a variety of neurodegenerative diseases whose etiology involve oxidative stress, including ischemic stroke. However, little experimental evidence exists linking TRPM2 channel activation to neuronal damage following cerebral ischemia. In the current study, we directly tested the hypothesis that TRPM2 channels are involved in ischemic injury using pharmacological inhibitors and lentivirus expressing short-hairpin RNA (shRNA) to knockdown expression of TRPM2. Our data show that TRPM2 channel involvement in ischemic outcome is male specific, not contributing to damage in female neurons or brain following ischemia.
Materials and methods
Experimental Animals
All experimental protocols were approved by the Institutional Animal Care and Use Committee at Oregon Health and Science University and conformed to the National Institutes of Health guidelines for the care and use of animals in research. All middle cerebral artery occlusion (MCAO) experiments were performed in a blinded randomized manner using male and female C57Bl/6 mice weighing 20 to 25 g. Culture experiments were performed on embryos obtained from pregnant C57Bl/6 mice on embryonic day 17 (E17).
Primary Cell Culture
Experiments were performed on sex-stratified mouse cortical neuronal cultures. Cortices were dissected from E17 C57BL/6 mice and embryos sexed as described previously (Ardeshiri et al, 2006). Briefly, E17 embryos were rapidly removed from timed pregnant mice under deep isoflurane anesthesia. The sex of each embryo was identified by laparotomy to inspect gonads and internal organs: uterine horns and ovaries in females and testes in males. The cortical tissue was dissected and placed in ice-cold dissection buffer (Hank's-buffered salt). The isolated cortices were then digested with papain (20 μg/mL; Worthington Biochemical, Lakewood, NJ, USA) and halted by addition of trypsin inhibitor (Sigma-Aldrich, St Louis, MO, USA). The digested pieces were triturated and filtered through cell sorting nylon mesh (70 μm; BD Biosciences, Bedford, MA, USA). Cell suspension was pelleted by centrifugation at 1,000 g and resuspended in neurobasal without Phenol red+B27 supplement (Invitrogen, Carlsbad, CA, USA). Cells were plated at a concentration of 2.5 × 105 cells per well (24-well plate) coated with poly-
Oxygen–Glucose Deprivation and Drugs
In vitro ischemia was induced by transferring cells after 10 to 11 days in vitro to glucose-free neurobasal-A medium and placing them into an anaerobic incubator for 2 hours. The anaerobic incubator contained an oxygen reacting catalyst to ensure the presence of <5 p.p.m. O2, kept at 37°C with 5% CO2 and 95% N2 (Coy Laboratory Products, Grass Lake, MI, USA). Cells were treated with maximal concentrations of four different compounds known to inhibit TRPM2 channels (Harteneck, 2005), N-(p-amylcinnamoyl)anthranilic acid (Kraft et al, 2006), 2-aminoethoxy diphenyl borate (Togashi et al, 2008), clotrimazole (CTZ) (Hill et al, 2004b), and flufenamic acid (Hill et al, 2004a) 15 minutes before oxygen–glucose deprivation (OGD) and maintained throughout OGD and reoxygenation. Reoxygenation was initiated by transferring cells to culture media in aerobic incubator and cell viability was determined 24 hours after initiation of reoxygenation (see below).
Neuronal Cell Death
Cortical neurons exposed to 2 hours OGD and 24 hours reoxygenation were assayed for cell death using the MTT assay. The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay is a standard colorimetric assay for measuring the activity of mitochondrial enzymes that reduce MTT to formazan, producing a purple color. After OGD, MTT was added to cells for 1 hour (20 μL/well). After 1 hour incubation, formazan accumulated in living cells was dissolved in 200 μL DMSO. The formazan was quantified by measuring optical density at 540 nm with microplate reader (Victor3, Perkin-Elmer, Waltham, MA, USA). MTT assay provides a value for control cells (100% survival), and we obtained cell death values by subtracting experimental numbers from control values to get percentage cell death.
Lentivirus Generation, Neuronal Infection, and Intrastriatal Microinjection
Third generation pseudotype lentivirus expressing shRNA and/or green fluorescent protein (GFP) was generated as previously described (Cheng et al, 2011). Probe sequence for shRNA targeted to the coding sequence of TRPM2 was: TTTGGCTCATGGATTCCCGAGAATACTCGAGTATTCTCGGGAATCCATGAGCTTTTTG.
Primary cortical neurons were infected with lentivirus by adding 3 to 5 μL of high titer virus (106 to 108 TU/mL) and confirmed by fluorescent microscopic observation of GFP-positive neurons (95% to 100% infection efficiency) before exposure to OGD. Knockdown of TRPM2 expression was confirmed by quantitative real-time polymerase chain reaction on ABI Prism 7000 using commercially available probe and primers (Applied Biosystems, Foster City, CA, USA) and standard Western blot analysis as previously described (Kelley et al, 2008), using anti-TRPM2 antibody from Novus Biologicals. Intrastriatal injection of lentivirus into the right striatum (0.5 mm anterior, 2.0 mm lateral, 3.5 mm deep) was performed under isoflurane anesthesia 2 to 3 weeks before MCAO, as previously described (Cheng et al, 2011). The morphology of striatum makes it ideal for delivery of virus, as the white matter (external capsule) surrounding the striatum provides containment of virus and striatum's consistent ischemic exposure via the MCAO model.
Middle Cerebral Artery Occlusion and Infarct Analysis
Methods are as previously published in mouse (Vagnerova et al, 2010). Cerebral ischemia was induced by 90 minutes reversible MCAO via intraluminal suture technique under isoflurane anesthesia. Adequacy of MCAO was confirmed by laser Doppler flowmetry measured over ipsilateral parietal cortex. All 87 mice had similar levels of occlusion throughout, reduced by >80% from baseline (see Supplementary Table 1). Physiological measurements were performed in separate groups of mice treated with vehicle or CTZ, by femoral arterial catheter to measure blood pressure and obtain blood gas measurements. At 24 hours reperfusion, brains were harvested for infarct volume analysis and immunohistochemistry. Clotrimazole (30 mg/kg) was administered via two subcutaneous injections (100 μL/10 g body weight) 5 minutes after occlusion and at the time of reperfusion. For CTZ-treated mice, brains were sliced into 2 mm thick coronal sections for staining with 2% triphenyltetrazolium chloride in saline (Vagnerova et al, 2010). For virus-injected brains, 6 μm sections through the entire striatum (500 μm apart) were stained with cresyl violet and infarct volume was calculated (Hattori et al, 2000). Infarct volume was measured by a separate blinded investigator using digital imaging and image analysis software. Infarct volumes of striatum, cortex (predominantly neocortex), and total hemisphere were measured separately. For immunohistochemistry, 6 μm sections were incubated at 4°C overnight in rabbit anti-GFP polyclonal antibody and mouse monoclonal anti-NeuN and visualized with fluorescent microscopy. Neuronal viability was measured in striatal sections at three levels (100 μm apart) by a blinded investigator by counting the number of viable NeuN-positive, GFP-positive neurons in the entire striatum of each section. Viable neurons were identified by larger oval to round nuclei while nonviable neurons exhibited shrunken, angular nuclei. The number of viable neurons was expressed as the average of three levels for each animal.
Statistical Analysis
All data are presented as mean±s.e.m. Each n represents an individual culture for in vitro experiments and an individual animal for in vivo experiments. All experiments were performed in a randomized blinded manner, with analysis and surgery performed by separate investigators. Statistical significance was determined using Student's t-test (unpaired, two-tailed, if P<0.05) for two groups and one-way analysis of variance with Newman–Keuls post hoc analysis for multiple groups. Two-way analysis of variance with Newman–Keuls post hoc analysis was used to determine influence of TRPM2 inhibitors and sex on neuronal viability. Statistical significance was set at P<0.05.
Results
Transient Receptor Potential M2 Inhibitors Protect Male but not Female Neurons Against Oxygen–Glucose Deprivation-Induced Cell Damage
To test the hypothesis that TRPM2 is involved in sex-specific neuronal cell death, neurons from male and female embryos were cultured separately in sex steroid-free medium. All four TRPM2 inhibitors tested significantly decreased neuronal cell death following OGD in male cortical neurons (Figure 1). In contrast, none of the inhibitors significantly altered survival of female cortical neurons following OGD (Figure 1), revealing that sex significantly interacts with treatment (two-way analysis of variance, P<0.05).
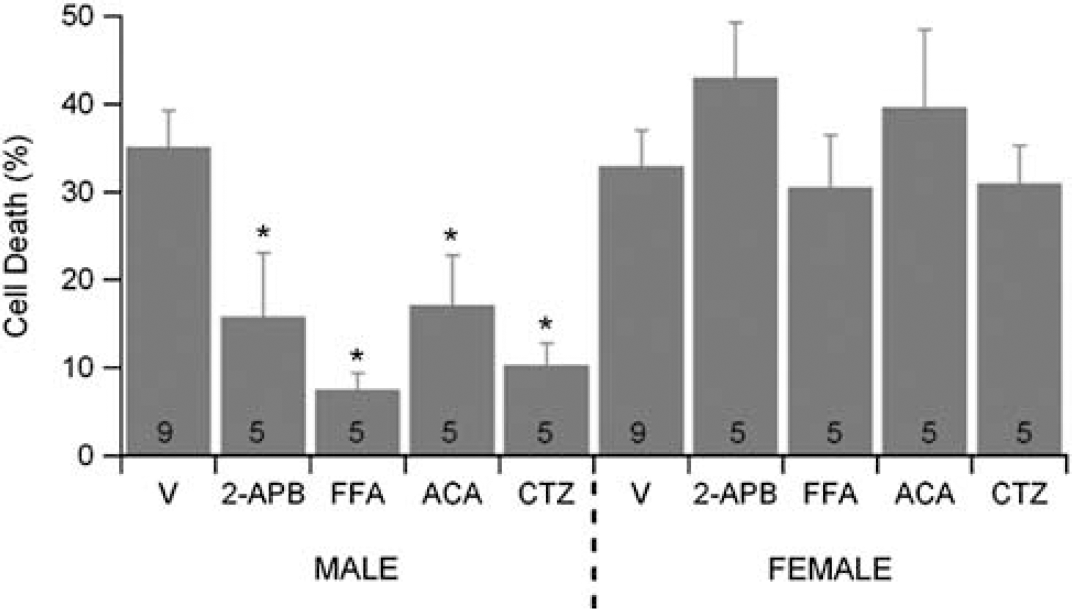
Transient receptor potential M2 (TRPM2) inhibition decrease oxygen–glucose deprivation (OGD)-induced cortical cell death in male neurons preferentially. Quantification of cell death 24 hours after 2 hours OGD in male and female cortical cultures. Neurons were exposed to each inhibitor 15 minutes before OGD and inhibitor maintained throughout OGD and reoxygenation. Data presented are mean±s.e.m. The numbers in each bar represent number of experiments and ∗ indicates P<0.05 compared with vehicle-treated cultures of same sex. ACA, N-(p-amylcinnamoyl)anthranilic acid; CTZ, clotrimazole; FFA, flufenamic acid; 2APB, 2-aminoethoxy diphenyl borate.
Transient Receptor Potential M2 Knockdown Protects Male Cortical Neurons from Oxygen–Glucose Deprivation-Induced Cell Death
Lentiviral vectors expressing shRNA targeted against the coding region of mouse TRPM2 were observed to significantly decrease expression in male and female neurons to a similar extent, reducing to 22.5%±4.1% (n=4) and 23.7%±3.8% (n=3) of control, respectively (Figure 2A). Empty virus had no effect on TRPM2 expression. Importantly, TRPM2-specific shRNA did not alter mRNA levels of the closely related protein TRPM7 in male or female neurons (data not shown). Western blot analysis confirmed efficient knockdown of TRPM2 protein following infection with shRNA89 expressing lentivirus in male and female neurons, reducing to ∼45% of control levels in both sexes (Figures 2B and 2C). Figure 2D illustrates that infection with shRNA producing lentivirus significantly reduced male cortical neuron cell death after OGD compared with neurons infected with control virus (n=5, P<0.05). In contrast, TRPM2 knockdown had no significant effect on survival of female cortical neurons following OGD. Neither virus altered cell survival in control neurons not exposed to OGD (data not shown).
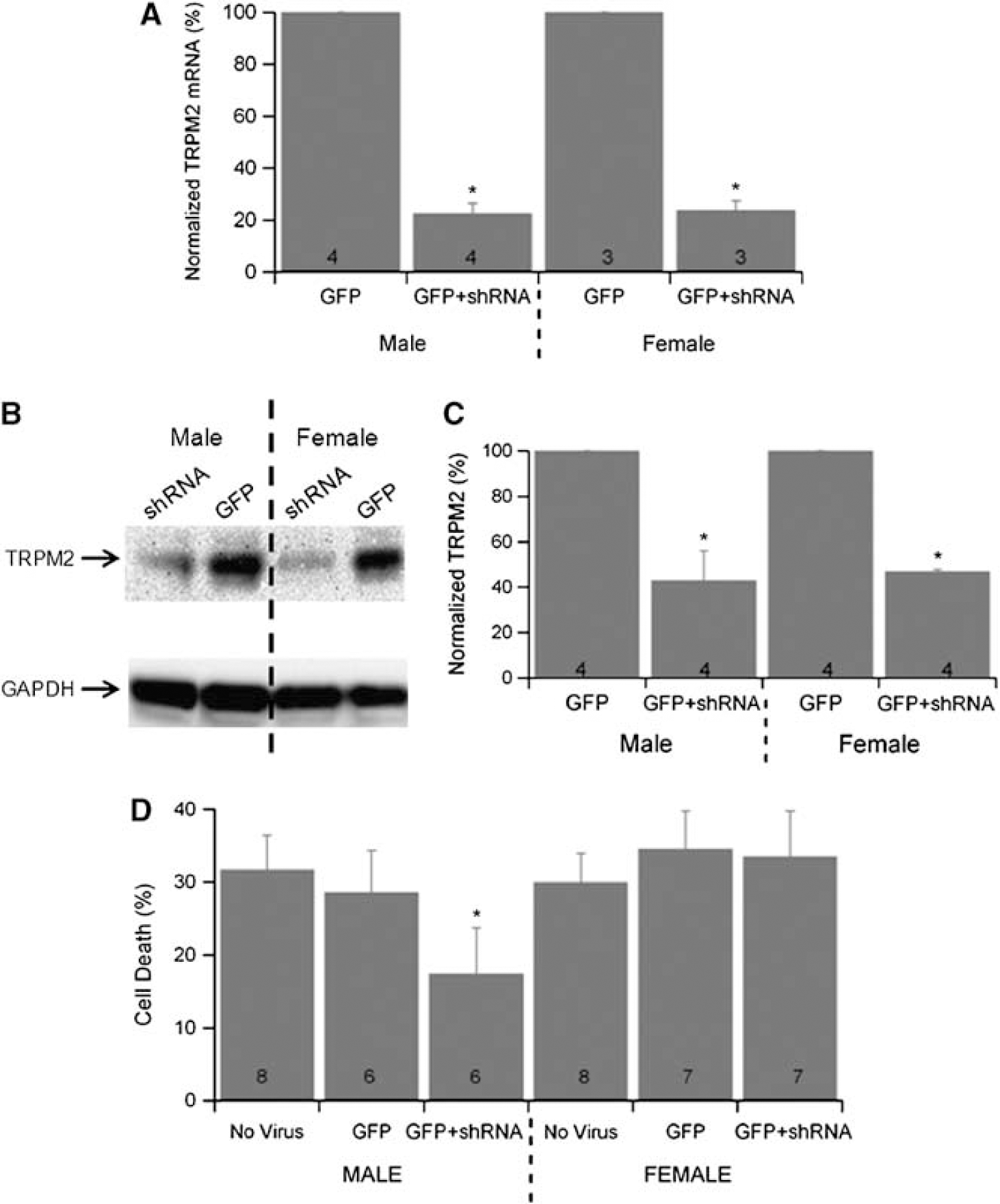
Transient receptor potential M2 (TRPM2) knockdown decreases cortical cell death in male neurons preferentially. (
Effect of Transient Receptor Potential M2 Channel Inhibitor Clotrimazole on Infarct Volumes in Male and Female Mice
Male and female mice were subjected to 90 minutes MCAO and infarct volume of cortex, striatum, and total hemisphere was analyzed. The TRPM2 inhibitor CTZ (30 mg/kg) was injected during ischemia and infarct volume was analyzed 24 hours after reperfusion. All mice had similar levels of occlusion throughout (Supplementary Table 1). Two mice were excluded for cerebral hemorrhage (one CTZ and one vehicle male) and two vehicle-treated male mice were excluded due to death before 24 hours time point. Male mice treated with CTZ had smaller total infarct volumes compared with vehicle-treated male mice (Figure 3; n=10, P<0.05). Similar findings were observed in cortical and striatal infarct volumes (Figure 3). In contrast, in female mice, CTZ had no effect on total, cortical, or striatal infarct volumes (Figure 3; n=10). To evaluate the physiological effects of CTZ, we measured blood pressure and arterial blood gases during ischemia and 30 minutes after reperfusion. There were no differences between sexes or treatments; all remaining within physiological limits (Table 1). In addition, intraocclusion cortical cerebral blood flow measured by laser Doppler flowmetry was similar in all groups.
Physiological parameters in male mice injected with vehicle or clotrimazole at 45 minutes of MCAO and 30 minutes after reperfusion (n=4 per group)
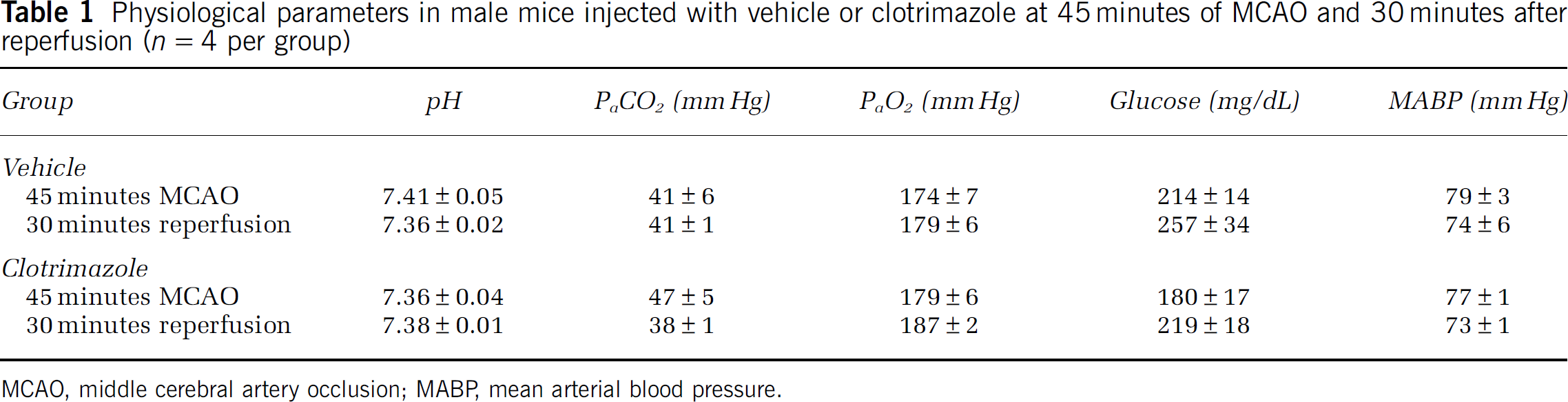
MCAO, middle cerebral artery occlusion; MABP, mean arterial blood pressure.
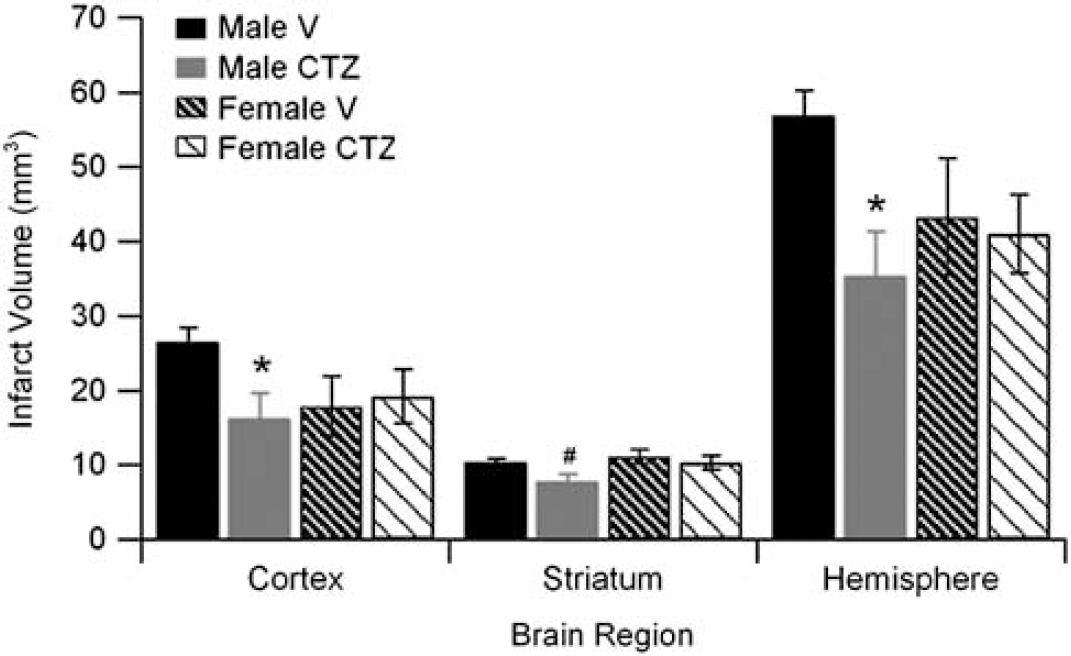
Clotrimazole (CTZ) reduces infarct volume in male brain. Quantification of infarct volume in vehicle- and CTZ-treated male and female mice (n=9 to 12 per group, mean±s.e.m.). ∗Different than vehicle, P<0.05.
Effect of Lentiviral-Mediated Transient Receptor Potential M2 Knockdown on Infarct Volume and Neuronal Survival in Male Mice
To confirm the role of TRPM2 in ischemic damage, we evaluated the effect of TRPM2 knockdown on ischemic outcome. Male and female mice were injected with high titer virus expressing shRNA targeted to TRPM2 (or control GFP virus). Supplementary Figure 1 illustrates our ability to infect a large portion of the striatum with lentivirus. Qualitative observation of several male and female mice injected with lentivirus did not reveal obvious sex differences in the number of neurons infected with virus. All mice had similar level of occlusion throughout (Supplementary Table 1). Two male virus (one control GFP and one GFP+shRNA)-injected mice were excluded for cerebral hemorrhage and three mice were excluded due to death before 24 hours time point (one shRNA-injected male, one shRNA-injected female, one GFP-injected female). Male mice treated with shRNA expressing lentivirus had significantly smaller striatal infarct volumes than control virus-injected male mice (Figures 4A–4C; n=9 in control and n=12 in shRNA, P<0.05). In contrast, no significant effect of cortical infarct volume was observed; 14.0±1.9 mm3 (56.6%±6.9%) in control virus and 11.7±1.2 mm3 (39.8%±5.8%) in shRNA. Lentivirus expressing shRNA had no effect on striatal infarct volumes in female mice (Figure 4C). Similarly, no effect was observed in female cortical infarct volumes; 14.5±1.6 mm3 (57.3%±3.8%) in control virus and 10.2±2.6 mm3 (39.5%±8.9%) in shRNA.
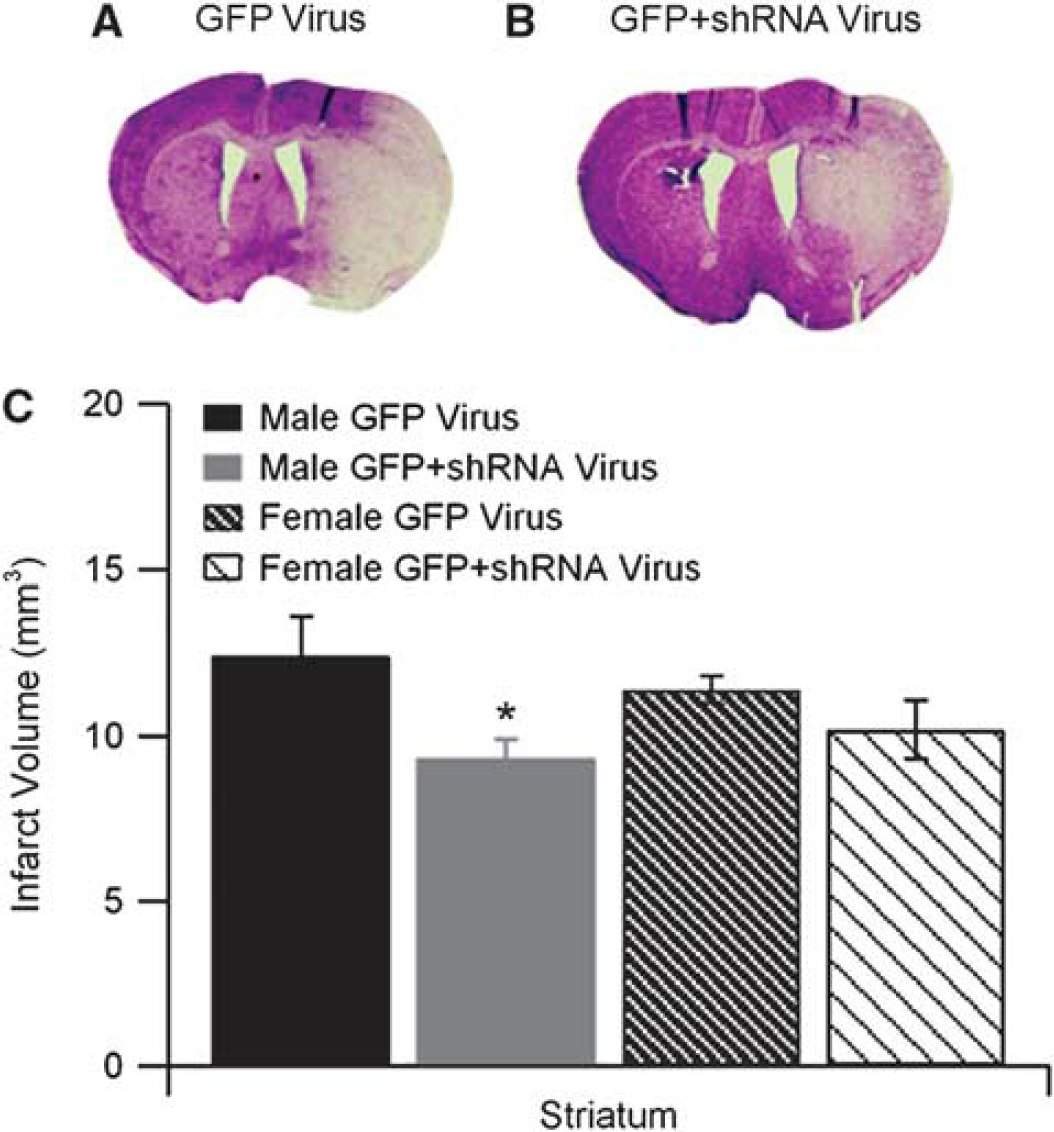
Short-hairpin RNA (shRNA) expressing lentivirus reduces infarct volume in male brain. Representative cresyl violet-stained brain slices from green fluorescent protein (GFP) (
To observe neuronal cytoprotection following TRPM2 knockdown in male mice, sections of striatum were double labeled with anti-GFP antibody (green) to detect infected cells and anti-NeuN antibody (red) to assess neuronal infection and survival. Male mice injected with control GFP virus had very few surviving GFP expressing neurons in the striatum (Figure 5A), consistent with loss of membrane integrity and leakage of cytosolic GFP, as described recently (Sun et al, 2009). In contrast, striatum injected with GFP+shRNA virus had significantly greater numbers of GFP-labeled neurons (Figures 5B and 5C), indicative of cytoprotection provided by TRPM2 channel knockdown. Consistent with the infarct volume analysis, female striatum injected with either GFP or GFP+shRNA virus had very few surviving neurons (Figure 5C).
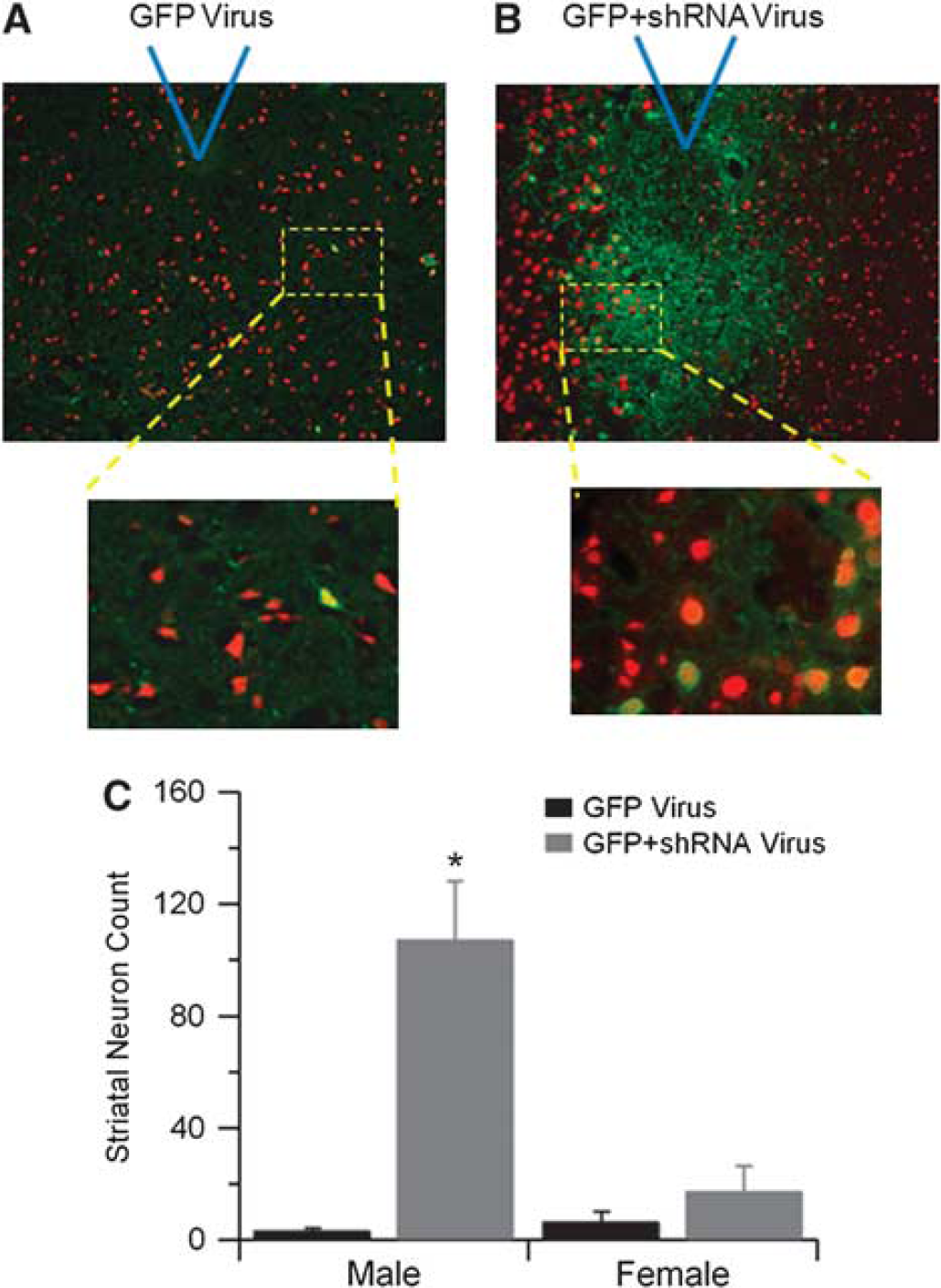
Short-hairpin RNA (shRNA) expressing lentivirus increases neuronal survival in male brain. Representative fluorescent micrographs of brain slices labeled with anti-green fluorescent protein (GFP) (green) to identify viral infected cells and double labeled with anti-NeuN (red) to identify neurons in slices from GFP (
Discussion
The results presented show that TRPM2 channel activity contributes to ischemic injury selectively in the male brain following experimental stroke. Further, we show that inhibition or genetic knockdown of TRPM2 channels decreases neuronal damage following in vitro ischemia (OGD) in male neurons, while having no effect on female neurons. This is a novel example of an important neuronal death mechanism that is engaged in a sexually dimorphic manner. Thus, inhibition of TRPM2 channels represents a potential therapeutic strategy to improve outcome following stroke in men.
To our knowledge, we are the first to directly test the hypothesis that TRPM2 channels contribute to ischemia-induced neuronal cell death in vitro and in vivo. It is interesting to note, however, that a role for TRPM2 in ischemia has been proposed in multiple review articles (MacDonald and Jackson, 2007; Simard et al, 2007; Szydlowska and Tymianski, 2010), due to their sensitivity to oxidative stress. Indeed, several studies have demonstrated a role for TRPM2 in mediating neuronal cell death using standard mixed-sex cultures, reporting neuroprotection with inhibition or silencing of TRPM2 channels following H2O2-induced oxidative stress in hippocampal (Lipski et al, 2006), striatal (Hill et al, 2006), and cortical neurons (Kaneko et al, 2006). Interestingly, we observe a sex-specific role for TRPM2 in neuronal response to in vitro ischemia (OGD) using TRPM2 inhibitors or shRNA-mediated knockdown of TRPM2. Similarly, we observe that TRPM2 inhibition with CTZ and shRNA virus decreased infarct size in male brain following experimental ischemia, while having no effect in female. Consistent with previous reports, we observed that females have smaller infarcts compared with males. This is unlikely to explain the lack of effect observed in females because it remains possible to provide further protection, as others have demonstrated protection in intact females with various compounds (Liu et al, 2009). Our observation that TRPM2 channels contribute to male ischemic outcome, but not to the female, may be linked to several different factors. Transient receptor potential M2 channels are activated by increased levels of intracellular ADPribose (ADPr) and/or by oxidative stress. Therefore, one possibility is that ischemia results in larger generation of ADPr in males versus females. In fact, others have shown that male cells are highly vulnerable to excessive oxidant production and subsequent over-activation of poly (ADPribose) polymerase (PARP), whereas female cell death involves caspase-dependent apoptosis (for review see Lang and McCullough, 2008; Herson and Hurn, 2010). Thus, male-specific over-activation of PARP may be key because PARP-generated ADPr activates TRPM2 following neuronal exposure to exogenous H2O2 (Fonfria et al, 2004), while TRPM2 channels may also be activated directly by H2O2 (Kolisek et al, 2005). Therefore, it is possible that TRPM2 is downstream of PARP activation following cerebral ischemia while also a sensor of oxidative stress experienced during reperfusion. In addition, several other enzymes are capable of metabolizing NAD+, including CD38, mono-ADP-ribosyl transferases, and ADPribose cyclases (for review see Hassa et al, 2006) to produce ADPr, which may contribute to postischemic TRPM2 channel activation. Similarly, it has recently been demonstrated that alternative metabolites of NAD+ stimulate TRPM2 channel activity, including cADPr (Kolisek et al, 2005) and O-acetyl-ADPr (Grubisha et al, 2006). Therefore, these alternative metabolic pathways may contribute to TRPM2 channel activation and male-specific response to ischemia. As such, TRPM2 channels may represent a common mediator of damage following perturbation of various metabolic pathways.
Unfortunately, no selective TRPM2 inhibitor has yet been described (Harteneck, 2005), making it difficult to conclusively link TRPM2 channel activity to neuronal viability following ischemia. Therefore, we used several inhibitors (N-(p-amylcinnamoyl)anthranilic acid, 2-aminoethoxy diphenyl borate, flufenamic acid, and CTZ) to demonstrate TRPM2 channel activation and OGD-induced neuronal cell death. Each compound was tested at a concentration known to maximally inhibit TRPM2 channel activity. Each compound has a different spectrum of nonoverlapping ‘off-target’ effects; therefore, we attribute comparable effects of these diverse inhibitors to their inhibition of TRPM2 channels. For example, 2-aminoethoxy diphenyl borate was initially characterized as an antagonist of the inositol 1,4,5-triphosphate (InsP3) receptor (Maruyama et al, 1997) and more recently has been reported to inhibit several members of the TRPC and TRPM families, including TRPC1,3,5,6 and TRPM2,3,7,8 (for review see Ramsey et al, 2006). N-(p-amylcinnamoyl)anthranilic acid is a well-characterized phospholipase A2 antagonist (Konrad et al, 1992; Harteneck et al, 2007), with minimal TRP channel targets, inhibiting only TRPC6, TRPM2, and TRPM8 (Harteneck et al, 2007). Importantly, CTZ has not been demonstrated to inhibit any TRP channels other than TRPM2, making this the most promising compound to proceed to in vivo studies. Our data in cultured cortical neurons demonstrate that each of these disparate TRPM2 inhibitors protect male neurons from OGD-induced cell death without altering outcome in female neurons. Similarly, we observe that CTZ provides male-specific protection against experimental stroke in intact animals. It remains a possibility that CTZ neuroprotection is mediated due to off-target effects. We consider this unlikely because the most well-characterized target for CTZ is the intermediate-conductance Ca2+-activated potassium channel (IKCa), which underlies a portion of endothelium-dependent vasodilation in brain arterioles and is likely a compensatory protective mechanism following ischemia (Cipolla et al, 2009), and therefore would be predicted to exacerbate damage. The consistent effect observed with each separate inhibitor in our in vitro experiments and CTZ in vivo, coupled with the effects of TRPM2 knockdown in vitro and in vivo are strong evidence that TRPM2 contributes to male-specific ischemic sensitivity.
Importantly, TRPM2 inhibition and knockdown was able to protect both male-specific neuronal cultures and increase neuronal survival in the brain of intact male animals, implicating neuronal TRPM2 as an important mechanism in experimental stroke. However, TRPM2 is expressed in other relevant cell types, including endothelial cells (Hecquet et al, 2008) and microglia (Kraft et al, 2004), as well as various peripheral immune cells, including macrophages and T cells (Sano et al, 2001; Gasser et al, 2006; Yamamoto et al, 2008). Therefore, the protective effect of pharmacological inhibition of TRPM2 with CTZ might be multicellular in origin and impact numerous cell types within the central nervous system. For example, increased TRPM2 expression has been demonstrated weeks after focal cerebral ischemia, and implicated microglial TRPM2 in outcome (Fonfria et al, 2006). While our data do not directly exclude inflammatory cell TRPM2 origin, we chose to analyze stroke damage early in the maturation process, that is 24 hours post-MCAO, at a time when relatively few peripheral immune cells have invaded the injured brain (Gelderblom et al, 2009). Lentiviral-mediated knockdown of TRPM2 decreased infarct volume and increased surviving neuron number, indicating that neuronal TRPM2 contributes to ischemic outcome. The choice of analyzing an early time point addresses our hypothesis of neuronal involvement; however, it remains important to interpret cautiously as it is possible that TRPM2 slows the rate of infarction rather than final infarct volume per se. Further work is warranted to elucidate the relative contribution of TRPM2 channels expressed in various cell types on ischemic outcome.
The current study provides the first evidence that TRPM2 channels contribute to ischemic damage, demonstrating that pharmacological inhibition and genetic knockdown of TRPM2 decrease damage preferentially in the male brain following experimental stroke. In summary, the data suggest that neuronal TRPM2 channels have a role in the progression of ischemic damage following stroke and may provide a new therapeutic approach to the treatment of stroke in men.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.