
Abstract
The aim of the present study was to evaluate the influence of bradykinin on microcirculatory changes and outcome after global cerebral ischemia (15 minute) in Mongolian gerbils. The cerebral microcirculation was investigated by fluorescent intravital microscopy. Survival and functional outcome was evaluated up to 4 d after ischemia. Animals were treated with the selective B1 and B2 receptor antagonists B 9858 and CP 0597, respectively, and the nonselective B1/B2 receptor antagonist B 9430. Leukocyte activation was significantly reduced by all antagonists as indicated by a significant decrease in the number of rolling (33 ± 20, 6 ± 8, 9 ± 10, and 13 ± 10) and adherent leukocytes (9 ± 7, 3 ± 4, 1 ± 1, and 2 ± 3 · 100 μm–1 · min–1 in controls and in animals treated with B1, B2, and B1/B2 antagonist, respectively). Arteriolar diameters were significantly reduced during reperfusion (35 ± 11 before and 27 ± 8 μm 40 minutes after ischemia) in animals treated with the B2 antagonist. The postischemic hypoperfusion, however, was not affected. Mortality was significantly higher in animals treated with the B1 and the B1/B2 antagonist. The authors concluded that bradykinin is involved in postischemic disturbances of cerebral microcirculation. The therapeutic effect of specific bradykinin receptor antagonists on functional outcome, however, remains unclear.
Kinins, in particular bradykinin and its metabolite des-Arg9-bradykinin, fulfill all criteria of a mediator involved in brain ischemia. First, all components of the system, including kallikreins (the proteolytic enzymes), kinins (the peptides), and kininases, have been described for the brain (Camargo et al., 1973; Kariya et al., 1981; Kizuki et al., 1994; Simson et al., 1986). Second, bradykinin levels as well as kininogen consumption increase after cerebral ischemia and reperfusion (Kamiya et al., 1993; Makevnina et al., 1994). Third, bradykinin synthesis inhibitors and bradykinin receptor antagonists can reduce secondary brain damage after cerebral ischemia and trauma (Kamiya et al., 1993; Maier Hauff et al., 1984; Relton et al., 1997).
The effects of bradykinin are mediated via two different bradykinin receptors, the B1 (inducible) and the B2 (constitutive) receptor (Hall, 1992). On binding of the agonist to the receptor, a G-protein is activated, resulting in production of prostaglandins and leukotrienes, mobilization of Ca2+, and activation of phosphokinase C (Bhoola et al., 1992; Regoli et al., 1980). Kinins thereby initiate the production of certain inflammatory mediators, that is, free radicals, lipid peroxidation, nitric oxide, eicosanoids, and cytokines (Holland et al., 1990; Whalley et al., 1987; Wolfe and Pappius, 1983). Via this pathway bradykinin may contribute to the induction of the inflammatory response after cerebral ischemia.
One of the most prominent parts of inflammation, the activation of leukocytes has been found in global as well as focal cerebral ischemia in various species (Barone et al., 1991; Clark et al., 1993; Garcia et al., 1994; Hallenbeck et al., 1986; Uhl et al., 2000). Until recently, it was assumed that this inflammatory reaction was delayed and intended to remove tissue debris from the necrotic area. But leukocyte activation has also been found in the first minutes and hours after cerebral ischemia, which implies that these cells may play a role in the progression from ischemia to brain infarction (Garcia et al., 1994; Uhl et al., 2000). In several studies attenuation of the brain damage after cerebral ischemia was observed by antileukocyte intervention, using either leukocyte depletion, adhesion molecule antibodies, or knockout mice deficient in an adhesion molecule (Bednar et al., 1991; Connolly Jr. et al., 1997; Zhang et al., 1995).
Leukocytes accumulate in the brain parenchyma after a cascade including margination, adhesion, and transmigration through the vessel wall. Many molecules are known to orchestrate activation of leukocytes and their extravasation, that is, integrins, selectins, cytokines, and chemokines. The localization of B1 and B2 receptors on the surface of endothelial cells (Raidoo et al., 1996; Wahl et al., 1996) and the localization of kininogens, kallikreins, and kinin receptors on the plasma membrane of neutrophils (Bhoola et al., 1992; Gustafson et al., 1989) imply an involvement of the kallikrein–kinin system in the interactions of these cells. First evidence for a role of bradykinin in the activation of leukocytes has been described in the peripheral organs (Ahluwalia et al., 1996; Hoffmann et al., 1996; Tayeh et al., 1998).
To date, it is not known whether bradykinin contributes to microcirculatory alterations including leukocyte adherence and vascular reactivity during reperfusion after global cerebral ischemia (GCI). The aim of the present study was to investigate the influence of bradykinin B1 and B2 receptor antagonists on microcirculatory changes, focusing on leukocyte–endothelium interactions in the early reperfusion period and their impact on the development of neurologic deficits and the viability of neurons after GCI.
MATERIALS AND METHODS
The study used 49 Mongolian gerbils (body weight 60 to 75 g; aged 12 to 16 weeks). Three to 4 animals were kept in Macrolon type 3 cages (Ehtet, Emmendingen, Germany) before ischemia and singly thereafter. The animals had free access to tap water and pellet food; room temperature was 22°C. The experiments, conducted according to institutional guidelines, were approved by the state government of Bavaria.
Four experimental groups were set up, receiving the B1, the B2, or the B1/B2 receptor antagonist or the vehicle. Ten animals were randomly assigned to each group. After surgical preparation, intravital microscopy was performed. Three baseline measurements were carried out at 20-minute intervals. Global cerebral ischemia was achieved by occlusion of both common carotid arteries for 15 minutes. On reperfusion, intravital microscopic measurements were performed at 5, 20, 40, 60, 90, 120, and 180 minutes. Thereafter, the integrity of the blood–brain barrier was tested. After microscopy the catheters were removed, the wounds were sutured, and the animals were allowed to wake up. During a 4-d survival period, the neurologic deficit and the body weight was obtained daily. On day 4 animals were killed and the brains were harvested for histology.
The surgical preparation has been described in detail (Uhl et al., 2000). In brief, anesthesia under spontaneous breathing was induced with 4% halothane and continued during the preparation at a concentration of 1.5% and during intravital microscopy before ischemia at 0.8%, 0.4% during ischemia, and 0.6% during reperfusion. Oxygen content of the inspired gas was 30%; the rate was 1 L/min. Body temperature was maintained at 37.0°C using a feedback-controlled heating pad. Polyethylene catheters were inserted into the tail artery for continuous blood pressure monitoring and into the femoral vein for application of fluorescent dyes and drugs. The common carotid arteries were dissected bilaterally and encircled with a monofil thread (Prolene 5–0; Ethicon, Norderstedt, Germany) without disturbing the blood flow. The skull was then fixed in a stereotactic frame (Model 51600; Stoelting Co., Wood Dale, IL, U.S.A). After a midsagittal skin incision from the forehead to the neck, the calvaria were exposed and a rectangular 4 × 4-mm cranial window was prepared over the left parietal hemisphere, with the dura mater left intact. During intravital microscopy the dura mater was rinsed continuously with physiologic saline at 37.0°C.
For intravital microscopy, the animals were placed on a computer-controlled microscope stage for repeated analysis of identical vessel segments. The intravital fluorescence microscope (Leitz, Wetzlar, Germany) was equipped with a 75 W xenon bulb and N2 and L3 Ploemopak filter blocks for epiillumination. Enhancement of microvessels was achieved by an intravenous injection of fluorescein isothiocyanate–labeled dextran (molecular weight: 150.000; Sigma, Taufkirchen, Germany) before the first measurement (bolus of 0.2 mL of a solution of 65 μmol/L). Leukocytes were stained in vivo before each measurement by intravenous injection of 0.05 mL of a rhodamine 6G solution of 0.2 mol/L (Merck, Darmstadt, Germany). The microcirculatory parameters were analyzed using a salt-water immersion objective ×25. Recordings lasted 10 seconds using the L3 filter block for assessment of diameter and were restricted to 30 seconds using the N2 filter block for quantification of leukocyte–endothelium interactions and an additional 10 seconds for measurement of the arteriovenous transit time to minimize the light dose to the brain surface. To test the integrity of the blood–brain barrier at the end of the experiment, 0.2 mL of sodium–fluorescein solution of 2.7 mmol/L (Sigma) were injected intravenously. Extravasation of the fluorescent marker was observed with the L3 filter and an objective with a magnification of ×10. For measurement of the arteriovenous transit time a bolus of rhodamine 6G (0.05 mL) was given intravenously and the lighting of an arteriole and a concomitant venule were recorded, using the N2 filter block. All regions of interest, comprising at least 1 capillary bed, 1 arteriole, and 3 venules, were recorded by a SIT-video camera (C 2400; Hamamatsu Photonics, Herrsching, Germany) and stored on S-VHS videotapes.
Arteriolar and venular diameters (μm), the number of rolling and adherent leukocytes in postcapillary venules (n · 100 μm–1 · min–1), the capillary density (cm–1), and the integrity of the blood–brain barrier (yes/no) were measured by using a computer-assisted microcirculation analysis system (CapImage; Ingenieurbüro Dr. Zeintl, Heidelberg, Germany). The leukocytes were classified according to their interaction with the venular endothelium as rolling or adherent leukocytes, as described previously (Uhl et al., 2000). The capillary density was defined as the length of capillaries per area, which were perfused with plasma tracer and blood cells. Arteriovenous transit time was measured using an image analysis system (IBAS 2.0; Kontron, Eching, Germany). For analysis of the arteriovenous transit time 22 images over a period of 6 seconds were digitized. Means of gray tones in an area of 40 × 40 pixels in the arteriole and the venule were calculated in each image. The calculation of the mean arteriovenous transit time was performed using the difference integral method as suggested by Rovainen et al. (1993).
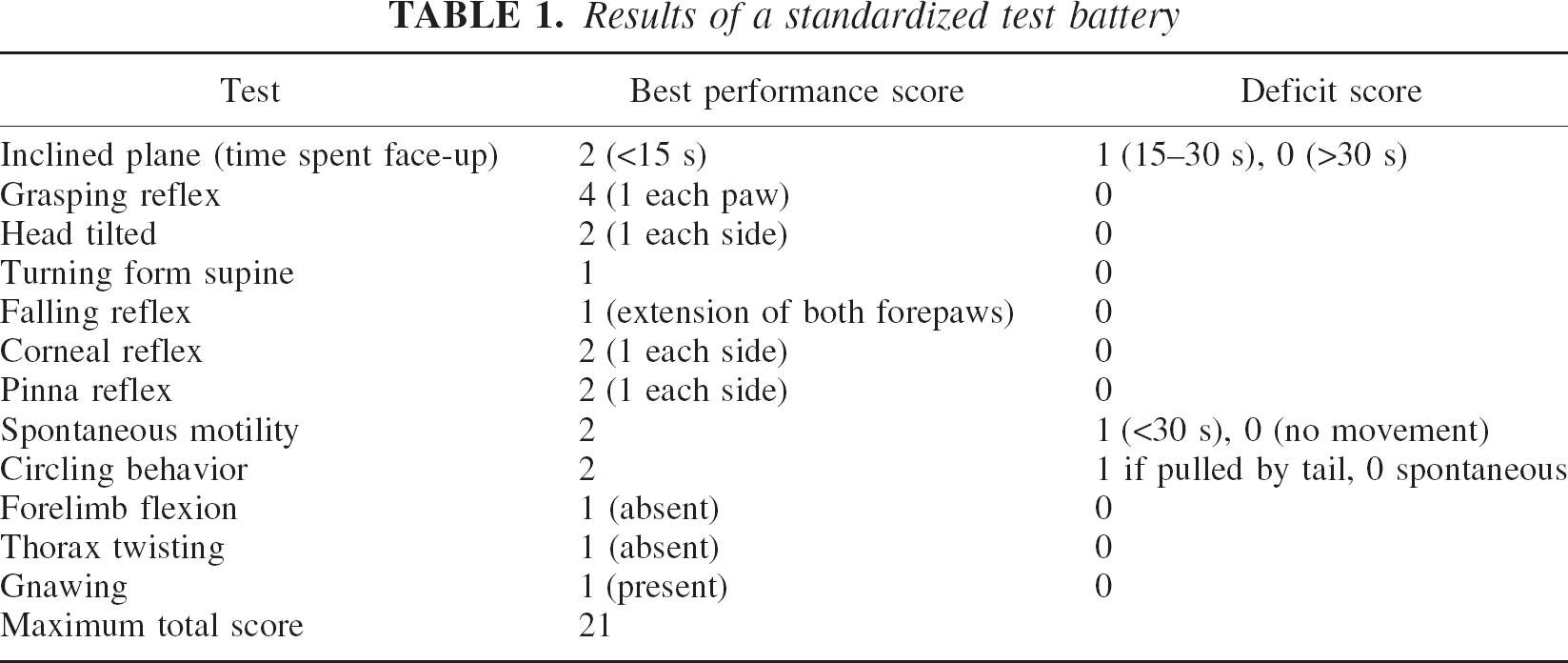
To quantify postischemic neurologic deficits animals were examined daily before and after ischemia by a blinded investigator using a standardized test battery containing 12 different tests (Table 1) (Glickman et al., 1970; McGraw, 1977).
Results of a standardized test battery
On day 4 after GCI, animals were killed and all brains were harvested for histologic evaluation. In deep halothane anesthesia transcardial perfusion was performed with phosphate-buffered paraformaldehyde solution (2%) after flushing the circulation with physiologic saline for 60 seconds at a pressure of 100 cm H2O. Brains were removed and stored in paraformaldehyde solution for at least 24 h until further processing. The brains were then dehydrated in ethanol and embedded in paraffin. Coronal slices of 5-μm thickness were cut 1.7 mm caudal and 0.5 mm rostral to the bregma and stained with cresyl violet. The density (n/mm2) of morphologically viable neurons from both hemispheres was quantified in the CA1, CA2, CA3 and CA3/CA4 sectors of the hippocampus, in the striatum, and in the parietal and frontoparietal neocortex by a blinded investigator according to the procedure and the identification criteria as described in detail previously (Stummer et al., 1994).
For blockade of the bradykinin B1 receptor, the antagonist B 9858 was administered. This antagonist shows high potency on B1 receptors and a very low potency on B2 receptors (Hanson et al., 1996). Inhibition of the bradykinin B2 receptors was achieved by administration of CP 0597, a potent, long-acting, and selective B2 receptor antagonist (Goodfellow et al., 1996). Blockade of the B1 and B2 receptors was obtained by application of B 9430, a long-acting and potent antagonist on both receptors (Burkard et al., 1996). The potency, selectivity, and duration of action of the antagonists have been tested in a model of cardiovascular response to agonists of the B1 and B2 bradykinin receptor in dogs. The intravenous dose of CP 0597 and B 9430 in blocking the hypotensive response to bradykinin (B2 receptor agonist) has been 50 ng/kg per minute for both antagonists. The intravenous doses of B 9858 and B 9430 in blocking the hypotensive response to des-Arg9-bradykinin (B1 receptor agonist) have been 50 and 220 ng/kg per minute, respectively. The duration of action was more than 4 h for all of the antagonists (Hanson et al., 1996). The compound CP 0597 has been used in a dose of 300 ng/kg per minute subcutaneously in a focal ischemia model in rats, where it has reduced the ischemic brain damage (Relton et al., 1997). The three antagonists were provided by Cortech (Denver, Colorado, U.S.A.). Treatment with the antagonists or the vehicle (phosphate-buffered solution, Dulbecco's; Life Technologies, Karlsruhe, Germany) was started by intravenous injection of 18 μg/kg dissolved in 0.1 mL phosphate-buffered solution, followed by continuous subcutaneous infusion by microosmotic pumps (Alzet, model 1007D; Alza Corp., Palo Alto, CA, U.S.A.). Primed pumps containing vehicle or one of the antagonists were implanted into the subcutaneous space of the animals back, releasing 300 ng/kg per minute at a rate of 0.5 μL/h until the end of the experiment on day 4.
Statistical evaluation of data was performed on a personal computer using Sigma Stat 2.0 (SPSS Science, Chicago, IL, U.S.A.). The Kruskal-Wallis one-way analysis of variance on ranks, followed by the Dunnett test, was used for analyzing differences among control and treatment groups. The Friedman test, followed by the Dunnett method, was used for detection of differences within each group. All data are presented as mean ± SD. A statistically significant difference was assumed at P < 0.05. Statistical analysis of survival was performed with the software Statistica (StatSoft, Tulsa, OK, U.S.A.) using the Mantel test followed by the Cox F test.
RESULTS
Mean arterial blood pressure (mm Hg) was 73 ± 9, 72 ± 5, 74 ± 5, and 73 ± 8 in the vehicle- and the B2-, B1/B2-, and B1 antagonist–treated animals, respectively, 40 minutes before ischemia. The infusion of the antagonists 15 minutes before ischemia did not alter the blood pressure in any of the groups. During occlusion of both carotid arteries, the blood pressure increased significantly (P < 0.05) in the groups treated with the different antagonists and returned to preischemic values on reestablishment of cerebral perfusion, with no changes during the period of microscopy (data not shown).
The diameter of pial arterioles remained unchanged during the preischemic phase. Infusion of the bradykinin antagonists did not alter the diameters under physiologic conditions. During ischemia the width of the arterioles in the antagonist-treated animals decreased and returned to baseline values shortly after reperfusion. Diameters in animals with B2 antagonist were significantly reduced between 20 and 120 minutes of reperfusion as compared with preischemic values. Vehicle-, B1/B2 antagonist-, and B1 antagonist–treated animals showed no change during the early reperfusion period.
Venular diameters remained unchanged during the preischemic phase. Infusion of the antagonists had no effect on the diameters. In animals treated with vehicle, B2-, and B1/B2 antagonist, diameters were significantly reduced from 20 to 180 minutes after ischemia and from 40 to 120 minutes in the B1 group. No significant differences among the four groups were found (Table 2).
Comparison of the four test groups
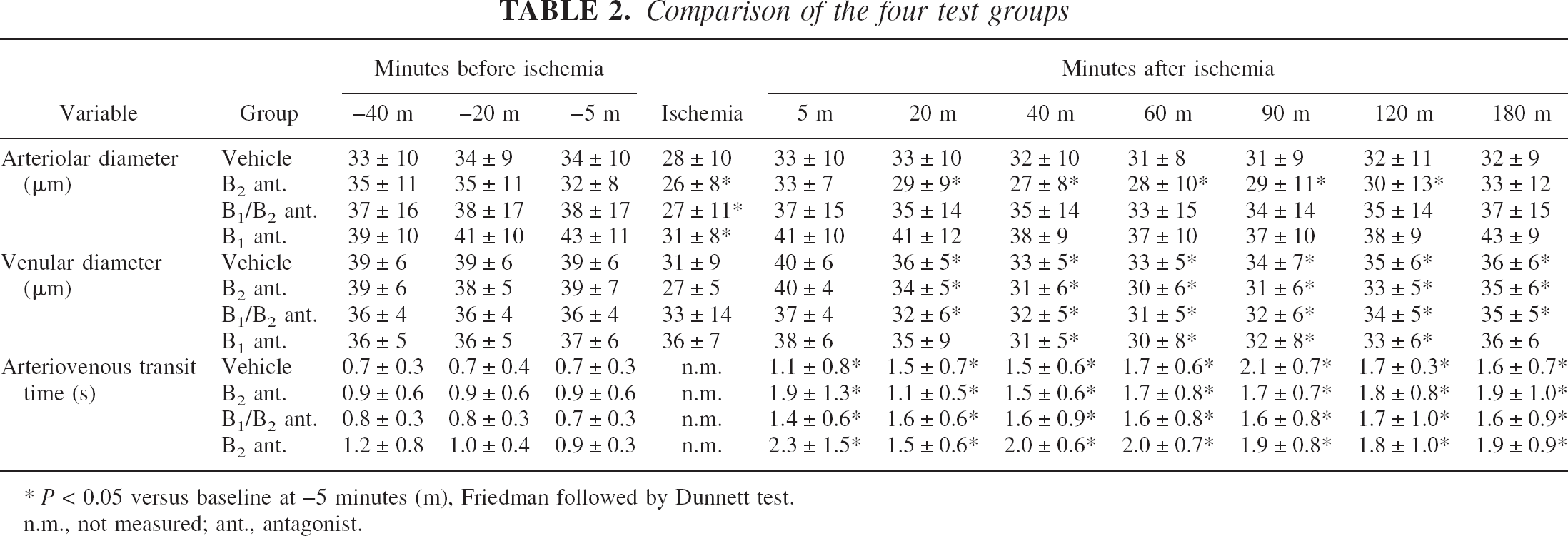
P < 0.05 versus baseline at −5 minutes (m), Friedman followed by Dunnett test.
n.m., not measured; ant., antagonist.
In all groups the number of rolling leukocytes (n · 100 μ–1 · min–1) increased continuously during reperfusion. The peak was reached at 3 h of reperfusion, with 33.0 ± 19.6 in the control animals, but was significantly lower in treated animals, with 9.3 ± 9.8 in the B2 antagonist group (P < 0.05), 6.2 ± 7.9 in the B1/B2 antagonist–treated animals (P < 0.05), and 12.9 ± 9.8 in the B1 antagonist–treated animals (P < 0.05; Figs. 1 and 2).
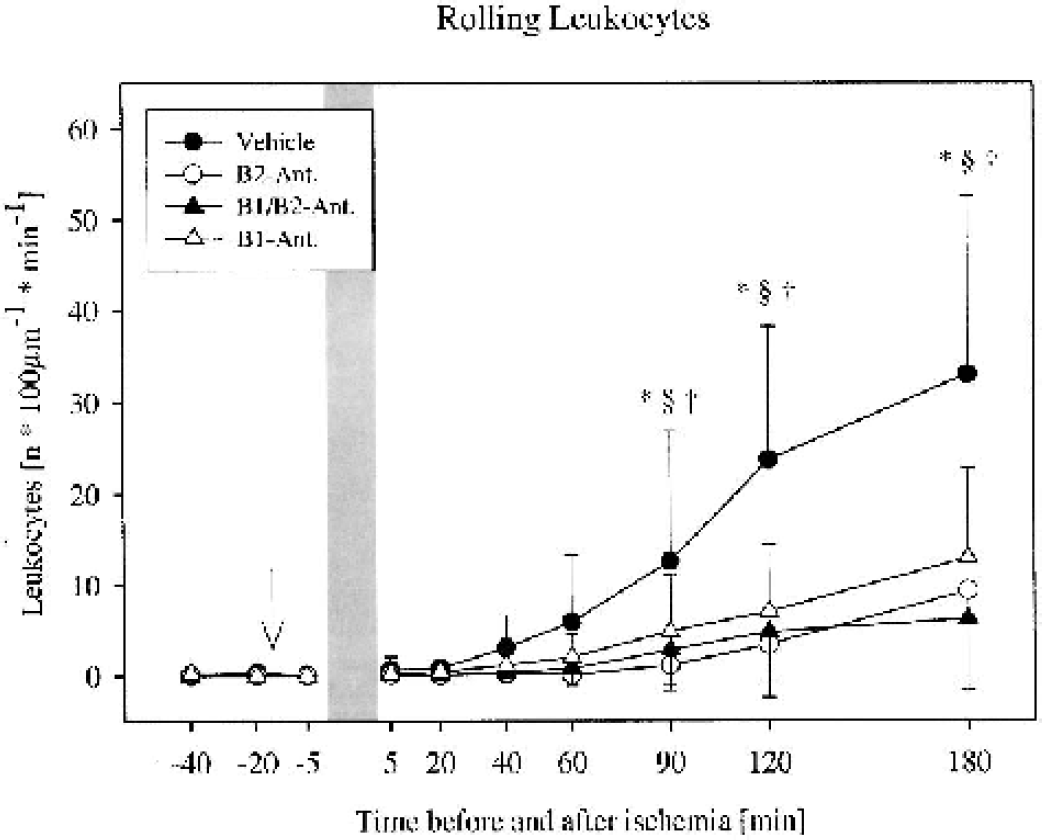
Frequency of leukocytes rolling along the venular endothelium before and after global cerebral ischemia in gerbils. An increase in the number of rolling leukocytes was observed in all 4 groups (n = 10 per group). Treatment with the bradykinin antagonists significantly reduced leukocyte rolling. *P < 0.05 vehicle versus B2 antagonist, §P < 0.05 vehicle versus B1/B2 antagonist, †P < 0.05 vehicle versus B1 antagonist, Kruskal-Wallis followed by Dunnett test. Mean ± SD. The arrow indicates the start of infusion of the antagonist or the vehicle. Bar = duration of ischemia.
Intravital fluorescence microphotographs of the Mongolian gerbil brain surface showing a pial venule during the control period before
Firm adherence of leukocytes increased significantly in control animals, but less so in animals treated with the B1/B2 antagonist and with the B1 antagonist. The total number of adherent leukocytes (n · 100 μ–1 · min–1) at 3 h was 8.5 ± 7.3 in control animals, but only 0.9 ± 0.7 in the B2 antagonist–treated animals (P < 0.05), 1.7 ± 2.5 in the B1/B2 antagonist–treated animals (P < 0.05), and 3.3 ± 3.9 in the B1 antagonist–treated animals (P < 0.05; Figs. 2 and 3).
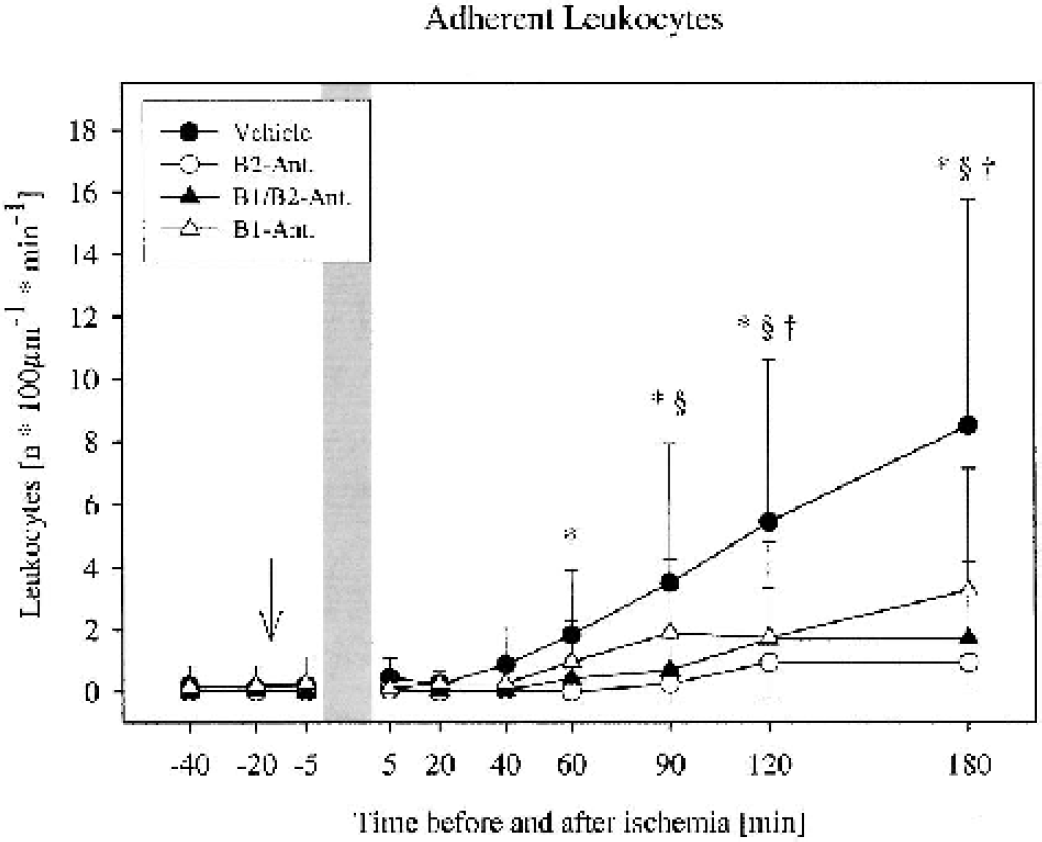
Frequency of leukocytes adherent to the venular endothelium before and after global cerebral ischemia in gerbils. An increase in the number of adherent leukocytes was observed in all 4 groups (n = 10 per group). Treatment with the bradykinin antagonists significantly reduced leukocyte adherence. *P < 0.05 vehicle versus B2 antagonist, §P < 0.05 vehicle versus B1/B2 antagonist, †P < 0.05 vehicle versus B1 antagonist, Kruskal-Wallis followed by Dunnett test. Mean ± SD. The arrow indicates the start of infusion of the antagonist or the vehicle. Bar = duration of ischemia.
The capillary density (cm–1) of 149 ± 20, 167 ± 39, 176 ± 46, and 162 ± 27 at 5 minutes before ischemia was slightly reduced during reperfusion to 138 ± 31, 150 ± 38, 147 ± 47, and 138 ± 47 at 3 h after ischemia in the vehicle-, the B2 antagonist-, the B1/B2-antagonist-, and the B1 antagonist–treated animals, respectively. However, no statistically significant difference, either among the groups or to preischemic values within each group, was found (data not shown).
The arteriovenous transit time as a measure for the local microvascular perfusion was significantly prolonged in all four experimental groups during the entire reperfusion period, indicating postischemic hypoperfusion. There was no difference among the four groups (Table 2).
Opening of the blood–brain barrier was not observed in any group 3 h after ischemia. The tracer sodium fluorescein remained intravascular during the observation period of 20 minutes.
The preischemic neuroscore (points) of 21 ± 0 was reduced to 13.4 ± 10.1, 14.4 ± 8.6, 5.0 ± 8.4, and 6.7 ± 9.2 at day 4 in the animals treated with vehicle, the B2 antagonist, the B1/B2 antagonist, and the B1 antagonist, respectively (P < 0.05 vs. preischemic values). No significant difference among the four experimental groups was detected. The body weight (g) of 66 ± 4, 69 ± 3, 66 ± 3, and 68 ± 4 before ischemia was reduced to 56 ± 4, 53 ± 6, 49 ± 1, and 52 ± 7 in the animals with treated with vehicle, B2 antagonist, B1/B2 antagonist, and B1 antagonist, respectively (P < 0.05 vs. preischemic values; only remaining animals at day 4 were considered). No significant difference among the four experimental groups was detected (data not shown).
Nine of 10 animals treated with B2 antagonist survived the 4-d observation period, whereas 7 in the control group survived. Additional blocking of B1 receptors resulted in an increased mortality (P = 0.03). Only 3 of 10 animals treated with the B1/B2 antagonist survived the 4 d, whereas B1 blockade alone resulted in survival of 4 animals of that group. Comparison of the B2 with the B1/B2 and the B1 antagonist showed significant reduction of survival in the B1/B2 and the B1 groups (P = 0.004 and P = 0.001, respectively, Fig. 4).
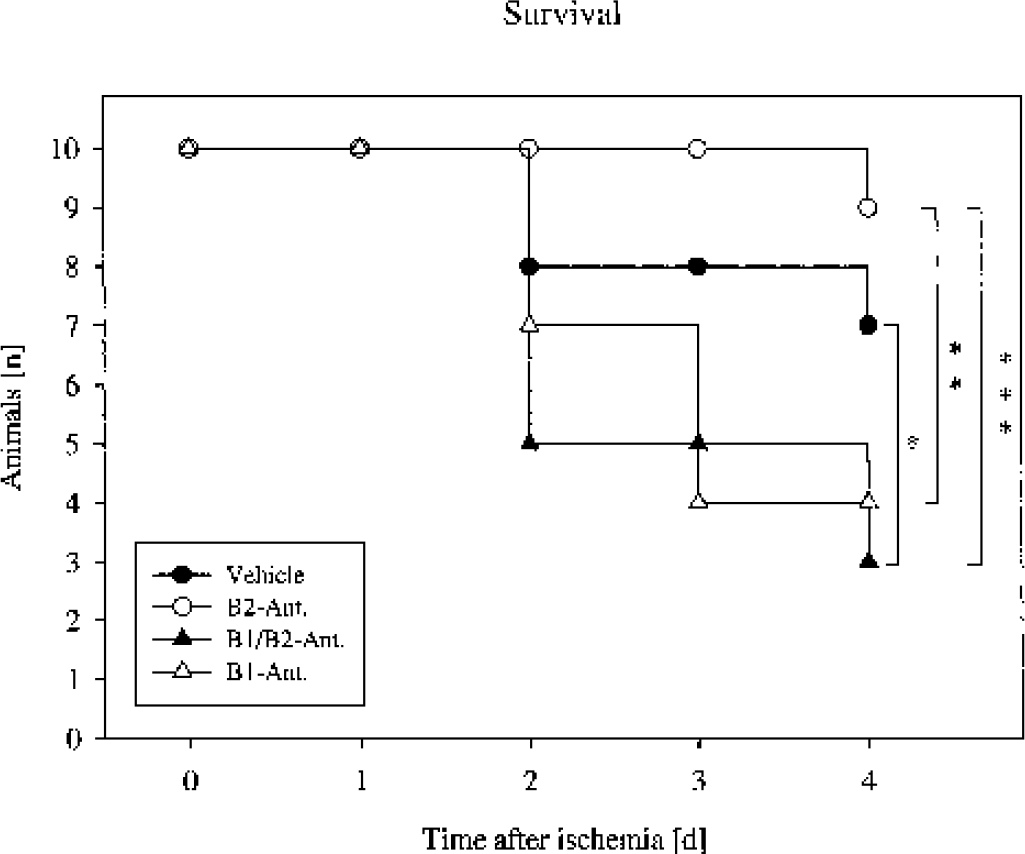
Kaplan-Meier survival plot of Mongolian gerbils after 15 minutes of global cerebral ischemia. Comparison of the vehicle-treated animals with the B1/B2 group showed a significant reduction of survival in the latter group. Comparison of the B2 with the B1/B2 and the B1 antagonist showed significant reduction in the B1/B2 and the B1 groups. *P = 0.03, **P = 0.001, and ***P = 0.004, Mantel followed by Cox F test.
In the groups treated with vehicle, B2 antagonist, B1/B2 antagonist, and B1 antagonist 6, 8, 3, and 4 brains, respectively, were analyzed. Only brains that were fixed by perfusion with paraformaldehyde were considered for morphometry. The density of viable neurons (n/mm2) in animals treated with vehicle, B2 antagonist, B1/B2 antagonist, and B1 antagonist in the CA 1 region of the hippocampus was 824 ± 203, 842 ± 253, 959 ± 254, and 830 ± 252, respectively. No significant differences in the number of viable neurons among the groups were observed in the different brain areas investigated.
DISCUSSION
The role of bradykinin as a mediator in secondary brain damage after ischemic and traumatic brain injury has been known for many years. Bradykinin has not only been shown to induce the opening of the blood–brain barrier but has also been found to be a potent vasoactive agent on cerebral vessels leading to contraction as well as dilation of vascular smooth muscle cells by the activation of B1 or B2 receptors (Unterberg et al., 1984a; Whalley and Wahl, 1983). Furthermore, bradykinin initiates the production of other mediators such as prostaglandins, arachidonic acid, cytokines, nitric oxide, and free radicals and thus may be a key mediator involved in the inflammatory reaction after cerebral ischemia.
Our data show for the first time that bradykinin is involved in the activation of leukocyte–endothelium interactions after GCI. By application of specific bradykinin-receptor antagonists it was possible to significantly reduce leukocyte rolling along and leukocyte adherence to the endothelium of postcapillary venules in the brain. A similar effect of bradykinin receptor antagonists has been described after ischemia–reperfusion of the pancreas (Hoffmann et al., 1996). The effect seems to be mediated via both bradykinin receptors, since we could not find any difference when the B1 or the B2 receptor was blocked selectively. Nonselective blocking of both receptors did not further enhance this effect. It has been widely accepted that rolling of leukocytes is dependent on interaction of the selectins and that firm adherence is dependent on interaction of integrins with their respective counterreceptors (Vestweber and Blanks, 1999). Whether the inhibition of leukocyte–endothelium interactions is the effect of a direct suppression of selectins or integrins by bradykinin antagonists or is caused indirectly via the suppression of the release of secondary mediators cannot be concluded from our data. First evidence for a direct action of bradykinin has been reported, since the expression of P-selectin on endothelial cells in vitro was stimulated by bradykinin (Tayeh et al., 1998). Numerous studies report the initiation of secondary mechanisms by bradykinin including the production of prostaglandins and platelet-activating factor, the release of cytokines such as tumor necrosis factor and interleukin-1 and free radicals (Hsu et al., 1989; Kontos et al., 1984; McIntyre et al., 1985; Tiffany and Burch, 1989). All of the aforementioned substances are known to have proadhesive properties. The significance of bradykinin for the induction of leukocyte–endothelium interactions is further supported by the presence of kininogens, kallikreins, and kinin receptors in or on the plasma membrane of neutrophils (Bhoola et al., 1992; Gustafson et al., 1989). Neutrophils provide a circulating platform for the components of the contact-phase system involved in the activation of bradykinin (Henderson et al., 1994). Owing to the short half-life of bradykinin of 30 seconds, it can be assumed that relevant concentrations of bradykinin may only be reached locally, as, for instance, during the interaction of leukocytes with the endothelium (Kariya et al., 1982). Since peptidic antagonists have been used in this study and the blood–brain barrier was found to be intact after 3 h of reperfusion, it has to be assumed that luminal, but not abluminal, bradykinin receptors have been blocked. This again supports the hypothesis that direct action on the endothelium and leukocytes, but not the brain tissue, was responsible for inhibition of leukocyte–endothelium interaction. The finding that antagonism of bradykinin inhibited leukocyte–endothelium interactions is consistent with in vivo observations that superfusion of the mesentery with bradykinin stimulates leukocyte adhesion as well as with in vitro observations that bradykinin promotes leukocyte adhesion to endothelial monolayers (Satoh et al., 1995; Shigematsu et al., 1999).
It is surprising, however, that reducing leukocyte–endothelium interactions by bradykinin antagonism did not result in an improved outcome or an increased number of vital neurons in vulnerable brain areas because, based on several animal studies, it has been postulated that activation of leukocytes contributes to the development of secondary brain damage after focal as well as global ischemia. Although antileukocyte intervention was found to result in reduced brain damage (Connolly et al., 1997; Zhang et al., 1994) in focal ischemia, the results of this therapy remain inconclusive in GCI (Grogaard et al., 1989; Schurer et al., 1991). Our data support the hypothesis that the early activation of leukocyte–endothelium interactions in GCI does not aggravate the neurologic deficit or the survival of the animals. Despite the fact that leukocyte rolling and adherence were inhibited by the bradykinin antagonists, the postischemic clinical course was not positively influenced by the treatment.
Our data are even more irritating because treatment with the three bradykinin antagonists inhibited similarly leukocyte–endothelium interactions but the selective B1 antagonist and the nonselective B1/B2 antagonist even increased mortality. Only the animals treated with the selective B2 antagonist showed a satisfying outcome. It is worth remarking that increasing neuronal loss in the hippocampus is well correlated with an increased mortality in gerbils after GCI (Weber et al., 1988). It could therefore be suspected that the neuronal loss of the nonsurviving animals was increased compared with the surviving animals. One might also speculate that the increased mortality of animals treated with B1 antagonist was caused by systemic side effects of the antagonist, but no evidence exists for this so far; chronic application of the antagonists B 9430 and B 9858 has not been described before. Since expression of the B1 receptor is inducible in the brain, another site of action of the selective bradykinin B1 receptor antagonists seems to be unlikely. Infusion of a different B1 antagonist (Lys-[Leu8]-des-Arg9-bradkyinin) for 5 d was not associated with side effects (Blais et al., 1997).
Finally, we could not find any differences in microvascular perfusion when the treated animals were compared with the control group. In all groups ischemia was followed by a prolonged time of hypoperfusion that was not influenced by blocking the bradykinin receptors. Only a transient reduction (2 h) of arteriolar diameters in animals treated with the B2 antagonist was observed as compared with the other groups in which arteriolar diameters returned to baseline values at the beginning of reperfusion. That this effect has any relevance with regard to the reperfusion injury is doubtful, since the animals treated with the B2 antagonist had a better overall outcome. The finding that an intravenously administered peptidergic B2 receptor antagonist reduced arteriolar diameters by 23% at 40 minutes of reperfusion, compared with preischemic values, is not consistent with former observations. Bradykinin is known as a potent vasodilator of cerebral arteries if the mediator is superfused over the cortex. No effect on diameters could be observed, however, when bradykinin was infused into the carotid artery, suggesting that bradykinin receptors are not expressed on brain endothelial cells under physiologic conditions (Unterberg et al., 1984b). In the present study peptid antagonists were used. Assuming that the blood–brain barrier was intact during ischemia and reperfusion, the antagonists should act only on the luminal membrane of the endothelial cell. It can be concluded, therefore, that stimulation of B2 receptors on the luminal surface of brain endothelial cells by bradykinin during early reperfusion after GCI dilates pial arterioles, since inhibition of the B2 receptor reduced arteriolar diameter. Because the nonselective antagonist had no effect on vessel diameter, one might speculate that antagonism of B1 receptors abolished the vasoconstriction elicited by the antagonism of B2 receptors. To address these postulates, further studies investigating the vasomotor response of cerebral vessels under pathologic conditions (i.e., ischemia) are necessary.
In conclusion, bradykinin is involved in the activation of leukocytes in the early reperfusion period after GCI. This effect is mediated via both bradykinin receptors, since blocking of either bradykinin receptor, B1 or B2, results in a significant reduction of leukocyte–endothelium interactions. In regard to vasoreactivity, however, there seems to be a functional difference between B1 and B2 receptors because blocking of the B2 receptor resulted in a decrease of arteriolar diameters, whereas the B1 and the B1/B2 receptor antagonist had no effect. Furthermore, blocking of the B1 receptor led to an increase of mortality, which was not observed after treatment with the B2 receptor antagonist. Whether this indicates a neuroprotective role of bradykinin after GCI mediated via the B1 receptor needs further evaluation.
Footnotes
Acknowledgments:
The authors gratefully acknowledge the excellent technical assistance of Miss M. Fürst.