
Abstract
Ischemic brain injury causes a local inflammatory response, involving the activation of resident brain cells such as microglia and the recruitment of infiltrating immune cells. Increasing evidence supports that plasticity of the myeloid cell lineage is determinant for the specific role of these cells on stroke outcome, from initiation and maintenance to resolution of post-ischemic inflammation. The aim of this review is to summarize some of the key characteristics of these cells and the mechanisms for their recruitment into the injured brain through interactions with platelets, endothelial cells and other leukocytes. Also, we discuss the existence of different leukocyte subsets in the ischemic tissue and, specifically, the impact of different myeloid phenotypes on stroke outcome, with special emphasis on neutrophils and their interplay with platelets. Knowledge of these cellular phenotypes and interactions may pave the way to new therapies able to promote protective immune responses and tissue repair after cerebral ischemia.
Introduction
Inflammation is recognized as an important contributor to the pathophysiology of stroke. The inflammatory response induces the release of reactive oxygen species (ROS) and promotes immune-derived mechanisms associated with cytotoxicity and brain damage.1,2 In particular, the innate immune system plays a pivotal role in the evolution of ischemic cerebral injury, as soluble mediators (i.e. cytokines and chemokines) and specialized cells, activated in the brain or recruited from the periphery, actively participate in the detrimental processes implicated in tissue damage.3,4 In this context, a vast number of experimental studies have explored the beneficial role of anti-inflammatory approaches to block/antagonize key pro-inflammatory pathways driven by cerebral ischemia. 2 However, the dualistic role exerted by several mediators of the immune reaction may explain why most anti-inflammatory approaches, conceived disregarding the potential beneficial function of the target, have failed to reach the clinical setting. 5 In addition, the fact that ischemic cerebral injury involves several mechanisms may explain why “single-target” therapies have resulted insufficient, a reason for which it has been proposed that therapeutic approaches should target multiple cell types to promote protection and recovery. 6 An important factor to consider is that a part of the inflammatory response is its resolution, an active process that involves the participation of myeloid cells with distinct phenotypes, which could reflect either the local conversion of cells from a pro-inflammatory to an anti-inflammatory phenotype or the recruitment of cells with different phenotypes and functions. In this context, whereas polarization of microglia/macrophages has been extensively investigated, 7 the roles of neutrophils are understudied. Increasing evidence shows the ability of neutrophils to switch towards a variety of phenotypes dependent on specific microenvironmental signals which are related to the spatio-temporal progression of ischemic brain damage.8–10 Another crucial issue which remains quite unknown is the important impact of the interaction of neutrophils with platelets in the onset of inflammation. Platelets are growingly considered a major actor of the immune response. Their binding to myeloid cells has been shown to enhance their function as well as recruitment to extravascular areas (rev. in Gomez-Moreno et al. 11 ).
The aim of this review is to summarize the roles that have been identified so far for hematopoietic stem cell (HSC)-derived myeloid cell subsets (neutrophils, monocytes and platelets) and resident microglial cells, with a special emphasis on neutrophils and their interaction with platelets in the stroke setting. We focus on the mechanisms that regulate their activation and recruitment to inflamed tissues and on how they contribute to stroke pathogenesis. Also, we highlight the important role of immune cells after stroke and the therapeutic relevance of their polarization as a potential novel target in stroke neuroinflammation. We also propose that potential cell-based treatments using the pleiotropic mechanisms conferred by their polarization towards non-inflammatory phenotypes may be an effective therapeutic strategy to the acute, subacute and chronic phases of this pathology.
Monocytes/macrophages
Monocytes are a population of mononuclear leukocytes that originate in the bone marrow from HSCs and then are released into the circulation as non-dividing cells. 12 These cells represent approximately 4 or 10% of total leukocytes in mice or human peripheral blood, respectively, with a considerable pool located in spleen and lungs, apart from their homing into inflammatory sites in response to specific chemokines. 13 Monocytes are immune effector cells, equipped with chemokine and pathogen recognition receptors (PRRs) that mediate migration from blood to tissues during infection. Their functions include the killing of pathogens via phagocytosis, the expression of inflammatory cytokines and enzymes and the subsequent production of ROS and reactive nitrogen species (RNS). 14 Under specific conditions, monocytes can stimulate or inhibit T-cell responses during cancer as well as infectious and autoimmune diseases. They are also involved in tissue repair and neovascularization. 15 They can also differentiate into inflammatory dendritic cells (DC) or macrophages during inflammation and constitute the resident pool of macrophages in the lamina propria of the intestine and skin16,17 in steady state. Monocyte migration to tissues and differentiation to inflammatory DC and macrophages is likely determined by the inflammatory milieu and the activation of pathogen-associated PRRs. 18
Two predominant subsets of circulating monocytes, “inflammatory” and “patrolling” monocytes, are found in the blood of rodents. Short-lived inflammatory monocytes are characterized by the expression of high levels of lymphocyte antigen 6 complex locus C1 (Ly6C), low to intermediate levels of CX(3)-C motif chemokine receptor 1 (CX3CR1) and the expression of the C–C chemokine receptor type 2 (CCR2): Ly6ChighCXC3CR1+CCR2+. This subset is actively recruited to inflamed tissues and requires CCR2 for mobilization and transmigration from the bone marrow to blood and subsequently for the transmigration into these sites of inflammation.19–22 In contrast, patrolling monocytes express Ly6C at low levels, high levels of CXC3CR1 and do not express CCR2 (Ly6ClowCXC3CR1brightCCR2). Their egress from bone marrow is Ccr2-independent but dependent on sphingosine-1 phosphate receptor 5 (S1pr5). 23 They have a longer half-life, crawl on the vessel luminal wall 24 and are recruited within the vasculature of inflamed and damaged tissues.25,26 To date, a specific precursor for patrolling monocytes has not been identified and Ly-6Chi monocytes are thought to give rise to Ly-6Clo monocytes either in the blood or in the bone marrow. 27 C/EBPβ-dependent control of Nr4a1 expression in monocytes has been proposed as the molecular mechanism that mediates the maturation of Ly-6Chi monocytes into Ly-6Clo. 28
Human monocytes have been classified into three subtypes based on the differential expression of CD14 and CD16: classical CD14++CD16− (also referred as CD14+ or CD14+CD16−), intermediate CD14++CD16+ (also referred as CD14+CD16+ or CD14+CD16int/low) and non-classical CD14+CD16++ (also referred as CD14lowCD16+ or CD14dimCD16+) monocytes. 29 The marker of human monocytes, CD14, is a glycoprotein and myelomonocytic differentiation antigen that functions as an accessory protein to toll-like receptor (TLR4). 30
Monocyte/macrophage lineages display a remarkable plasticity and are able to change their phenotype in response to the microenvironment, giving rise to different populations of cells with distinct functions. 31 In damaged tissues, as in ischemic stroke, peripheral-blood monocytes could have the ability to differentiate into macrophages and further polarize into several subtypes with specific functions including the production of inflammatory molecules and phagocytic activity (Figure 1). 32 To easily classify them and based on in vitro studies, two main activation states in macrophages, designated as M1 (classically activated) and M2 (alternatively activated) macrophages, have been proposed. M1 subset is mainly associated with cytotoxicity and is thought to play an important role in the killing phase of the inflammatory response, producing pro-inflammatory cytokines and ROS upon stimulation with interferon-γ (IFNγ) and lipopolysaccharide (LPS), whereas polarization of macrophages towards an M2 phenotype, induced by interleukin-4 (IL-4), IL-10 or transforming growth factor-β (TGF-β), promotes tissue repair and trophic functions.31,33–35 Moreover, associated markers to each polarization state have been used to distinguish between these two macrophage activation states: in mice, the inducible nitric oxide synthase (iNOS) for the M1 macrophages, and the mannose receptor (CD206), arginase 1 (Arg1), chitinase-like 3 (Chil3 or Ym1) and IL-10 for the M2 ones.36–38
In the context of cerebral ischemia, growing evidence suggests that monocytes/macrophages can contribute to both brain injury39,40 and repair (Figure 1).41,42 Monocytes massively infiltrate into brain infarct early after stroke.43,44 In ischemic stroke patients, the number of peripheral monocytes is significantly increased in blood,45–47 and a significantly positive correlation between monocyte count and NIHSS score on admission has been reported. 48 Urra et al.45,46 also found that monocytes are major players in the prognosis and risk of infection after acute stroke. Ly6Chi monocytes, which are the predominant CCR2-expressing monocyte cell type, requiring CCR2 for extravasation from bone marrow into blood and subsequently for transmigration into sites of inflammation, 22 appear to be a prominent cell subset recruited from the circulation into the ischemic brain in mice.49–51 As to the cytokines/chemokines involved, CCL2 (also known as monocyte chemoattractant protein 1, MCP-1) is the most potent activator of CCR2 signalling in mice, leading to monocyte transmigration after stroke. The implication of the CCL2/CCR2 axis in mediating monocyte infiltration into the inflamed tissue has also been shown in other neuroinflammatory conditions such as EAE or traumatic brain injury.49,52,53 CCR2 knock-out mice display an increased number of monocytes in the bone marrow, a decreased number of circulating monocytes and a subsequent decrease in ischemic tissue, thus explaining that the absence of this chemokine receptor is protective against cerebral ischemia/reperfusion injury. 54 There is, however, also evidence showing that CCR2+ monocytes may play an important role in anti-inflammatory and recovery processes after cerebral ischemia in mice models. CCR2+ monocytes have been associated to the maintenance of the neurovascular unit integrity and have been implicated in mediating neuroblast migration from neurogenic regions to damaged areas of the brain after stroke.41,55 In agreement with a pro-repairing role of CCR2+ monocytes after stroke, monocyte depletion during the first week after cerebral ischemia leads to impaired recovery of the sensorimotor function and drastically decreases the tissue expression of anti-inflammatory genes, including TGFβ, CD163, and Ym1. 32 Also, recently, the use of CX3CR1GFP/+CCR2RFP/+ bone marrow chimeric mice showed that CCR2+Ly6Chi monocytes are the primary monocytic cell infiltrating during the acute inflammatory response to brain ischemia. This is followed by CX3CR1+ cell accumulation at the border of the infarct core 14 days after MCAO. In addition, this study suggests the possibility that CCR2+ monocytes undergo a phenotypic transformation into CX3CR1+CCR2− cells following infiltration to the ischemic tissue because accumulation of CX3CR1+Ly6Clo monocytes was absent in the brains of CCR2-deficient mice, which exhibit deficiency in CCR2+Ly6Chi recruitment, but not in NR4A1−/− chimeric mice, which lack circulating CX3CR1+Ly6Clo monocytes. 51 These findings strengthen the idea of the presence of different effector functions driven by monocytes/macrophages after CNS injury that play a pleiotropic role in mediating brain injury and repair. In addition, monocyte infiltration into the ischemic tissue follows a spatiotemporal pattern whose consequences are currently not well understood.
In this regard, immunoregulatory and anti-inflammatory cytokines released after stroke, such as IL-4, IL-10 and TGF-β, could influence monocyte/macrophage activation, fate and functionality. 31 In the context of cell activation, only a few experimental studies have evaluated the therapeutic benefits of drugs acting by increasing the M2/M1 ratio in stroke. Among these, Amantea et al. 56 have demonstrated that acute treatment with azithromycin attenuates blood–brain barrier (BBB) damage and cerebral ischemic damage in rodents subjected to MCAO, with a significant amelioration of neurological deficits up to seven days after the insult. Even more, chronic metformin treatment was reported to promote functional recovery and tissue repair via AMPK-dependent turning microglia/macrophages toward an M2 phenotype in mice after MCAO, 57 and up-regulation of M2 markers has also been shown to underlie neuroprotection by exendin-4, used in clinic against type 2 diabetes, when administrated in young healthy and in aged diabetic/obese after MCAO. 58 As it will be discussed below, the differential role of infiltrating monocytes/macrophages vs. resident microglia in these settings remains to be accurately established. Moreover, phenotypic characterization of myeloid subsets using the M1/M2 paradigm of activation could not reflect the real diversity and functionality of these cells in the context of brain inflammation as it relies on the assumption that a macrophage genetic program is recapitulated by the culture of monocyte-derived macrophages (MDMs). In this regard, identification of specific myeloid subsets with specific functions could be more useful to understand the diversity of the myeloid compartment in the context of stroke pathogenesis.
Microglia
Microglia derives from an extraembryonic erythromyeloid precursor and persist in adult mice independently of haematopoiesis.59,60 Therefore, they are not normally replaced by bone marrow-derived cells, 61 strongly suggesting that these cells have an elaborate repertoire of brain-specific functions that may not be appropriately taken over by peripheral monocytes/macrophages. Microglial cells are the first immune responders in the CNS. Upon cerebral injury microglial processes quickly and autonomously converge on the site of the damage (Figure 1).
An increasing number of studies suggest that resident microglia are functionally distinct from HSC-derived macrophages and play unique roles within the CNS.22,62–64 Since the lack of reliable microglia-specific markers has made very difficult to discriminate between microglia and infiltrated MDMs in the inflamed brain, an essential step to allocate differential functions to these cell types requires being able to conclusively distinguish them from each other. One of the most common approaches is the observation that, in contrast to macrophages, microglia expresses low or intermediate levels of the leukocyte marker CD45. This allows the characterization of microglia by flow cytometry using the levels of expression of CD45 in CD11b+ cells, a myeloid marker similarly expressed in both resident and non-resident populations.65–67 Although, by flow cytometry, MDMs can be differentiated by the expression level of CD45 and Ly6C, 68 however, these cells down-regulate the expression of Ly6C after entry into the CNS. 51 Different in vivo strategies have been established to attempt this discrimination after stroke, such as adoptive transfer studies of infiltrating cells using bone marrow chimeric mice with labelled bone marrow cells (DsRed; red fluorescent reporter) replaced after irradiation, to characterize the phenotype of macrophages entering the ischemic cortex. 69 The main limitation of this technique is that it might harm the CNS by affecting the integrity of the BBB, 70 so it is important to protect the heads of recipient mice from irradiation by head shielding. 49 Complementary data on the recruitment of peripheral myeloid cells into the CNS have been obtained by parabiosis. However, the rate of chimerism in the parabiosis model is generally low compared to irradiation, which might lead to an underestimation of myeloid cell engraftment.52,71 Also, in situ CFSE (carboxyfluorescein succinimidyl ester)-labelling of blood-derived cells has been extensively used in different models of cerebral ischemia,43,44,62,72 but this molecule labels all proliferating blood cells and may not be ideal for purifying cells for gene expression analysis. One of the best approaches in the recent years has been the use of genetically modified mouse models such as the LysM-EGFP knock-in transgenic mouse, in which EGFP is inserted into the lysozyme M locus labelling neutrophils and hematogenous macrophages but not microglia.73–76 This EGFP labelling helps to distinguish MDMs that have entered the damaged CNS from microglia that are LysMEGFP-negative and was confirmed using microglia-specific antibodies. 75 Zarruk et al. 77 used for the first time LysM-EGFP knock-in mice to separate MDMs from microglia by FACS and purified cell populations for expression analysis after ischemic stroke. They performed a PCR-array screen of 84 inflammatory genes revealing that pro-inflammatory chemokines and cytokines were predominantly upregulated in macrophages but down-regulated in microglia in the ischemic brain. These results show clear differences in the inflammatory expression profiles of microglia and macrophages 72 h post-ischemia which may shape repair and pro-regenerative mechanisms after stroke. A very good model to specifically label microglia is the Cx3cr1-ertCre. 78
As previously indicated for macrophages, an M1- or M2-like activation has also been suggested to occur in microglia. In vitro studies in which primary microglial cultures were exposed to LPS or IFNγ showed the induction of an M1-like phenotype with reduced phagocytic activity and expression of IL-1β, TNFα, iNOS or IL-6, while IL-4 drives microglia towards a M2-like phenotype with increased Arg1, Ym1 and resistin-like α (FIZZ1) expression. 79 Although M1/M2 activation can also take place in microglial cell cultures, it must be taken into account that, as previously indicated for monocytes, brain microglial gene expression signature could be lost in primary microglial cell cultures as well as in microglial cell lines such as BV2. 53 Considering this limitation, in vivo M1/M2 activation of microglia has been proposed as a potential therapeutic option in different diseases.79–81
In stroke, modulation of the M1 and M2 phenotype in microglia/macrophages (Figure 1) has been linked to detrimental and neuroprotective actions, respectively (see, for instance, see Han et al. 82 ). Further studies on different CNS pathologies such as traumatic brain injury or spinal cord injury also indicate that microglia shift from a transient M2 phenotype to become M1-like phagocytes.36,83 However, a major caveat of these studies relies on the characterization of the brain myeloid cells that express a limited array of M1/M2 markers that could not reflect the current activation state of the cell. In this regard, recent studies on AD using single-cell transcriptomics identified a novel microglial type associated with neurodegenerative diseases (disease-associated microglia or DAM) with a unique potential to restrict neurodegeneration 84 that indicates a high degree of specialization in response to pathogenic stimuli of this myeloid subset that falls beyond the M1/M2 dichotomy.
Neutrophils
Within the leukocyte populations, neutrophils play a key role in the innate immune response. They act as a fundamental defence in diseases associated with infections but also in sterile inflammation (as in stroke, cancer, autoimmune diseases, etc.). Their role in defence against pathogens has been extensively studied. Their ability to phagocytose and release molecules into their environment is well known. 85 In addition, in response to different stimuli, neutrophils release neutrophil extracellular traps (NETs), which are large web-like structures composed of histones, DNA, and granule proteins, such as elastase and MPO. 86 Although NETs primary function is to trap, restrain, and neutralize invading microbes, 87 their role in diseases associated with sterile inflammation is increasingly demonstrated.
The bone marrow produces 1010 neutrophils per day. There, they will mature acquiring a high content in granules equipped with various proteins involved in the modulation of the immune response. 88 Following maturation, neutrophils are released to the bloodstream where their half-life has been estimated to be 6 to 12 h, both in mice and humans. 85 After that time, they are thought to undergo spontaneous apoptosis being then eliminated in different organs through phagocytosis. 89 Besides circulating in the bloodstream, neutrophils can also be found in marginated pools in tissues such as the spleen and the lungs. 90 Importantly, neutrophil biology can be altered by different situations. For example, their life span increases when an inflammatory process occurs. This process, called priming, expands neutrophils half-life when exposed to certain cytokines. 91 It has been recently demonstrated that neutrophils are able to travel back to the bloodstream after they have infiltrated into an injured tissue. This process, so-called reverse transmigration, could explain such increased life span. 92
In response to inflammatory stimuli, neutrophils activate and contribute to the resolution of inflammation, to inflammation-associated immunopathology or to the modulation of the adaptive immune responses. Ischemic damage leads to neuroinflammation and cell death. Necrotic cells release pro-inflammatory cytokines that will induce subsequent neutrophil infiltration to the ischemic tissue. Neutrophils are the first leukocytes to be recruited into the parenchyma, 93 reaching the peak of infiltration between 48 and 72 h after the insult. 94 This process is facilitated by the characteristic disruption of the BBB that takes place after stroke (Figure 2). 95 The presence of neutrophils in the ischemic tissue contributes to stroke-associated brain injury, oedema and also to BBB disruption. Many mechanisms underlie these effects, such as the synthesis/release of cytokines and chemokines and the production of ROS. In the bloodstream, neutrophils respond to cytokines such as TNFα, IL1β, IL6 and IL8. Also, they can be activated by DAMPS (damage-associated molecular patterns) like HMGB1 or HSP72 via the TLR family (Figure 2). 96 Aside from the above mentioned, other molecules can modulate neutrophil migration, such as the leukotriene B4, that is required to recruit neutrophils to the lesion site. 97
The specific role of neutrophils in brain ischemia is currently under debate (Figure 2). Classically, neutrophils have been shown to be mainly detrimental, by mechanisms that will affect outcome, severity and infarct volume. For instance, neutrophils account for the no-reflow phenomenon, increasing tissue damage. 98 Even more, they can contribute to tissue injury through the release of elastase which damages the parenchyma and the production of ROS that contribute to BBB disruption. 99 In this context, as we will discuss below, the formation and release of NETs within blood vessels may contribute to the formation of heterotypic aggregates, thus contributing to inflammation and thrombosis. However, in spite of their implication in tissue damage, there is not a clear correlation between the number of circulating neutrophils and the extent of the ischemic lesion. 100 In this context, neutrophils are instrumental for the resolution of inflammation and their phagocytic activity contributes to the elimination of the necrotic tissue. In addition, other factors, such as the existence of neutrophil subsets with different roles in stroke pathogenesis, may account for possible beneficial actions of these cells. In this manner, the view of neutrophils as a homogeneous population is changing and nowadays it is believed that there are neutrophil subpopulations that exhibit different characteristics with anti-inflammatory, angiogenic or pro-resolutive properties. One of the first studies to describe the heterogeneity of neutrophil populations was published in the context of cancer and showed the existence of tumour-associated neutrophils (TANs), a subset of neutrophils that was named, by analogy to the macrophages M1/M2 paradigm, as N2 neutrophils or pro-tumorigenic. 101 On the other side of this dichotomy, these authors found a population of neutrophils within the tumour that exhibited a pro-inflammatory profile and that was named as N1. The shift towards one or the other phenotype was determined by the environment. For example, the presence of IL-10, TGFβ, IL-13 and IL-4 would induce N2 polarization, whereas the presence of IFNβ, IFNγ and IL-12 would favour N1 polarization. 102 A similar heterogeneity in the neutrophil compartment has been described by our group in the setting of stroke (Figure 2). Immunofluorescence characterization of the expression of prototypical M2 markers in ischemic tissue revealed that neutrophils located in the lesion differentially expressed some of these markers. Moreover, this phenotypic heterogeneity was modulated by PPARγ activation with the agonist rosiglitazone, that skewed neutrophils towards an M2-like or N2 phenotype which was associated with neuroprotection and resolution of inflammation after experimental stroke induced by pMCAO. 103 Interestingly, this neutrophil subset was more efficiently cleared by microglial phagocytosis than the other subset. Through this mechanism, debris was promptly removed from the inflamed tissue by phagocytosis, contributing to the restoration of tissue homeostasis and improving the final outcome. In addition, one of the most accepted views proposes that there are different populations of neutrophils depending on their age: fresh neutrophils, recently released from the bone marrow, display a set of surface markers different to those present in aged neutrophils, close to be cleared away from the bloodstream.104,105 It has also been reported that neutrophil pro-inflammatory activity correlates positively with their ageing, while in circulation, a process driven by the microbiota via TLRs and MyD88-mediated signalling pathways. 106 Consistently, our group has recently observed that a member of the TLR family, TLR4, one of the main actors for the initiation of inflammation in acute stroke, 107 also plays a major role in the modulation of the neutrophil phenotype (García-Culebras et al., unpublished).
Platelets
Platelets, or thrombocytes, are small non-nucleated cell fragments derived from a myeloid progenitor that circulate in the bloodstream. They are well-known for their role in thrombosis and haemostasis. Notably, an increasing number of studies indicate that platelets also play a critical part in inflammation (Figure 3).108,109
Under physiological conditions, platelets circulate in a quiescent state; however, when they become activated, platelets are able to express and release different inflammatory mediators, such as cytokines, chemokines and surface molecules110–113 to recruit leukocytes to the site of inflammation or injury (Figure 3). Many of these molecules are stored in platelet granules, of which three different types, dense-granules, α-granules and lysosomes, have been described, each of which is rich in certain chemicals that have an important role in platelet function.
Released from dense granules, ADP and ATP play a key role in inflammation and ischemia-reperfusion injury during platelet activation; 114 serotonin has been implicated in neutrophil rolling and adhesion to inflamed endothelium. 115 Moreover, inorganic polyphosphates (PolyP), also released upon platelet activation, have been suggested to cause neutrophil accumulation and increase vascular permeability,116,117 although their role has recently been questioned. 118 The regulation of neuroinflammation by modulating the expression of platelet adhesion receptors interacting with leukocytes or by releasing cytokines that affect leukocyte function is mediated by proteins stored in α-granules.109,119,120 Platelet factor 4 (PF4), the most abundant protein secreted from α-granules, acts as a chemoattractant for monocytes and neutrophils. 121 PF4 also promotes neutrophil granule release and adhesion to endothelial cells, mediated by L-selectin and leukocyte function antigen 1 (LFA-1). 122 PF4 induces cytokine release from monocytes including CXCL8, CXCL3, IL-1α, IL-1β, IL-6, IL-19, TNFα, CCL2, CCL3 and CCL22. 123 The chemokine RANTES (regulated on activation normal T cell expressed and secreted) is released either directly or in microparticles which can be immobilised on activated endothelium and promote monocyte recruitment by P-selectin.124,125 T cells can also induce platelet RANTES release promoting T cell recruitment to the endothelium. 126 Among other platelet-derived chemokines, macrophage inflammatory protein- (MIP-) 1α is a chemoattractant for monocytes, macrophages, T-cells and neutrophils, and is involved in transendothelial migration at sites of inflammation. 127 When platelets become activated, they also release IL-1 which has a major role in vascular inflammation. 128 IL-1 is known to promote neutrophil adhesion to endothelial cells and mediate vascular NO production.129,130 Platelet-secreted IL-1α causes endothelial activation which leads to the expression of ICAM-1 and VCAM-1 adhesion molecules on endothelial cells, CXCL1 release, and neutrophil transendothelial migration. 131 Another constituent of platelet α-granules is P-selectin, 132 an important adhesion molecule which, in response to activating signals, is translocated to the surface of platelets and binds to P-selectin glycoprotein ligand-1 (PSGL-1) on immune cells. 133 In vitro studies have suggested that leukocytes can roll and adhere to immobilized platelets adherent in the vessel wall causing a pro-inflammatory state. 134 This result was corroborated in the cerebral ischemia setting by an in vivo study revealing that platelets modulate leukocyte recruitment via P-selectin in a mouse tMCAO model (Figure 3). 135 In addition, our group has described in a mouse pMCAO model that, at early stages of inflammation, neutrophils recruited to the injured vessel use their PSGL-1 domain to scan for the presence of activated platelets, as a primary mechanism for the initiation of the inflammation process. 136
Specifically, platelet–neutrophil crosstalk leading to the formation of NETS is being increasingly recognised as a driver of inflammation and thrombosis. In this context, cell activation appears to be bidirectional.
137
Activated platelets can stimulate neutrophils to release NETs,86,138 for instance, through P-selectin and PSGL-1 binding.
139
In addition, neutrophils-derived NETs can activate platelets, a process in which TLR-dependent mechanisms may be involved (Figure 3).140,141 NET formation, by leading to the formation of heterotypic aggregates within blood vessels, is likely to have a large impact on the outcome of inflammatory processes. Specifically, signs of NET formation have been described in perivascular spaces, in the brain parenchyma nearby blood vessels, and in the lumen of capillaries in the ischemic brain in the tMCAO model.
142
Importantly, the administration of DNase I, which degrades NETs, has been shown to have a protective in vivo effect in murine models after ischemic stroke.
143
To date, NETs have been detected in venous and arterial thrombosis in patients.140,144 Recent studies showed that thrombus NETs content is associated with poor outcome in cerebral ischemia and may be responsible for reperfusion resistance, including mechanical or pharmacological approaches with intravenous t-PA.145,146 Even more, Vallés et al.
147
not only found that NETs were significantly elevated in the plasma of acute ischemic stroke patients when compared to healthy subjects, but also that NETs levels were increased in patients who were over 65 years of age or with a history of atrial fibrillation (AF), cardioembolic stroke, high glucose levels, and severe stroke scores at admission and discharge. However, data are still scarce and further studies are required to establish their role in stroke patients.
Circulating and infiltrated monocytes and resident microglia participate actively in an immune-inflammatory scenario after stroke. In this environment, these cells are able to respond to different signals, modulate their response and change their phenotype (M1/M2 polarization). Different mechanisms could be proposed as possible targets for modulating the neuroinflammation process.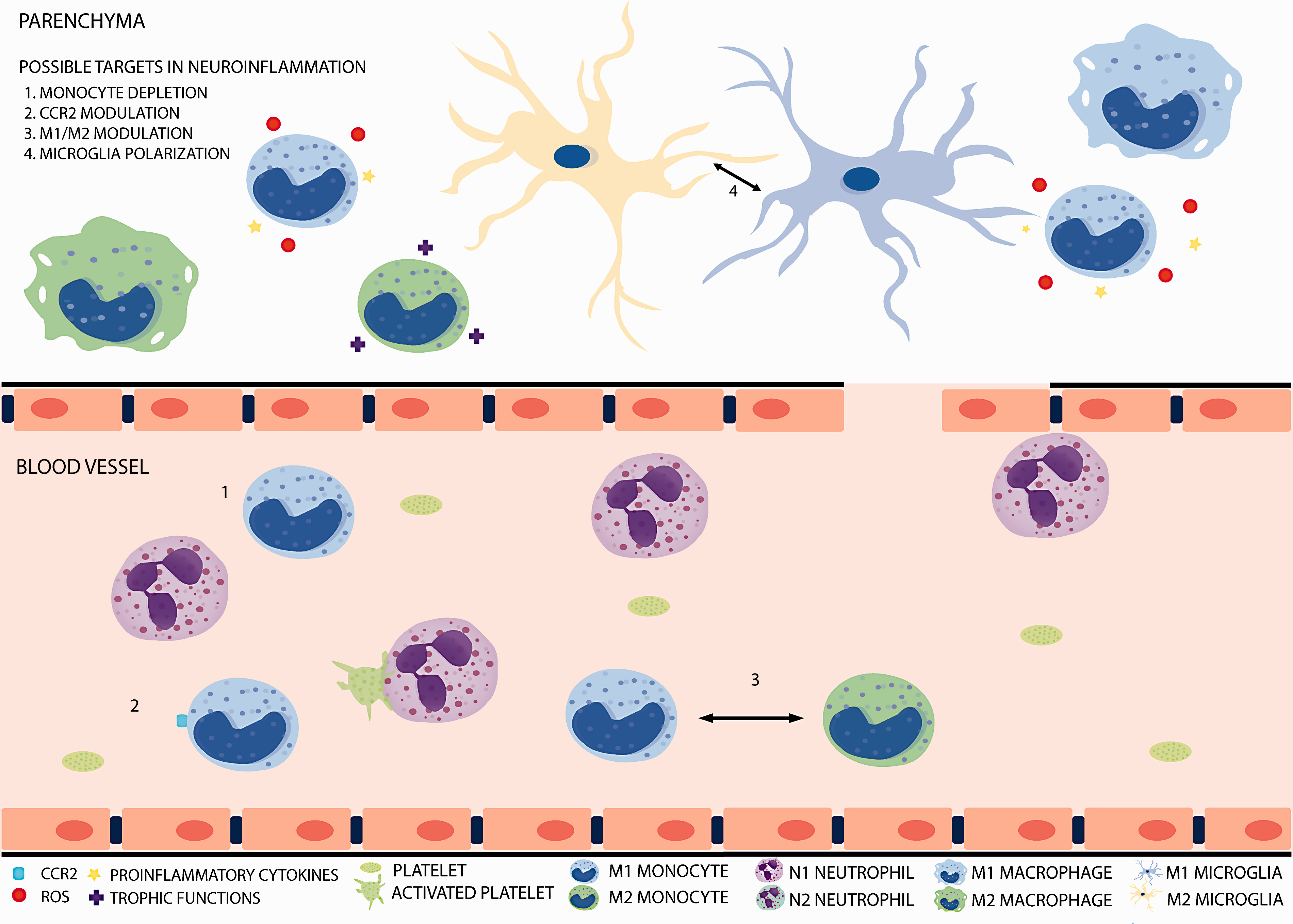
Modulation of neutrophil function as a novel therapeutic target in neuroinflammation
All the aforementioned data evidence the high degree of plasticity of the myeloid system. Monocytes, neutrophils and microglia have been described to play different functions and to be composed by heterogeneous cell populations with specialized functions that could be exploited to investigate novel therapeutic targets for the treatment of brain damage associated to neuroinflammation after stroke. The importance of monocytes/macrophages and microglial cells in this context has been thoroughly reviewed. 148 Importantly, growing evidence supports the therapeutic interest of neutrophils and their interaction with platelets.
The inhibition of neutrophil recruitment to the ischemic tissue is a strategy that could be theoretically approached by targeting the signalling pathways involved (Figure 2). In this context, combined pharmacological inhibition of CXCR1 and CXCR2 reduced ischemic brain injury and improved outcome in a rat stroke model,
149
although this improvement did not occur when only CXCR2 was blocked.
150
CCR2 loss of function using CCR2−/− mice reduced the infarct volume and neutrophil infiltration but this target is not neutrophil-specific as it also acts on monocyte recruitment to the ischemic tissue.
54
A promising target for the early phases of acute stroke at the level of neutrophil recruitment to the ischemic tissue is the inhibition of the P-selectin-PSGL-1 interaction that leads to platelet–neutrophil adhesion, a process that is neuroprotective in experimental models of stroke.
136
Neutrophils, under inflammatory conditions, activate and contribute to the inflammatory process and its resolution. They are able to skew to different phenotypes (including N1/N2 polarization), produce and release different cytokines, form platelets–neutrophil complexes, adhere to the endothelium and infiltrate into damaged tissues. All these steps are potential targets to modulate neuroinflammation.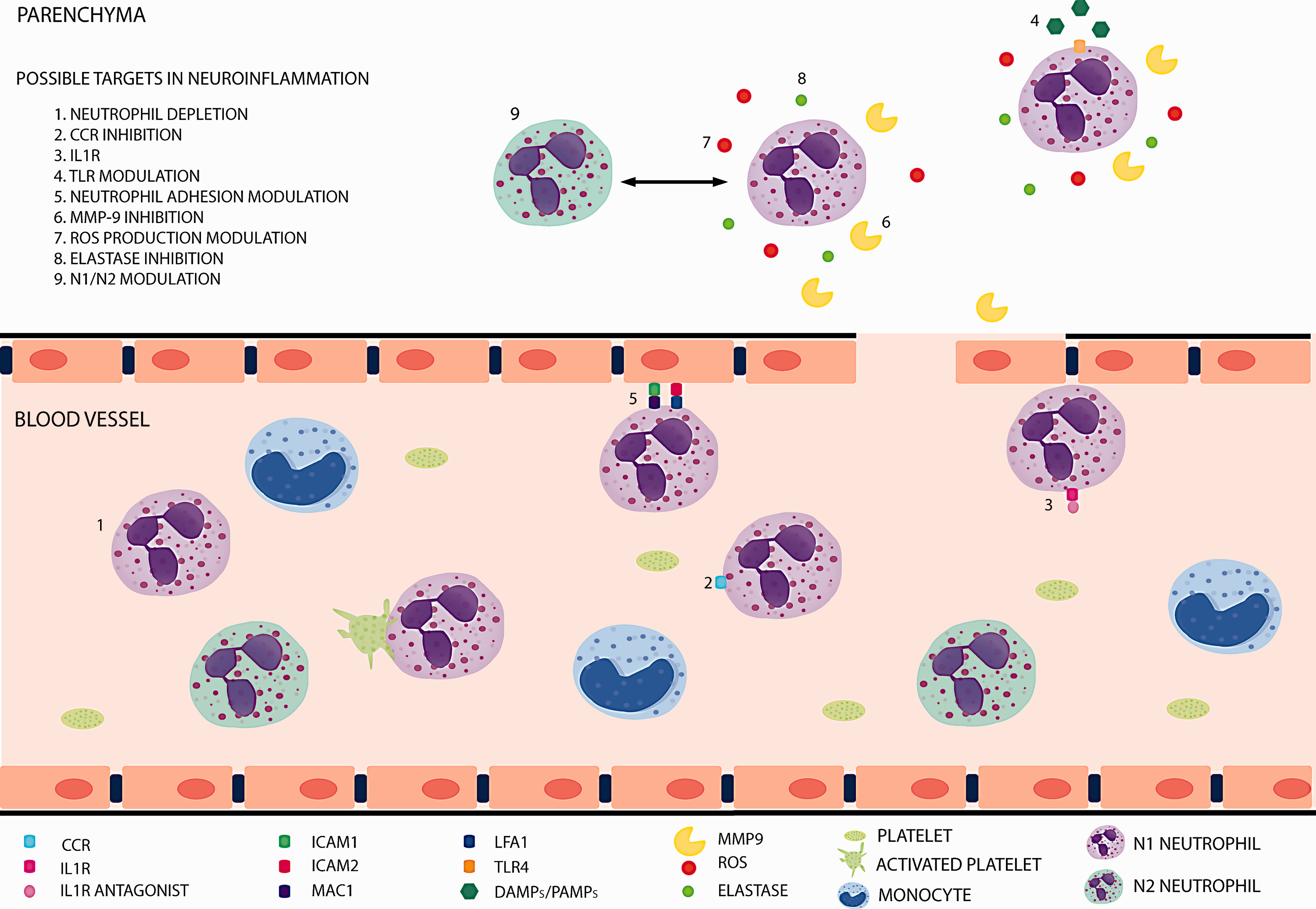
A related potential approach is to impede neutrophil infiltration (Figure 2). This process is mediated by integrins and selectins. The blockade of ICAM-1 showed some promising results in animal models but failed to be effective in patients.151,152 The effect of blocking LFA1 (CD11a/CD18) and Mac1(CD11b/CD18), which mediate neutrophil adhesion through ICAM1 and ICAM2 in endothelial cells, has also been explored.153–155 Again, the results have been different depending on the timing and the model used. Inhibiting LFA1 and Mac1 have been tried out in patients with no success.156,157 It should be noted that the infiltration of neutrophils into the ischemic tissue is considered controversial. Some authors even claim that neutrophils do not infiltrate into the parenchyma but remain in the perivascular spaces, 158 an effect that could depend on the ischemia model used, transient or permanent occlusion, as the presence of neutrophils within the brain parenchyma has also been shown. 103 A crucial issue that needs to be emphasised is the relevance of reperfusion for post-stroke inflammatory response. An interesting possibility is that neutrophils have different fates and/or roles depending on their final location, either within the vasculature or into the parenchyma of the ischemic tissue. In relation with the intravascular location, an additional aspect to be considered is the interaction of neutrophils with platelets and the subsequent release of NETs which may lead to vascular “thromboinflammatory” injury. 159 The understanding of these processes may provide new opportunities for the development of novel therapeutic strategies in ischemic stroke that remain to be studied.
Neutrophil-mediated detrimental effects are also possible targets (Figure 2). Neutrophils contribute to the disruption of the BBB through the release of proteases (such as MMP-9 and elastase) and ROS. MMP-9 has been correlated with BBB disruption and haemorrhagic transformation both in animal models and patients,
160
and elastase promotes the degradation of the basal lamina and the extracellular matrix.
99
Pharmacological and genetic approaches to block these molecules have showed promising results in animal models.161,162 However, this potential therapy remains to be proved beneficial in patients, especially considering the potential reparative actions of MMP-9, also known to promote angiogenesis/neurogenesis in the chronic phase of stroke (see, for instance, Lee et al.,
163
Barkho et al.
164
and Hao et al.
165
).
Platelets switch between a quiescent state to an activated one under inflammatory conditions and release different inflammatory mediators from their storage compartments into circulation. They are capable of interacting with neutrophils through P-selectin. This neutrophil–platelet interaction is bidirectional. Activated platelets can induce NETs formation and, also, NET components can activate platelets. Theoretically, these steps could be manipulated in order to modulate the neuroinflammation process.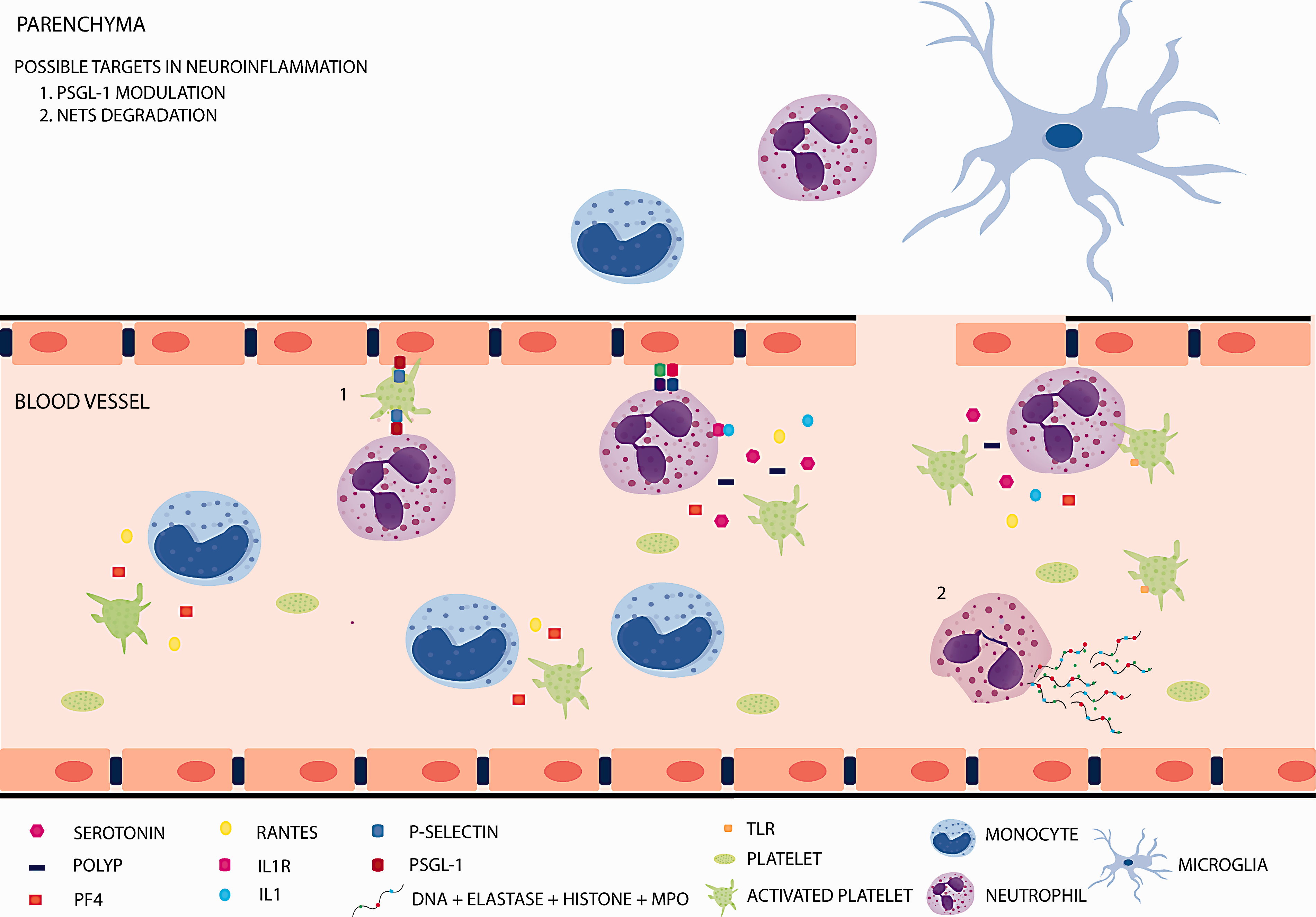
Finally, the modulation of neutrophil phenotype towards phenotypes with pro-resolving properties in stroke is an interesting novel therapeutic possibility (Figure 2), aiming to specific targets such as, for instance, PPARγ and TLR receptors.103,106
In summary, current knowledge underlines the importance of the identification of the myeloid cell subsets directly associated to neuroprotection after stroke. In addition, it is also essential to characterize specific states of activation in myeloid cells associated to brain ischemia and to explore the signalling pathways by which these states determine stroke progression and outcome, as well as the mechanisms and consequences of platelet–leukocyte interactions. The comprehension of these processes may pave the way to novel therapeutic avenues to stop the cytotoxic phase of the inflammatory response and to promote neuroprotection and tissue repair after ischemic stroke. Furthermore, a better characterization of the myeloid cell subsets that orchestrate the response to brain ischemia and their dynamics may help us to design specific treatments within specific therapeutic windows with the aim to increase the benefits of the modulation of cell–cell interactions in the immune response after stroke.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from Spanish Ministry of Economy and Competitiveness SAF2015-68632-R (MAM) and SAF2016-81716-REDC (MAM), from Instituto de Salud Carlos III and cofinanced by Fondo Europeo de Desarrollo Regional (FEDER) «Una manera de hacer Europa» PI17/01601 (IL) and RD16/0019/0009 (IL), and from Regional Madrid Government B2017/BMD-3688 (IL).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.