
Abstract
Tauroursodeoxycholic acid (TUDCA), a hydrophilic bile acid, is a strong modulator of apoptosis in both hepatic and nonhepatic cells, and appears to function by inhibiting mitochondrial membrane perturbation. Excitotoxicity, metabolic compromise, and oxidative stress are major determinants of cell death after brain ischemia-reperfusion injury. However, some neurons undergo delayed cell death that is characteristic of apoptosis. Therefore, the authors examined whether TUDCA could reduce the injury associated with acute stroke in a well-characterized model of transient focal cerebral ischemia. Their model of middle cerebral artery occlusion resulted in marked cell death with prominent terminal deoxynucleotidyl transferase-mediated 2′-deoxyuridine 5′-triphosphate-biotin nick end labeling (TUNEL) within the ischemic penumbra, mitochondrial swelling, and caspase activation. Tauroursodeoxycholic acid administered 1 hour after ischemia resulted in significantly increased bile acid levels in the brain, improved neurologic function, and an approximately 50% reduction in infarct size 2 and 7 days after reperfusion. In addition, TUDCA significantly reduced the number of TUNEL-positive brain cells, mitochondrial swelling, and partially inhibited caspase-3 processing and substrate cleavage. These findings suggest that the mechanism for in vivo neuroprotection by TUDCA is, in part, mediated by inhibition of mitochondrial perturbation and subsequent caspase activation leading to apoptotic cell death. Thus, TUDCA, a clinically safe molecule, may be useful in the treatment of stroke and possibly other apoptosis-associated acute and chronic injuries to the brain.
Ursodeoxycholic acid (UDCA) is a hydrophilic bile acid that has been shown to improve clinical and biochemical indices in a wide variety of liver diseases (Beuers et al., 1998). After oral administration, UDCA is rapidly conjugated with glycine or taurine to form glycoursodeoxycholic acid or tauroursodeoxycholic acid (TUDCA), respectively. Despite its clinical efficacy, the cytoprotective mechanisms by which UDCA improves liver function are not entirely understood. However, we have recently shown that UDCA and its conjugated derivatives play a unique role in modulating the apoptotic threshold in both hepatic and nonhepatic cells, including neuronal cells (Rodrigues et al., 1998a, 2000a,b). These molecules interact with the mitochondrial membrane to prevent several apoptotic events, including production of reactive oxygen species, translocation of the proapoptotic protein Bax, release of cytochrome c, caspase activation, and cleavage of the nuclear enzyme poly(ADP-ribose) polymerase (PARP) (Rodrigues et al., 1998b, 1999, 2000a). Finally, we have recently shown that TUDCA is neuroprotective in a 3-nitropropionic model of Huntington disease (Keene et al., 2001).
The mechanisms of neuronal cell death from acute stroke are complex and appear to involve the interplay of a number of factors, including oxidative stress, excitotoxicity, and activation of certain cell-cycle genes (Osuga et al., 2000). In addition, cerebral ischemia followed by reperfusion usually results in delayed neuronal cell death that is characteristic of apoptosis (Linnik et al., 1993; MacManus et al., 1993; Kihara et al., 1994; Li et al., 1995). In fact, Bcl-2 and Bcl-x L expression are increased in neurons that survive ischemia (Chen et al., 1997), whereas Bax expression is enhanced in neurons that ultimately die in the ischemic area (Isenmann et al., 1998). Caspase-3, an important apoptotic executioner in many cell types, is also activated after transient cerebral ischemia (Chen et al., 1998). In addition, caspase-1 activation may play a pivotal role in ischemia-induced neurodegeneration (Hara et al., 1997a; Schielke et al., 1998).
Caspase inhibitors afford significant neuroprotection in several different models of cerebral ischemia (Loddick et al., 1996; Hara et al., 1997b; Cheng et al., 1998; Endres et al., 1998; Ma et al., 1998; Rabuffetti et al., 2000). In addition, it has recently been shown that erythropoietin crosses the blood–brain barrier and prevents neuronal apoptosis from cerebral ischemia through the activation of certain protein kinases (Brines et al., 2000; Sirén et al., 2001). Paradoxically, administration of a cyclin-dependent kinase inhibitor dramatically reduced neuronal death in ischemia-reperfusion cerebral injury (Osuga et al., 2000). Finally, a number of studies have shown that modifications of mitochondrial function are a potentially important consequence of global cerebral ischemia (Sims and Zaidan, 1995). In particular, it has been shown that mitochondrial respiratory capacity can be significantly impaired during focal ischemia and is a key modulator of tissue injury (Kuroda et al., 1996; Nakai et al., 1997; Kristién and Siesjö, 1998).
It is well established that mitochondria are essential subcellular organelles in modulating some of the identified apoptotic pathways involved in cell death (Green and Reed, 1998; Kroemer and Reed, 2000; Kokoszka et al., 2001). In particular, during apoptosis normal electron transport and oxidative phosphorylation are disrupted with concomitant collapse of mitochondrial transmembrane potential and cytochrome c release (Greenlund et al., 1995; Liu et al., 1996; Vander Heiden et al., 1999). Also, there is translocation of proapoptotic Bax protein from cytosol to mitochondria that promotes cytochrome c release, though the mechanism is not entirely understood (Rossé et al., 1998). Once released into the cytoplasm, cytochrome c activates caspases that cleave specific intracellular substrates, such as PARP, which are critically involved in the processes that lead to cell death (Liu et al., 1996; Du et al., 1997; Kluck et al., 1997).
The purpose of this study was to further characterize the role of apoptosis in a well-characterized ischemia-reperfusion stroke model of cerebral injury, and to determine whether TUDCA can inhibit cell death by apoptosis. Our results indicate that TUDCA administered intravenously 1 hour after middle cerebral artery occlusion (MCAO) protected against cell death and markedly reduced infarct size. In addition, it significantly decreased mitochondrial membrane perturbation and the downstream activation of caspases and substrate cleavage associated with apoptosis. We conclude that TUDCA is a unique, nontoxic endogenous molecule that could potentially be a powerful therapeutic agent for the management of acute stroke.
MATERIALS AND METHODS
Rat middle cerebral artery occlusion model
All animals received humane care in compliance with the institute's guidelines as outlined in the Guide for the Care and Use of Laboratory Animals prepared by the National Academy of Sciences and published by the National Institutes of Health (NIH publication no. 86–23, revised 1985). Male Sprague-Dawley rats (300–350 g; Harlan Sprague-Dawley, Inc., Indianapolis, IN, U.S.A.) were anesthetized with an intramuscular injection of a mixture of ketamine and xylazine. After induction, animals were ventilated through a face mask with 10% to 20% oxygen. The arterial P
An incision was made in the midline of the neck and the left carotid bifurcation was exposed. The internal carotid artery was followed rostrally, and the pterygopalatine branch was identified and ligated. The common carotid artery was then occluded, and the branches of the external carotid artery were dissected and divided. The occluder, 4–0 nylon suture with a silicone-coated tip of 0.28 mm in diameter (Sherwood Davis & Geck, St. Louis, MO, U.S.A.), was then advanced from the external carotid artery into the lumen of the internal carotid artery until it blocked the origin of the middle cerebral artery (Yanaka et al., 1996). Cerebral blood flow was monitored in the ipsilateral hemisphere by a laser-Doppler flowmeter (Moor Instruments, Devon, England, U.K.) and successful occlusion defined by a 75% decrease in baseline flow. One hour later, the suture was completely withdrawn to allow reperfusion, which was confirmed by laser-Doppler flowmetry. After surgery, the rats were placed under a heating lamp and monitored for 1 hour after reperfusion and then allowed free access to food and water for 2 or 7 days.
Tauroursodeoxycholic acid (Calbiochem-Novabiochem, San Diego, CA, U.S.A.) was dissolved in 0.15-mol/L NaHCO3 (pH 7.4) at 400 mg/mL. Then, 400 mg/kg TUDCA solution or an equivalent volume of vehicle was administered intravenously during or 1 hour after reperfusion. Lower doses of 64 and 180 mg/kg TUDCA were also administered 1 hour after ischemia in a subgroup of animals. Physiologic monitoring, including blood pressure and body temperature, were performed in both the vehicle- and TUDCA-treated animals.
Bile acid analysis of plasma and brain
Normal controls and rats with MCAO were killed 24 hours after TUDCA or vehicle injection. Blood was collected, clotted, spun, and the plasma was removed and frozen at −20°C. In addition, brains that were removed after 5% chloral hydrate anesthesia and thorough transcardial phosphate-buffer perfusion were flash-frozen and stored at −70°C. Bile acids were assessed by gas chromatography after organic solvent extraction, purification on Lipidex 1000 (Packard Instruments B.V., Groningen, The Netherlands), liquid-solid extraction, hydrolysis, isolation by lipophilic anion exchange chromatography and conversion to methyl ester-trimethylsilyl ether derivatives (Setchell et al., 1997). Bile acid concentrations were expressed as nanomoles per gram tissue or as micromoles per liter plasma.
Neurologic testing
Neurologic deficits characterized by left-sided hemiparesis and right Horner syndrome were used as positive criteria. Rats were examined for changes 2 days after occlusion and rated on a standard scoring scale from 0 (normal) to 4 (severe) (Longa et al., 1989). Briefly, failure to extend the left forepaw when suspended vertically was graded as 1, circling to the left was graded as 2, falling to the left was graded as 3, and not walking spontaneously or exhibiting a disturbance in consciousness was graded as 4. Rats with convulsions, sustained disturbances in consciousness, or no neurologic deficits were excluded from the study.
Infarct size
The animals were killed 2 or 7 days after reperfusion, and their brains were immediately removed for serial cryostat sectioning or processing as seven consecutive 2-mm coronal sections. Infarction volumes were quantitated using Sigmascan Pro Image analysis software (SPSS Science, Chicago, IL, U.S.A.) on 2-mm sections stained with 2% 2,3,5-triphenyltetrazolium chloride or unbiased stereologic volumetric analysis of 20-μm cryostat sections stained with hematoxylin. Volumetric analysis was performed according to the Cavalieri method (Howard et al., 1998). The areas of lesion were determined in 11 hematoxylin-stained 20-μm-thick sections, 900-μm apart, from each animal. Volumes (mm3) were calculated according to the formula V = T × a/p × ∑Pi, where V is the volume of interest, T is the distance between sections (mm), a/p is area associated with each point (mm2), and Pi is points hitting object in each section.
TUNEL staining and electron microscopy
At 2 and 7 days after reperfusion, rats were killed for terminal deoxynucleotidyl transferase-mediated 2′-deoxyuridine 5′-triphosphate-biotin nick end labeling (TUNEL) labeling of apoptotic cells. Brains were quickly removed and frozen in powdered dry ice for cryostat sectioning as described previously. Brain sections 10 μm in thickness were then fixed with 4% formaldehyde, postfixed with acetate-ethanol solution at −20°C, and washed with phosphate buffer (pH 7.4). An Apoptag in situ apoptosis detection kit (Intergen, Purchase, NY, U.S.A.) was used for TUNEL staining according to the manufacturer's recommendations. In brief, the same serial brain section for each animal was incubated with equilibration buffer. Terminal deoxynucleotidyl transferase and digoxigenin-deoxynucleotidyl triphosphate were added to the sections and incubated at 37°C for 1 hour. Slides were then treated with antidigoxigenin-peroxidase solution for 30 minutes, colorized with diamino benzidine substrate, and counterstained with 0.5% methyl green. The number of TUNEL-positive cells was counted on a computer screen grid from at least three random fields (400x) within the penumbra for each animal.
For electron microscopy, samples from both hemispheres were fixed overnight at 4°C in 0.1-mol/L cacodylate buffer (pH 7.2) containing 6% glutaraldehyde. Samples were then rinsed with 0.1-mol/L PIPES buffer (pH 7.3) followed by a 20-minute postfix incubation at room temperature in 2% cacodylate-buffered OsO4. The tissue was then dehydrated using increasing concentrations of ethanol, infiltrated with propylene oxide, and embedded in Epon 812/Araldite 502 resin (Electron Microscopy Sciences, Fort Washington, PA, U.S.A.). Sections 70 to 100 nm in thickness were cut using glass knives on a Reichert Ultracut S ultramicrotome (Vienna, Austria), collected on 200-mesh copper grids (Electron Microscopy Sciences), and stained with Reynold lead citrate for 5 minutes, followed by a 25-minute stain in 80% uranyl acetate in 50% methanol. The morphology of nuclei and mitochondria were studied using a JEOL-100 CX electron microscope (JEOL U.S.A., Peabody, MA, U.S.A.) at 80 kV. Photographs were taken with Kodak ESTAR thick base electron microscope film (Eastman Kodak, Rochester, NY, U.S.A.). The number of normal-appearing mitochondria was counted from at least six random photographs at a magnification of 26,000x for each treatment group.
DEVD-specific caspase activity
On day 7 after reperfusion, animals were killed and their brains were removed, immediately flash frozen, and stored at −70°C. The brains were dissected and tissue from the ischemic and corresponding contralateral areas was isolated. The tissues were then homogenized in isolation buffer containing 10-mmol/L Tris-hydrochloride buffer (pH 7.6) 5-mmol/L magnesium chloride, 1.5-mmol/L KAc, 2-mmol/L dithiothreitol, and protease-inhibitor cocktail tablets (Complete; Roche Molecular Biochemicals, Indianapolis, IN, U.S.A.). General caspase activity was determined by enzymatic cleavage of chromophore p-nitroanilide (pNA) from the substrate N-acetyl-Asp-Glu-Val-Asp-pNA (DEVD-pNA). The proteolytic reaction was carried out in isolation buffer, containing 50 μg cytosolic protein and 50-μmol/L DEVD-pNA. The reaction mixtures were incubated at 37°C for 1 hour, and the formation of pNA was measured at 405 nm using a 96-well plate reader. Protein concentrations were determined using the Bio-Rad protein assay kit (Bio-Rad Laboratories, Hercules, CA, U.S.A.) according to the manufacturer's specifications.
Western blot analysis
Caspase-3 and PARP protein levels were determined from cytosolic and total homogenates separated by 15% and 8% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), respectively. Blots were probed with either primary rabbit polyclonal antibody reactive with the p11 and p20 subunits and the p32 precursor form of caspase-3 at a dilution of 1:200, or with primary rabbit polyclonal antibody reactive to PARP p116 full length and p85 subunits at a dilution of 1:2,000 (Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.), and subsequently incubated with secondary antirabbit antibody conjugated with horseradish peroxidase. Finally, membranes were processed for caspase-3 and PARP detection using the ECL system (Amersham Life Science, Arlington Heights, IL, U.S.A.).
Densitometry and statistical analysis
The relative intensities of the protein bands detected by Western blot were analyzed using a Bio-Rad model GS-700 imaging densitometer (Bio-Rad Laboratories). The percent changes in protein concentrations were calculated based on the corresponding controls. Statistical analysis was performed using InStat version 2.1 (GraphPad Software, San Diego, CA, U.S.A.) for the analysis of variance and Bonferroni multiple-comparison tests.
RESULTS
Tauroursodeoxycholic acid accumulates in brain after intravenous infusion
Bile acids were assayed to determine if intravenously administered TUDCA produced increased concentrations in the brain after MCAO. In fact, a single dose of TUDCA resulted in a significant increase in brain UDCA from almost undetectable levels to 0.15 ± 0.11 nmol/g (P < 0.01). This change was associated with a more than 12-fold increase in UDCA concentrations in plasma compared with vehicle-injected controls (P < 0.001). In addition, among the rats receiving TUDCA, there was a significant difference in brain total bile acid concentrations between the occluded animals and normal controls (0.42 ± 0.28 nmol/g vs. 0.26 ± 0.08, P < 0.05), suggesting increased transport across the blood–brain barrier. Almost 60% of the UDCA present in the brain was found in its taurine-conjugated form, 25% as the glycine-conjugated state and less than 20% in the unconjugated state. No significant differences in other major endogenous bile acids (deoxycholic, chenodeoxycholic, and cholic acid) were detected among any of the groups.
Tauroursodeoxycholic acid decreases infarct volumes
The infarct sizes of the brains were determined by volumetric analyses and found to be almost 50% larger in the vehicle-injected, compared with TUDCA-treated rats 2 days after reperfusion (Fig. 1). In fact, similar results were observed when TUDCA was administered either 1 or 2 hours after ischemia (P < 0.05). At 7 days after reperfusion, the same reduction in infarct volume was also observed (49.0 ± 10.1 vs. 24.2 ± 10.9 mm3 in vehicle vs. treatment group, respectively; P < 0.05), suggesting that the effect by TUDCA was not merely to delay the onset of tissue damage. Moreover, functional neurologic deficits 2 days after occlusion were significantly improved in rats treated with TUDCA (P < 0.05), indicating that infarct sparing may be accompanied by improved neurofunction. In fact, all rats treated with TUDCA circled to the left (grade 2), whereas all but one of the vehicle-injected controls fell to the left (grade 3). Although brain swelling may have contributed partly to the larger infarcts observed 2 days after reperfusion compared with 7 days, it did not affect the ratios of the vehicle- to TUDCA-treated animals. In fact, all the rats performed slightly better during neurologic testing by day 7, though the difference between the two groups remained the same (data not shown). Finally, lower doses of 64 and 180 mg/kg resulted in levels of protection as high as 30% to 40% 2 days after reperfusion (P < 0.05).
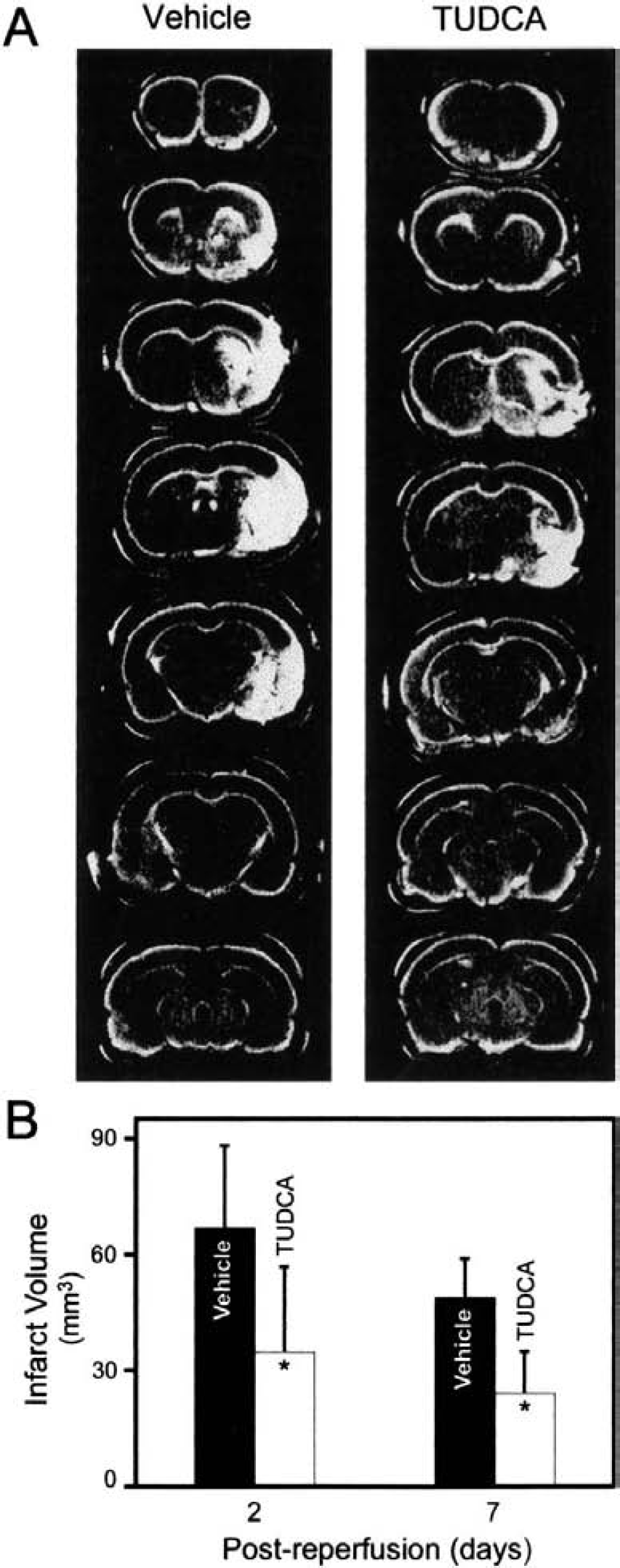
Tauroursodeoxycholic acid (TUDCA) reduces infarct volume after middle cerebral artery occlusion in the rat.
Tauroursodeoxycholic acid prevents apoptosis and preserves mitochondrial integrity
TUNEL staining was used to identify cells with nuclear DNA fragmentation within the ischemic penumbra. After 7 days of reperfusion, only occasional TUNEL-positive cells were observed in the contralateral hemisphere in both experimental groups (Fig. 2). In contrast, TUNEL staining was markedly increased in the ipsilateral hemisphere of vehicle-injected rats (P < 0.001). Tauroursodeoxycholic acid treatment reduced the number of TUNEL-positive cells by approximately 80% (P < 0.01), similar to levels observed in the contralateral hemisphere. In addition, when TUDCA was administered 2 hours after ischemia, it reduced apoptosis by more than 50% (P < 0.01), as determined by TUNEL staining. Finally, after 2 days of reperfusion, the number of TUNEL-positive cells per field was 65.5 ± 9.1 in the ipsilateral hemisphere, but only 34.8 ± 3.3 in TUDCA-treated rats (P < 0.001). We also examined mitochondrial morphology by transmission electron microscopy. In the contralateral hemisphere, the number and morphology of mitochondria appeared normal in both animal groups. After 7 days, swollen mitochondria with membrane disruption were virtually absent in contralateral hemispheres, but were readily detectable in the ipsilateral regions (Fig. 3A). In addition, there were a number of cells with shrunken nuclei and condensed chromatin (data not shown). In contrast, there were significantly more normal-appearing mitochondria throughout the entire hemisphere with TUDCA treatment (Fig. 3B) compared with vehicle (9.0 ± 2.1 vs. 1.9 ± 0.5 per field, respectively). These findings suggest that changes consistent with apoptosis appear to occur after ischemia-reperfusion injury and they are partially prevented by TUDCA.
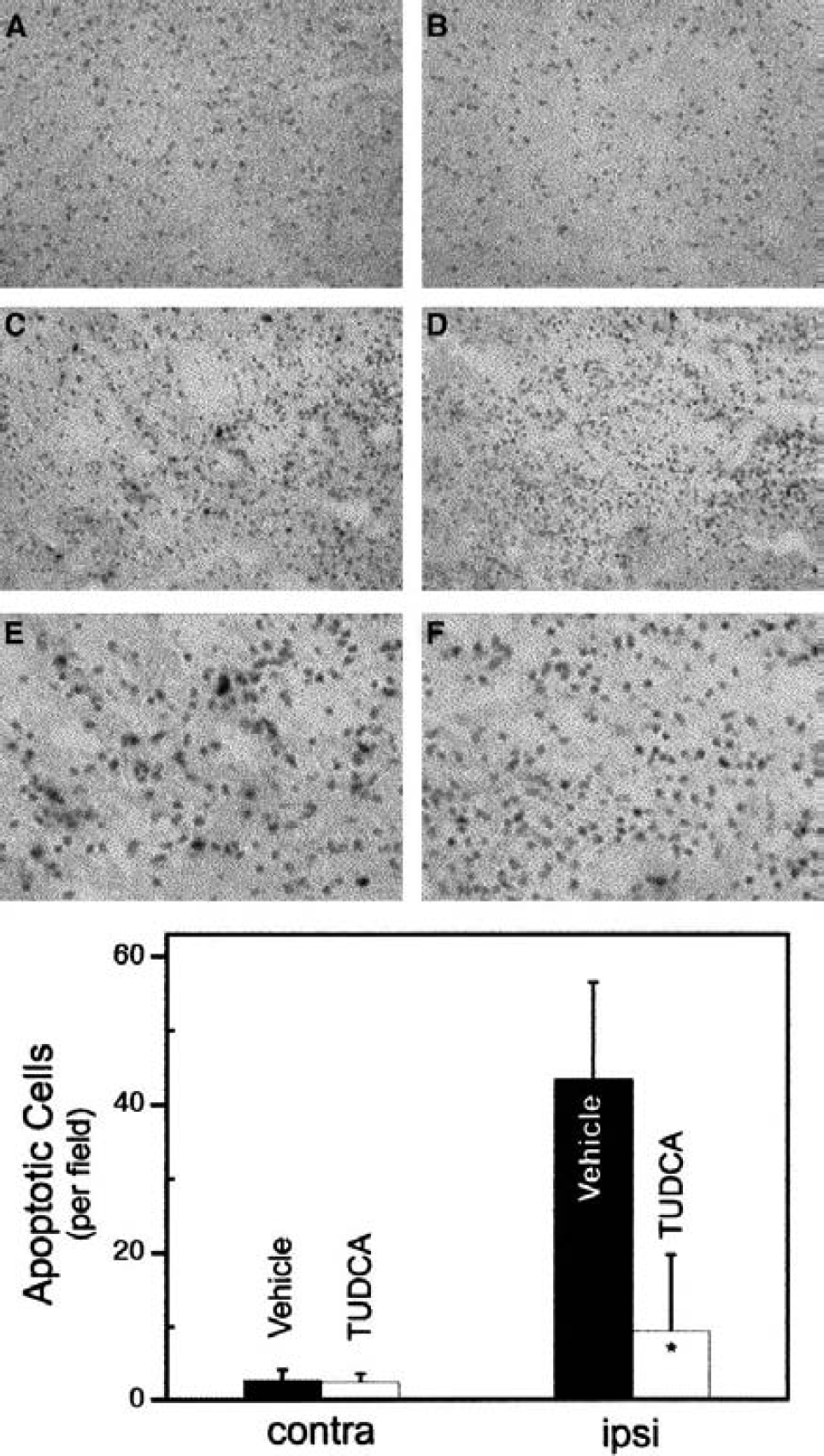
Tauroursodeoxycholic acid (TUDCA) reduces the number of TUNEL-labeled cells in the hemisphere ipsilateral to middle cerebral artery occlusion (MCAO). Seven days after reperfusion, tissue sections were analyzed for TUNEL labeling (darker staining) within the ischemic penumbra. Rare TUNEL-positive cells were seen in the contralateral hemisphere in vehicle-
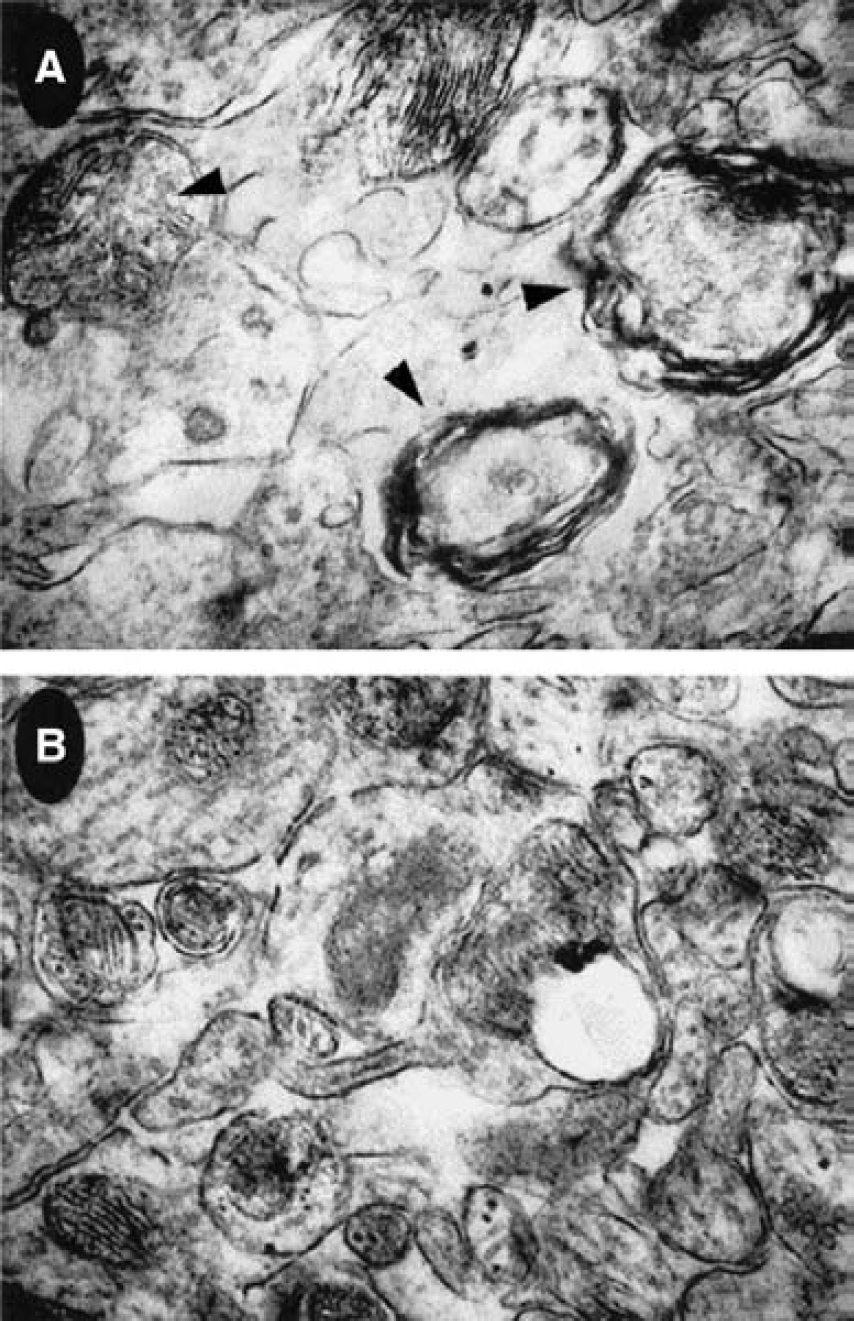
Tauroursodeoxycholic acid (TUDCA) prevents mitochondrial disruption in cells within the ipsilateral hemisphere of middle cerebral artery occlusion (MCAO). Seven days after reperfusion, electron micrographs of brain tissue in vehicle-injected animals
Tauroursodeoxycholic acid prevents caspase activation and poly(ADP-ribose) polymerase cleavage
We next examined caspase activity in both the ipsilateral and contralateral hemispheres after MCAO. Seven days after reperfusion, protein extracts were prepared from relevant brain tissues and incubated with DEVD-pNA, a preferred substrate for caspase-3–like enzymes. There was no significant increase in caspase activity in the contralateral hemisphere compared to normal brain tissue. In contrast, caspase activity was notably increased in the ipsilateral portion of the brain in vehicle-injected rats (P < 0.05) (Fig. 4A). Tauroursodeoxycholic acid reduced caspase activity in the lesioned hemisphere by approximately 60% (P < 0.05). In fact, there were no statistical differences between the TUDCA-treated ipsilateral and both the vehicle and TUDCA contralateral regions.
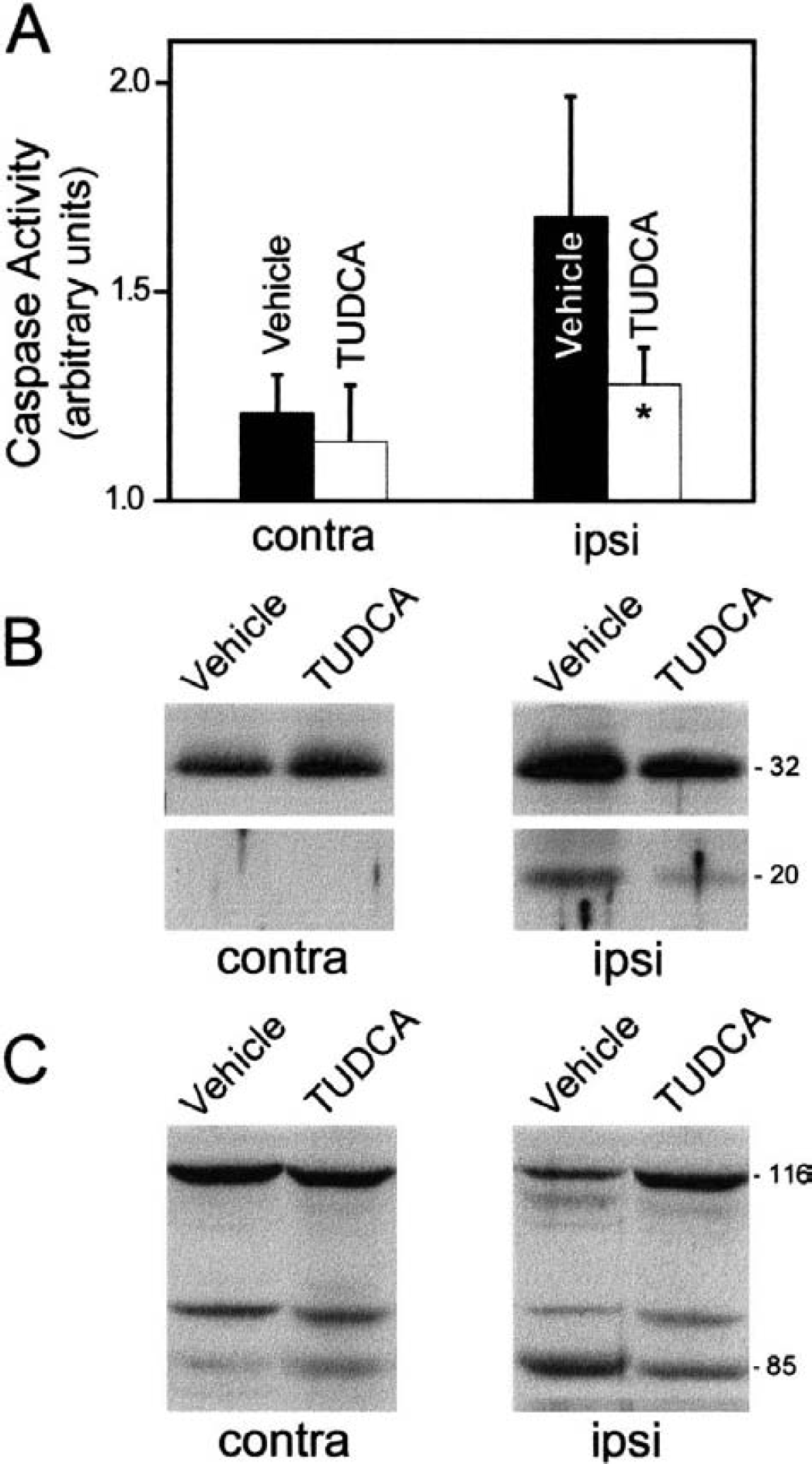
Tauroursodeoxycholic acid (TUDCA) prevents activation of DEVD-specific caspases, procaspase-3 cleavage, and degradation of PARP in the ipsilateral hemisphere of middle cerebral artery occlusion (MCAO).
We also examined procaspase and caspase-3 protein levels and PARP cleavage in the damaged brain tissue after MCAO (Figs. 4B and 4C). In vehicle-treated animals, there was a 2.7-fold increase in procaspase-3 levels in the hemisphere ipsilateral to the occlusion (1.0 ± 0.2 vs. 2.7 ± 0.6, P < 0.01). Similarly, the 20-kd active form of caspase-3 was also markedly increased in the lesioned hemisphere from almost undetectable levels in the contralateral regions. Tauroursodeoxycholic acid treatment resulted in a significant reduction in procaspase (1.6 ± 0.6, P < 0.05) and caspase-3 protein levels from vehicle-injected rats. Finally, we also assessed whether intravenous injection of TUDCA inhibited the cleavage of PARP, an endogenous substrate for caspase-3. By Western blot analysis, there was significantly less cleavage of PARP in brain extracts from TUDCA- compared with vehicle-treated rats in the ipsilateral hemisphere. In fact, full-length PARP decreased almost 50% in vehicle controls (1.0 ± 0.1 vs. 0.5 ± 0.2) compared with only 19% in TUDCA-treated rats (1.0 ± 0.2 vs. 0.8 ± 0.1, P < 0.05). Thus, TUDCA appears to protect against both anatomical and biochemical manifestations of cell injury.
DISCUSSION
Oral administration of UDCA and TUDCA has been used clinically in the management of a variety of hepatic diseases associated with cholestatic syndromes, such as primary biliary cirrhosis (Beuers et al., 1998; Invernizzi et al., 1999). In addition, it has recently been reported that these molecules can also protect the liver during ischemic insult (Ono et al., 1995; Hertl et al., 1999; Falasca et al., 2000). The cytoprotective mechanism of UDCA and TUDCA results, in part, from their ability to inhibit apoptosis in hepatic cells by preventing mitochondrial depolarization and reactive oxygen species production (Rodrigues et al., 1998a,b). More important, the antiapoptotic properties of these molecules are independent of the inducing agent and cell type, suggesting a common antiapoptotic mechanism. Specifically, UDCA prevents apoptosis by modulating mitochondrial membrane perturbation, opening of the mitochondrial permeability transition pore, Bax translocation, and cytochrome c release (Rodrigues et al., 1999, 2000a,b). In addition, UDCA may also regulate apoptosis through additional mechanisms, independent of its effects on mitochondria. Thus, by acting as a general modulator of cell survival, UDCA and TUDCA may play an essential role as therapeutic agents in the management of nonliver diseases, in which increased levels of apoptosis contribute to disease pathogenesis. Interestingly, it has recently been reported that a nitric-oxide derivative of UDCA protects against Fas-mediated liver injury by inhibiting caspase activity (Fiorucci et al., 2001).
Until recently, it was assumed that cell death after ischemic injury resulted from excitotoxicity-induced necrosis caused by overstimulation of neuronal glutamate receptors (Choi, 1992). However, it has now been reported that after ischemic injury, cells exhibit several features of apoptosis including chromatin condensation, DNA fragmentation, TUNEL labeling, and activation of caspases (Choi, 1996). Consistent with these findings, we detected significantly increased caspase-3 processing and PARP cleavage, and increased numbers of TUNEL-positive cells in the hemisphere ipsilateral to MCAO. In addition, we also observed disrupted and swollen mitochondria with complete loss of cristae that is characteristic of apoptosis occurring through the mitochondrial pathway.
It is now well established that caspases have a prominent role in apoptosis (Salvesen and Dixit, 1997). Several experimental approaches, including the use of caspase inhibitors and genetically engineered mice, have been used to show the key role of caspases in neuronal cell death after in vivo ischemic insults (Loddick et al., 1996; Hara et al., 1997a,b; Cheng et al., 1998; Endres et al., 1998; Ma et al., 1998; Schielke et al., 1998; Rabuffetti et al., 2000). In fact, mitochondria are critical effectors of both the function and activation of caspases (Green and Reed, 1998; Kroemer and Reed, 2000). On stimulation, proapoptotic molecules such as cytochrome c and procaspases are released from the intermembrane space of mitochondria into the cytosol. Cytochrome c then binds Apaf-1, which then interacts with and activates procaspase-9, (Li et al., 1997) which in turn activates caspase-3. Interestingly, it was recently reported that caspase-9 is located in the mitochondria of neurons and released into the cytosol in an animal model of transient global cerebral ischemia (Krajewski et al., 1999). Moreover, cytosolic redistribution of cytochrome c has been observed in neuronal cultures on reoxygenation (Tamatani et al., 2000) and after transient or permanent focal cerebral ischemia in rats and mice (Fujimura, et al., 1999, 2000).
Intravenous administration of TUDCA 1 or 2 hours after ischemia in the MCAO stroke model significantly reduced the infarct volume at 2 days and partially improved neurologic function in rats. More important, our results indicate that neuroprotection persisted for at least 7 days after reperfusion. Bile acid analysis by gas chromatography confirmed increased levels of TUDCA in the brains of rats after intravenous infusion of TUDCA, suggesting that the blood–brain barrier is, at some level, permeable to this endogenous nontoxic molecule. Moreover, this is the first demonstration that administration of a bile acid can act as a neuroprotective agent in an ischemia-reperfusion injury model. Using TUNEL staining, we recently determined that TUDCA significantly reduced apoptosis by more than 50% after brain injury in a blunt head trauma rat model (P < 0.05) (Rodrigues et al., unpublished data, August 2000).
In the setting of MCAO, TUDCA appears to afford neuroprotection by preventing apoptotic cell death. Acting at the mitochondrial membrane, TUDCA prevents mitochondrial swelling and disruption of the outer mitochondrial membrane. Membrane stability inhibits cytochrome c release, and thereby modulates cytochrome c–mediated downstream events, such as caspase activation and PARP cleavage. In fact, we have recently shown that TUDCA partially prevented apoptosis induced by 3-nitropropionic acid in rat neuronal RN33B cells (Rodrigues et al., 2000a). Incubation with TUDCA prevented both depolarization and channel-forming activity of the mitochondrial membrane. In addition, it directly inhibited reactive oxygen species production, which can significantly contribute to collapse of the membrane potential. TUDCA markedly decreased mitochondrial release of cytochrome c, which probably accounted for the observed reduction of DEVD-specific caspase activity and PARP cleavage. It appears that TUDCA exhibits unique properties in modulating not only apoptosis but also a variety of cell functions. For example, TUDCA may play key regulatory roles in signal transduction pathways by modulating cytosolic calcium levels and in gene expression by regulating transcription (Rodrigues and Steer, 2001).
Tauroursodeoxycholic acid may also modulate apoptosis by suppressing mitochondrial perturbation through pathways that are independent of the permeability transition. Indeed, TUDCA is a potent inhibitor of membrane disruption in isolated mitochondria incubated with recombinant Bax protein, in part, by preventing Bax binding to the outer membrane. Using spin labeling techniques and spectroscopic analysis of isolated mitochondria, Bax protein appears to directly increase polarity and fluidity of the membrane lipid core, and decrease protein spin label mobility, indicative of Bax insertion into the mitochondrial membrane. Tauroursodeoxycholic acid partially prevents the binding of Bax and subsequent disruption of membrane lipid and protein structure, and the loss of detectable paramagnetism of spin label characteristic of oxidative damage (Rodrigues et al., unpublished data, June 2000). Similar membrane-stabilizing effects were also evident when isolated mitochondria exposed to amyloid β-peptide fragment (25–35 amino acids) were pretreated with TUDCA (Rodrigues et al., 2001).
Our findings that TUDCA can reduce the mitochondrial perturbation associated with acute stroke may provide a new paradigm for preventing pathologic cell death in ischemic injury. Mitochondria may, in fact, represent a more attractive target for the prevention of apoptosis than caspases. For example, mitochondria appear to be upstream of caspase activation and are responsible for other apoptotic processes, including collapse of the transmembrane potential, reactive oxygen species production, and peroxide formation. To this end, recent reports indicate that combined therapy with an inhibitor of excitotoxic cell death, which reduces ischemia-associated increases in cytosolic Ca2+ and mitochondrial damage, and a caspase inhibitor provides synergistic protection in stroke models (Ma et al., 1998; Schulz et al., 1998).
In conclusion, our data show that the administration of a single dose of TUDCA resulted in substantial neuroprotection in a MCAO stroke model. In addition, the neuroprotective effect by TUDCA was relatively long lasting, suggesting that this bile acid does not simply delay cellular death. Rather, by interfering with key events in apoptosis, including mitochondrial perturbation and caspase activation, the process can be significantly reduced in the ischemic areas of the brain. Our findings suggest that this natural and essentially nontoxic bile acid can suppress certain events in the cell death pathway, and may be effective in the management of stroke and possibly other acute and even chronic injuries to the brain.
Footnotes
Acknowledgments:
The authors thank Betsy T. Kren for critical review of the manuscript, Martin Wessendorf and the Electron Microscopy Facility, the Biomedical Imaging and Processing Laboratory, and Dora Brites for use of the gas chromatographer.