
Abstract
Inflammatory processes have been implicated in the pathogenesis of brain damage after stroke. In rodent stroke models, focal ischemia induces several proinflammatory chemokines, including monocyte chemoattractant protein-1 (MCP-1). The individual contribution to ischemic tissue damage, however, is largely unknown. To address this question, the authors subjected MCP-1-deficient mice (MCP-1−/−) to permanent middle cerebral artery occlusion (MCAO). Measurement of basal blood pressure, cerebral blood flow, and blood volume revealed no differences between wild-type (wt) and MCP-1−/− mice. MCAO led to similar cerebral perfusion deficits in wt and MCP-1−/− mice, excluding differences in the MCA supply territory and collaterals. However, compared with wt mice, the mean infarct volume was 29% smaller in MCP-1−/− mice 24 hours after MCAO (P = 0.022). Immunostaining showed a reduction of phagocytic macrophage accumulation within infarcts and the infarct border in MCP-1−/− mice 2 weeks after MCAO. At the same time point, the authors found an attenuation of astrocytic hypertrophy in the infarct border and thalamus in MCP-1−/− mice. However, these effects on macrophages and astrocytes in MCP-1−/− mice occurred too late to suggest a protective role in acute infarct growth. Of note: at 6 hours after MCAO, MCP-1−/− mice produced significantly less interleukin-1β in ischemic tissue; this might be related to tissue protection. The results of this study indicate that inhibition of MCP-1 signaling could be a new acute treatment approach to limit infarct size after stroke.
In human stroke and animal stroke models, various brain cells express proinflammatory mediators such as cytokines and chemokines (for review see Stoll et al., 1998; Asensio and Campbell, 1999). Targeting those molecules may provide an extended time-window for treatment because inflammatory processes progress over many hours after onset of brain ischemia (Barone and Feuerstein, 1999). For most of the ischemia-induced cytokines and chemokines, the association with brain injury relies merely on descriptive expression studies. Among those is the CC-chemokine MCP-1. A wide variety of cell types induce MCP-1, which activates as well as attracts monocytes by binding to G-protein–coupled receptors CCR2 or CCR9 (Gu et al., 1999). In the human CNS, MCP-1 was found, for example, in microglia associated with mature senile plaques in brain tissue from patients with Alzheimer's disease (Ishizuka et al., 1997), in stimulated microglia from elderly human brain (Lue et al., 2001), and in cerebrospinal fluid from patients with stroke (Losy and Zaremba, 2001). Numerous studies in animal models of neurologic diseases revealed induction of MCP-1 both at the transcriptional and translational levels (for review see Asensio and Campbell, 1999). Regarding acute brain injury and stroke in particular, excitotoxic lesions in the neonatal rat brain resulted in a dramatic upregulation of both MCP-1 mRNA and protein within 8 to 12 hours (Galasso et al., 2000a). In addition, CCR2 protein, the receptor for MCP-1 was detected (Galasso et al., 2000a).
After focal brain ischemia, early MCP-1 induction has been described in several publications in rats and mice (Gourmala et al., 1997; Wang et al., 1995; Kim et al., 1995, Che et al., 2001). A pathogenetic role of MCP-1 for the manifestation of tissue damage after brain ischemia, however, has not yet been directly shown. MCP-1 deficient mice (MCP-1−/−) previously demonstrated that MCP-1 was crucial for monocyte recruitment in several inflammatory conditions (Lu et al., 1998). In addition, these mice provided evidence of an essential role of MCP-1 for accumulation of macrophages in disease models such as atherosclerosis (Gu et al., 1998) and asthma (Gonzalo et al., 1998). MCP-1−/− mice were markedly resistant to experimental autoimmune encephalitis after active immunization with concomitant drastically reduced recruitment of macrophages to the CNS (Huang et al., 2001). To explore the role of MCP-1 in ischemic brain injury, we therefore investigated infarct volume and cellular responses after permanent middle cerebral artery occlusion in MCP-1−/− mice.
MATERIALS AND METHODS
MCP-1-deficient mice (MCP-1−/−)
Homozygous MCP-1−/− mice were obtained from Dr. B. Rollins, Dana Farber Cancer Institute, Boston, and used to establish a breeding colony. The disruption of the MCP-1 gene for this mouse strain has been described previously (Lu et al., 1998). The MCP-1−/− mice were backcrossed eight times into a C57BL/6 background and were then bred as a homozygotic MCP-1−/− line. Therefore, C57BL/6 mice obtained from Jackson Laboratories were used as wild-type (wt) controls. Male or female animals weighing 20 to 30 g were used for the experiments (wt, n = 55; MCP-1−/−, n = 47). Blood pressure was measured in some animals (wt, n = 4; MCP-1−/−, n = 3) for 30 minutes via a catheter inserted into the femoral artery and a Digi-Med System (Micro-Med, Louisville, KY, U.S.A.) under the anesthetic conditions used for MCAO as described earlier (Wiessner et al., 1999). In these animals, brain vasculature was visualized by intracarotid injection of prestained latex beads as described previously (Wiessner et al., 1999).
Surgical procedures
All experimental procedures were approved by the “Kantonales Veterinäramt, Basel.” Unilateral and irreversible occlusion of the MCA (wt, n = 40; MCP-1−/−, n = 35) was performed as described previously (Wiessner et al., 1999; Wiessner et al., 2001). Briefly, animals were anesthetized with 2% isoflurane in 70/30 (by volume) nitrous oxide/oxygen mixtures, and the MCA was occluded by bipolar electrocoagulation. Body temperature was maintained at 37°C with a feedback heating system during surgery and until animals regained consciousness. Thereafter, animals were kept in heated cages for 2 hours where body temperature was checked frequently. After this, animals were returned to their home cages and allowed free access to food and water.
Magnetic resonance imaging
Magnetic resonance imaging experiments were carried out on a 4.7-Tesla, 15-cm-bore Spectrospin DBX (Bruker, Karlsruhe, Germany) equipped with a self-shielded gradient system. A 25-mm inner diameter birdcage probe has been used as radiofrequency transceiver.
Bolus tracking perfusion MRI was done as described previously (Rudin et al., 1997; Wiessner et al., 2001) in some nonoperated animals to characterize basal cerebral blood flow, cerebral blood volume and integrity of the blood–brain barrier (wt, n = 5; MCP-1−/−, n = 3). In addition, from each MCAO group, animals were randomly selected and subjected to bolus tracking perfusion MRI immediately (wt, n = 12; MCP-1−/−, n = 9) or 6 hours (wt, n = 9; MCP-1−/−, n = 10) after pMCAO. A transverse slice of 1.5-mm thickness 4 mm posterior to the rhinal fissure was recorded in 500 milliseconds using a T2*-weighted snapshot pulse sequence. For each animal, 40 consecutive images were acquired and 50 μL Endorem (Laboratoire Guerbet, Aulnay-sous-Bois, France) was infused intravenously after the tenth image during 2 seconds using a spectrometer-controlled infusion pump. The tracer concentration–time curves were fitted for each pixel using a γ-variate model function (IDL software package; Research Systems Inc., Boulder, CO, U.S.A.), allowing the computation of parameter images displaying cerebral blood volume (CBV ∝ time integral of the γ-variate function), mean transit time (= first moment of the tracer transport function), or relative cerebral blood flow (cerebral blood flow = CBV/mean transit time). These parameter images were further analyzed using image analysis software Analyze (Biomedical Imaging Resource, Mayo Foundation, Rochester, MN, U.S.A.). Ischemic tissue was segmented in cerebral blood flow images by applying a threshold value of approximately 40% of the normal value on the contralateral side. Ischemic area was expressed as percentage area of the ipsilateral hemisphere. Unaffected CBV and flow values were the calculated means from the nonischemic tissue.
Blood–brain barrier integrity was assessed by measuring the relaxation enhancement caused by the extracellular contrast agent Dotarem (Laboratoire Guerbet). A T1-weighted gradient-echo pulse sequence has been used with repetition delay of TR = 45 milliseconds, echo delay TE = 3.7 milliseconds, in-plane resolution (pixel dimension) = 97 μm × 97 μm, slice thickness = 1 mm, 8 averages. T1-weighted images were recorded before (S0) and 10 minutes after the intravenous administration of 40 μL Dotarem (S). The relative Dotarem concentration in the tissue was estimated as [Dotarem] ∝ (S/S0) − 1, assuming a linear relationship between the relaxivity R1 = 1/T1 and the Dotarem concentration.
T2-weighted MRI was used to determine infarct volume 24 hours after pMCAO (wt, n = 22; MCP-1−/−, n = 20). Measurements were performed as described in detail elsewhere (Wiessner et al., 2001). Briefly, mice were anesthetized with 1% to 1.5% isoflurane delivered via facemasks and positioned with their heads in 20-mm resonators. Each mouse was then subjected to one imaging cycle, in which 13 contiguous T2-weighted coronal sections of the brain with a thickness of 1.0 mm were taken using a RARE (rapid acquisition with relaxation enhancement sequence) (optimized parameters: TR = 3,000 milliseconds; effective TE = 86 milliseconds; in-plane resolution = 86 μm × 86 μm). The total measuring time was 5 minutes. Quantitative morphometric evaluation of single MRI sections was performed using a semiautomatic image analysis software (Analyze) on a Silicon Graphics (Mountain View, CA, U.S.A.) O2 computer. The damaged tissue was segmented by setting an intensity threshold. Infarct volume was calculated based on the damaged area in each slice and the distance between sections. One animal from the MCP-1−/− group was identified as an outlier by Chauvenet's criterion and was excluded from analysis. Statistical comparison was done with Student's t-test, with P < 0.05 regarded as being statistically significant.
Immunohistochemistry
Brains were removed 6 hours (n = 3/group), 24 hours (wt, n = 10; MCP-1−/−, n = 7), 4 days (wt, n = 3; MCP-1−/−, n = 4), or 2 weeks (n = 9/group) after pMCAO and frozen in Tissue-Tek (Sakura Finetek, Torrance, CA, U.S.A.). For control, brains from nonoperated animals (n = 3/group) and sham-operated animals with 2 weeks survival time (n = 3/group) were sampled. Coronal tissue sections (10 μm) were cut on a cryostat (Leica, Nussloch, Germany). Sections were fixed for 15 minutes in ice-cold 4% paraformaldehyde. The primary antibodies used included rat anti-mouse FA/11 (CD68) (mAb; Serotec, Oxford, U.K.), bovine anti-human glial fibrillary acidic protein (GFAP) (Dako, Copenhagen, Denmark), HB199, a polyclonal antiserum to neutrophils produced in house (Anthony et al., 1998), and anti-mouse NeuN (mAb; Chemicon, Temecula, CA, U.S.A.). Immunohistochemistry was done as described previously (Rupalla et al., 1998; Brown et al., 1998). Briefly, the primary antibodies were revealed using biotinylated secondary antibody, the ABC kit (Vector Laboratories, Burlingame, CA, U.S.A.), and diaminobenzidine as the chromogen (brown precipitate). Nonspecific Fc binding was prevented by preincubation with normal serum and nonspecific peroxidase activity was eliminated by using methanol-H2O2 peroxidase solution. Immunohistochemical staining was scored by a blinded investigator (P.M.H.) on a scale ranging from −1 to 4 (−1, loss of basal staining; 0, basal staining found in cage control wild-type brains; 1-4, increasing levels of staining intensity and morphologic changes toward hypertrophic astrocytes or phagocytic monocytes). NeuN-positive neurons were counted in areas bordering infarcts (0.26 to 0.35 mm2) from animals with 2-week survival in coronal sections (bregma 1.0 mm, and bregma −1.5 mm) and expressed as cells/mm2. Statistical analysis was done with Mann-Whitney rank sum test (FA/11 and GFAP) or Student's t-test (NeuN) and a P < 0.05 considered to indicate significance.
Interleukin-1β and interleukin-6 enzyme-linked immunosorbent assay
Six hours after pMCAO, the parietal cortex from the right and left (nonlesioned) hemisphere was snap frozen. Tissue was homogenized on ice in phosphate buffer (PB) (0.5 mol/L NaCl, 2.5 mmol/L NaH2PO4, 7.5 mmol/L Na2HPO4, pH 7.2), containing 100 mmol/L amino-n-caproic acid, 10 mmol/L Na2EDTA, 5 mmol/L benzamidine hydrochloride, 0.2 mmol/L aminoethylbenzenesulfonylfluoride hydrochloride. IL-1β enzyme-linked immunosorbent assay was done as described previously (Walsh et al., 2001) and was provided by National Institute of Biological Standards Council. For coating, a sheep anti-mouse IL-1β Ab (S5/150799/JW) (1 mg/mL in PB/0.1% Tween-20) was used. Blocking was done with 1% albumin (chicken egg grade; Sigma, St. Louis, MO, U.S.A.) in PB at room temperature. The second antibody, biotinylated sheep anti-mouse IL-1β antibody (S329/B4/190799/JW), was diluted 1:1,000 in PB/1% normal sheep serum. A similar protocol was followed for mouse IL-6 enzyme-linked immunosorbent assay (R&D Systems, Minneapolis, MN, U.S.A.). Coating antibody was rat anti-mouse IL-6, diluted to 2 μg/mL in PB. Blocking was done with 1% bovine serum albumin/PB. The second antibody was biotinylated goat anti-mouse IL-6 diluted to 200 ng/mL. For both enzyme-linked immunosorbent assay kits, streptavidin–horseradish peroxidase (Dako, Copenhagen, Denmark) and tetramethylbenzidine substrate (Pharmingen, San Diego, CA, U.S.A.) was used for development. More than 90% of recombinant IL-1β and IL-6 standard added to naïve mouse brain homogenate (spike) were detected in the assay (recovery). Protein concentrations were determined by Bradford's method (Bio-Rad, Hemel Hempstead, U.K.).
RESULTS
Basal cerebral blood flow and characteristics of MCP-1−/− mice
Mean arterial blood pressure under anesthetic conditions used for pMCAO surgery was 102.1 ± 9.9 mm Hg (n = 4) in wt and 95.4 ± 2.0 mm Hg (n = 3) in MCP-1−/− mice. These values were not significantly different. Visual inspection of the anatomy of intracranial arteries (circle of Willis, MCA) did not reveal obvious differences among the strains.
When a bolus of a MRI contrast agent (e.g., Endorem) is injected into the circulating blood stream, the transit of the bolus through the brain can be followed by fast imaging techniques (for a review of the technique see Hossmann and Hoehn-Berlage, 1995). Thus, bolus tracking perfusion MRI was used for semiquantitative determination of relative cerebral blood volume and flow (CBV and cerebral blood flow, respectively). The mean transit time of the tracer (Endorem) in MCP-1−/− mice (3.5 ± 0.4 seconds) was not significantly different from wt animals (3.4 ± 0.6 seconds). The same was found for relative cerebral blood flow in the cortex and striatum (Fig. 1A) and relative cerebral blood volume (Fig. 1B). Differences in blood–brain barrier characteristics might influence edema formation after pMCAO and, thus, were examined by calculating tissue uptake of the tracer Dotarem ([Dotarem] α S/S0–1). Dotarem does not cross the intact blood–brain barrier, whereas it leaks into brain parenchyma at a location where the blood–brain barrier is impaired. No differences in tissue uptake were found in wild-type versus MCP-1−/− mice (Fig. 1C). From all these results it was concluded that MCP-1 deficiency did not cause gross anatomic changes in brain angioarchitecture, or major changes in basal blood pressure, cerebral blood flow, and blood–brain barrier integrity that would confound interpretation of results in the pMCAO stroke model.
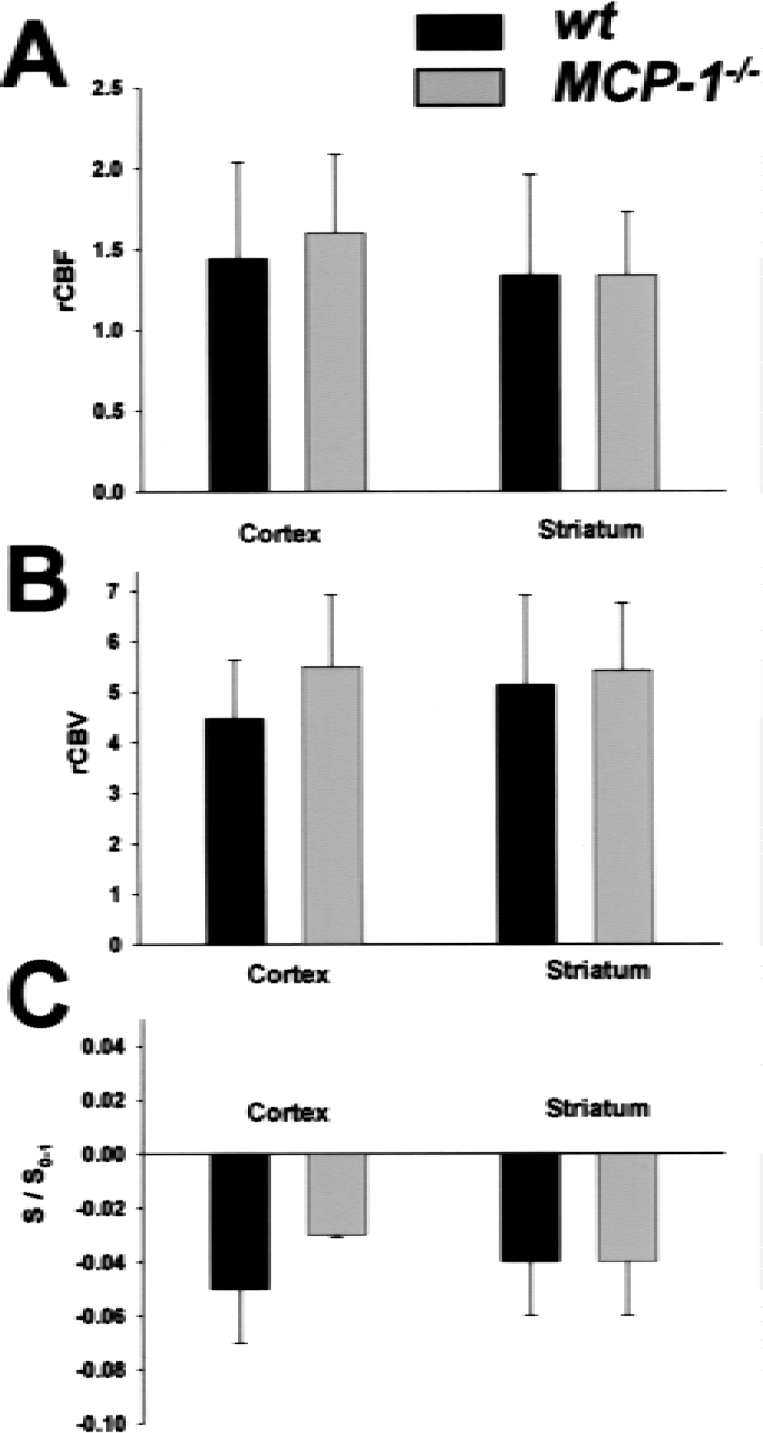
Bolus tracking magnetic resonance imaging did not reveal differences in basal cerebral blood flow, cerebral blood volume, and blood–brain barrier properties among wild-type (wt) and MCP-1−/− mice.
Middle cerebral artery occlusion and infarct volumes
The supply territory of the middle cerebral artery may vary substantially between mouse strains (Maeda et al., 1998) and thus, can be the underlying reason for any observed differences in infarct volume between wild-type and transgenic/KO mice. Therefore, ischemia after pMCAO was investigated by bolus tracking perfusion MRI. In addition to a semiquantitative determination of regional blood flow (see earlier herein), cerebral blood flow maps calculated by pixel-wise evaluation of perfusion MRI allows a topographic assessment of the blood flow deficit after pMCAO in the brain section selected for analysis. To characterize pMCAO-induced ischemia, the percentage of the hemisphere with a blood flow disturbance (cerebral blood flow smaller than 40% of intact hemisphere) was calculated from the MRI data. The ischemic area as determined this way was similar in wild-type and MCP-1−/− animals, either when measured immediately or 6 hours after pMCAO (Fig. 2). In addition, no significant differences in the extent of blood flow reduction within the MCA supply territory and in cerebral blood flow values in the intact hemisphere were found (data not shown). These results were in line with the basal characterization (see earlier herein) and showed that the initial ischemic impact in wild-type and MCP-1−/− animals was comparable.
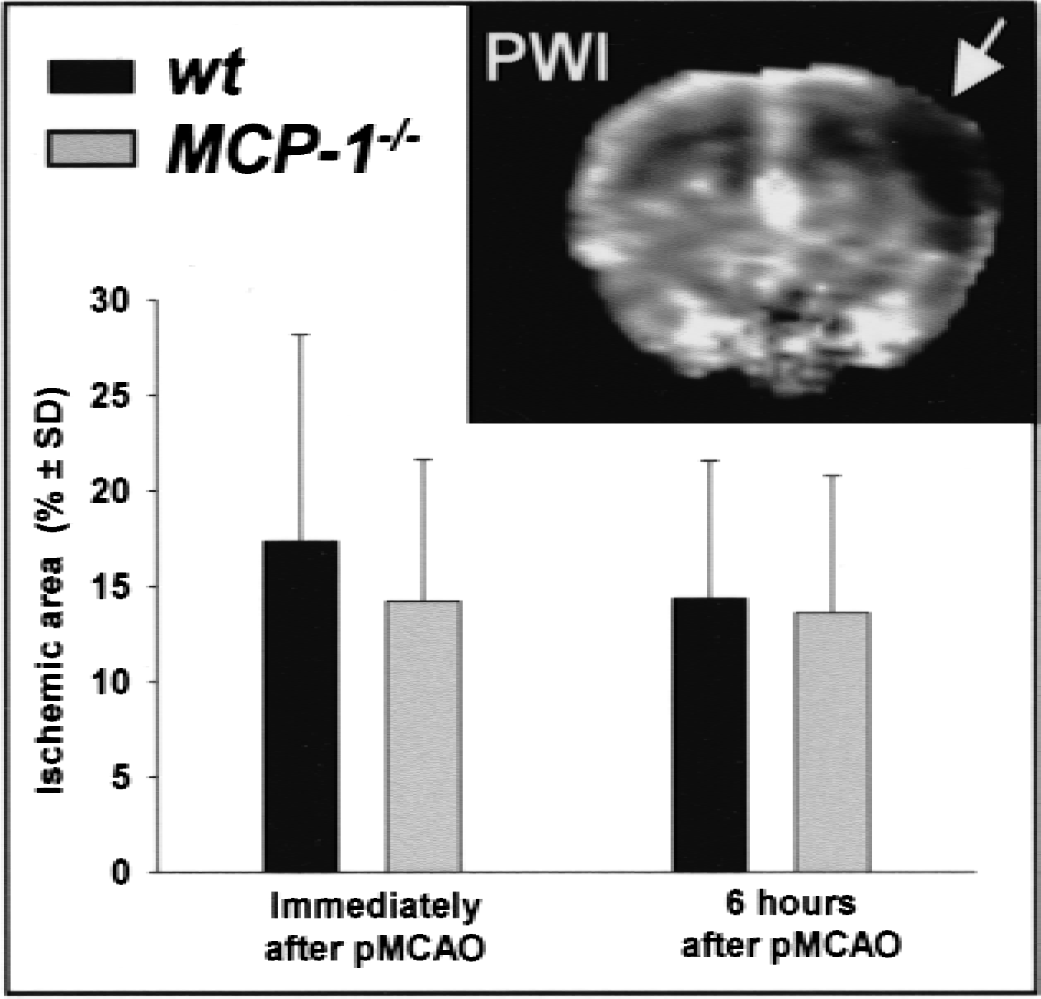
Focal ischemia after permanent middle cerebral artery occlusion (pMCAO) was similar in wt and MCP-1−/− mice. Blood flow was determined immediately (wt, n = 12; MCP-1−/−, n = 9) or 6 hours (wt, n = 9; MCP-1−/−, n = 10) after artery occlusion with bolus tracking MRI in a transverse slice of 1.5-mm thickness 4 mm posterior to the rhinal fissure (example in upper right corner of the figure). The perfusion deficit was quantified as percentage of affected area per hemisphere using an arbitrary threshold of 40% signal intensity compared with the nonischemic hemisphere (mean). Significant differences among the strains were not observed.
One day after pMCAO, vasogenic edema accompanying infarct development was sharply demarcated on T2-weighted MRI and could be used for lesion volume determination (Fig. 3A). It has previously been shown that lesion volumes derived from T2-weighted MRI highly correlate with volumes as determined by conventional histologic methods (for review see Rudin et al., 1999). MRI visible lesions were confined to cortical regions (Fig. 3A). Overall, the mean infarct volume was 30.2 ± 12.7 μL in wt mice (n = 22) and 21.5 ± 10.1 μL in MCP-1−/− mice (n = 19). Thus, mean infarct volume was 29% smaller in MCP-1−/− mice (P = 0.022) (Fig. 3B).
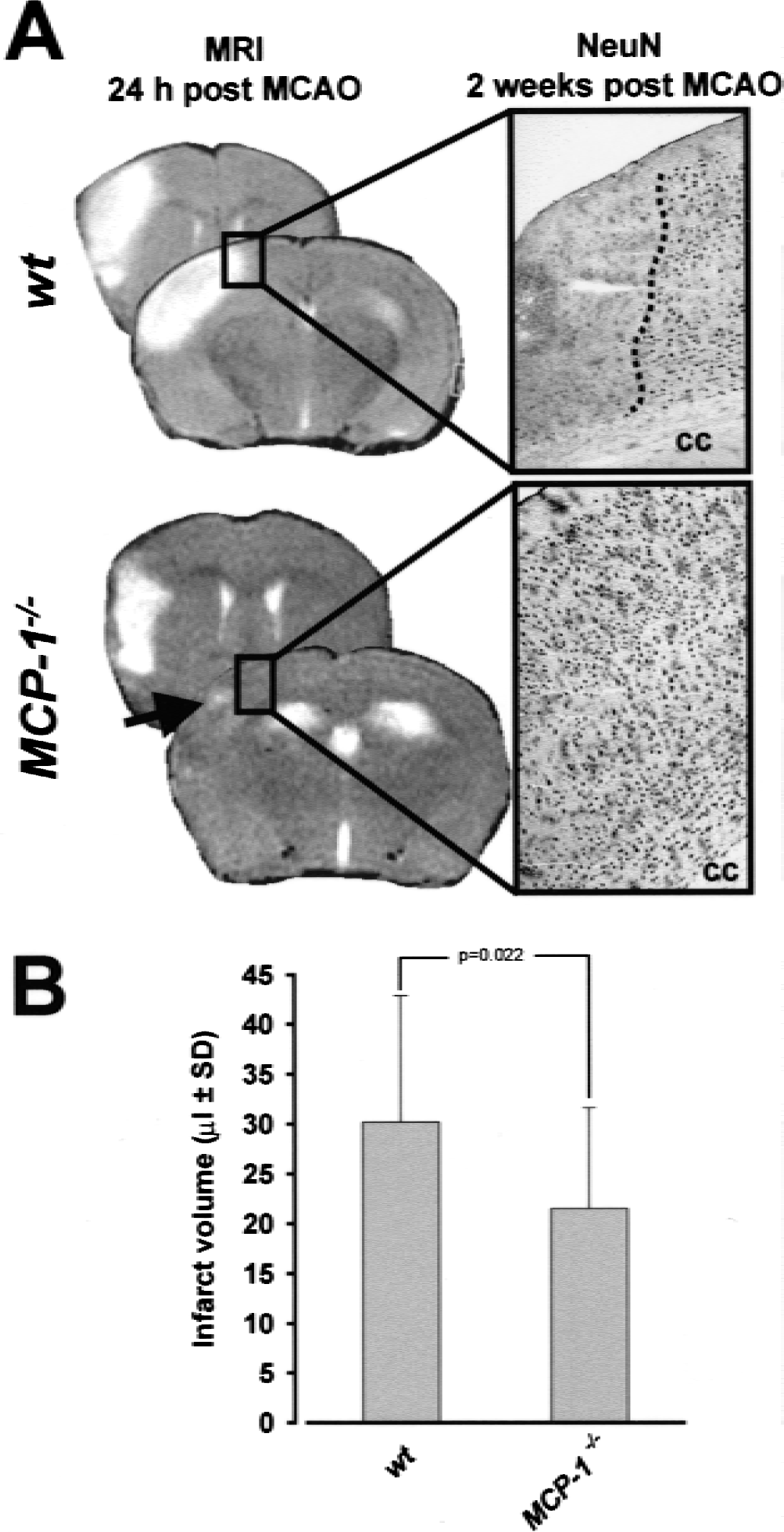
Compared with wild-type animals, infarcts were significantly smaller in MCP-1−/− mice.
In some animals, infarct morphology as determined noninvasively by T2-weighted MRI at 24 hours was directly compared with morphologic damage in histologic specimens from the very same brains removed for analysis 2 weeks after pMCAO (Fig. 3A, right column). Histologic infarcts were exclusively found in cortical areas displaying T2-signal hyperintensity at 24 hours after pMCAO in both wild-type and MCP-1−/− mice, indicating that no further infarct growth aside from tissue swelling caused by edema formation had occurred after 24 hours. In addition, this observation indicates that infarct volume reduction in MCP-1−/− mice measured at 24 hours did not reflect a delay of infarct growth, but a persisting reduction of infarcted tissue (at least for up to 2 weeks). Possible differences in delayed selective neuronal loss in infarct border areas were investigated by counting NeuN-stained neurons in brains sampled 2 weeks after pMCAO. For that purpose, a region of interest with a width of 0.42 mm and a total area of 0.26 to 0.35 mm2 was defined in the immediate vicinity of the infarct border or in a corresponding area when no infarct was present in the tissue section. Neuronal cell densities amounted to 1,066 ± 207 cells/mm2 and 1,038 ± 129 cells/ mm2 at bregma 1.0 mm in wt (n = 5) and MCP-1−/− mice (n = 5), respectively. At bregma −1.5 mm (corresponding to the boxed area in Fig. 3), NeuN density was 1,397 ± 166 cells/mm2 in wt mice and 1,377 ± 94 cells/mm2 in MCP-1−/− mice. Thus, no significant differences in neuronal density in the infarct border area were observed between wt and MCP-1−/− mice.
Microglia/macrophage responses after permanent middle cerebral artery occlusion
The monoclonal antibody FA/11 recognizes a membrane sialoglycoprotein widely expressed on microglia and macrophages (Gordon et al., 1992). The antigen is present in resident microglia/macrophages, but its level of expression is enhanced on stimulation. Compared with cage control and sham-operated animals, a slight increase of FA/11 immunoreactivity in the ischemic parietal cortex (lesion, Fig. 4A) was observed at 6 hours and 24 hours after MCAO in both wt and MCP-1−/− animals. By morphologic criteria, the increase in staining intensity occurred on brain resident microglia with enlarged cell body and a thickening of cell processes. At 4 days, staining intensity and numbers of FA/11-positive cells with morphologic characteristics of phagocytic macrophages increased within lesion areas. This response appeared more pronounced in wt mice without being significantly different from MCP-1−/− mice. Two weeks after pMCAO, staining intensity as well as number of FA/11-positive cells were significantly larger in wt animals (Fig. 4A) as compared with MCP-1−/− mice, especially toward the inner boundary of the lesion (Fig. 4B).
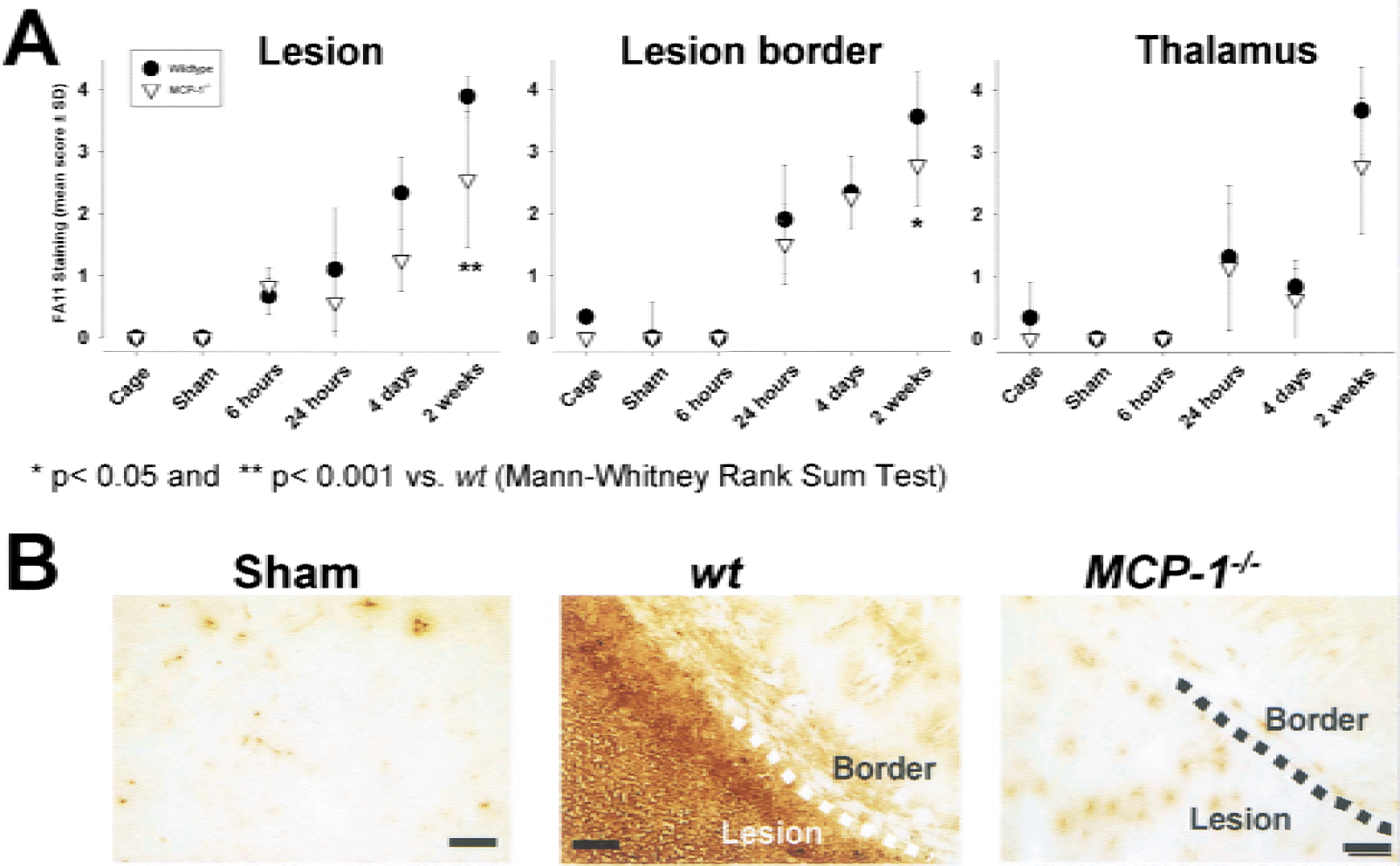
Infarct-associated phagocyte recruitment commencing 4 days after permanent middle cerebral artery occlusion (pMCAO) was significantly reduced in MCP-1−/− mice.
At the lesion border, an increase of staining was found from 24 hours after pMCAO onwards, but was not different among wt and MCP-1−/− mice at 24 hours and 4 days. At 2 weeks after MCAO, FA/11 staining was clearly distinguishable between wt and MCP-1−/− mice, reflected in a significantly lower staining score. In line with previous findings in C57BL/6 mice (Rupalla et al., 1998), microglia/macrophage responses were observed in the ipsilateral thalamus of wt mice from 24 hours after MCAO onwards, which were most prominent 2 weeks after the lesion (Fig. 4). Thalamic FA/11 staining was indistinguishable between wt and MCP-1−/− mice at 24 hours and 4 days after MCAO, but appeared to be less prominent in MCP-1−/− mice at 2 weeks in some animals. This effect, however, was not significant in the overall mean score.
Astrocytic responses after permanent middle cerebral artery occlusion
Immunohistochemical staining of GFAP was used to characterize astrocytic reactions at 6 hours, 24 hours, 4 days, and 2 weeks after pMCAO. Within ischemic or infarcted areas, no increase of GFAP immunoreactivity was observed at any time point in wt animals, and the same was found in MCP-1−/− mice (Fig. 5A). When compared with cage control or sham-operated animals, a minor increase in GFAP immunoreactivity was observed at the lesion border in animals surviving 6 hours and 24 hours, but was not different between wt and MCP-1−/− mice. At 4 days after MCAO, a pronounced astrocytic response was found in wt animals that appeared weaker in MCP-1−/− mice without reaching significance. GFAP immunoreactivity at the lesion border further increased at 2 weeks (Figs. 5A and 5B). In contrast to wt animals, astrogliosis in MCP-1−/− mice at 2 weeks was significantly (P < 0.01) attenuated in this region (Figs. 5A and 5B). In the thalamus, we observed in this study a pronounced increase in GFAP immunoreactivity at 2 weeks after MCAO in wt mice. In MCP-1−/− mice, this response at 2 weeks after MCAO was significantly weaker (Figs. 5A and 5B).
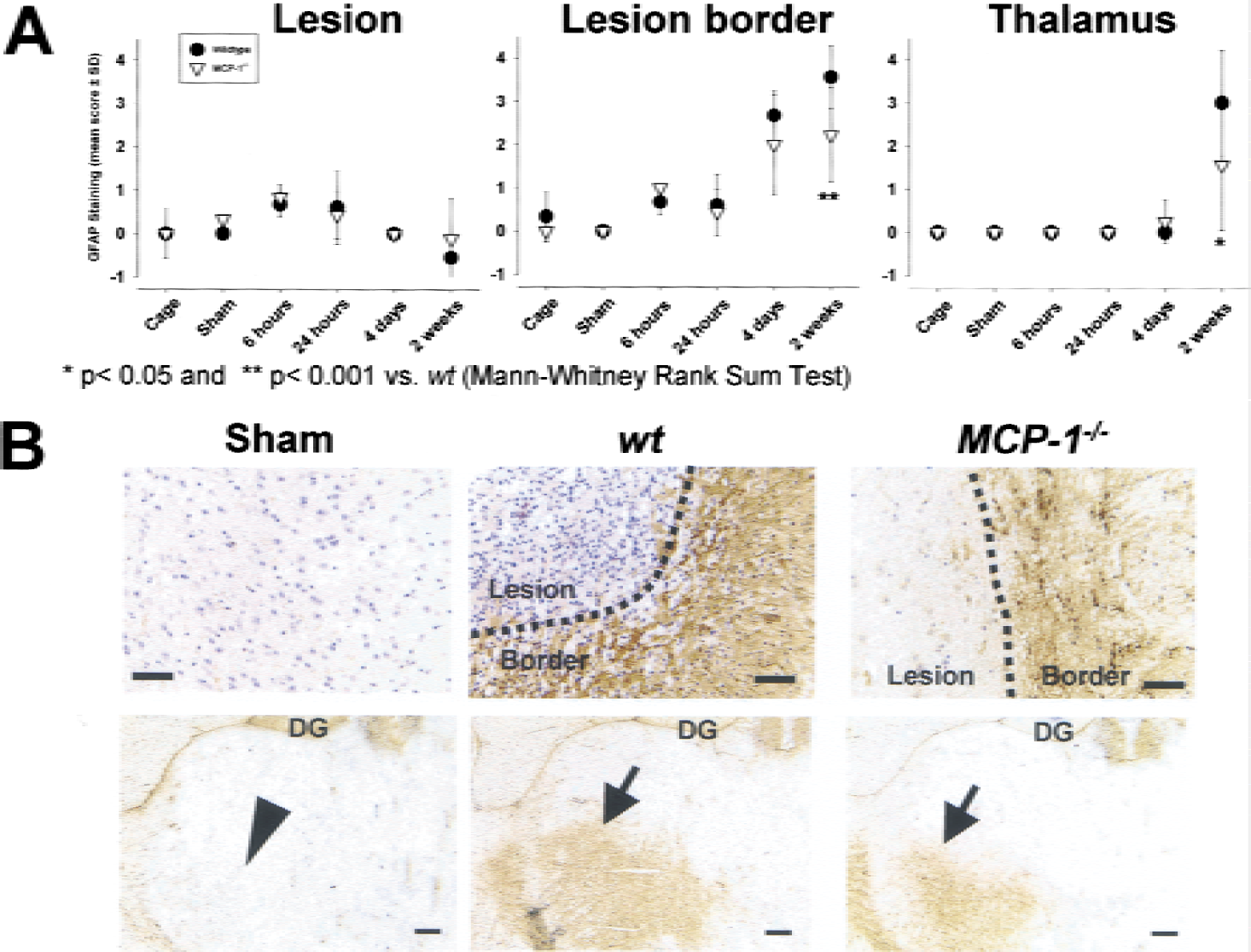
Astrocytic hypertrophy in the infarct border and thalamus was significantly attenuated in MCP-1−/− mice in the postacute phase after permanent middle cerebral artery occlusion (pMCAO) (>4 days).
Neutrophils in MCP-1−/− mice after permanent middle cerebral artery occlusion
Using the neutrophil specific antibody HB199 (Anthony et al., 1998), we examined the extent of neutrophil infiltration at 24 hours after pMCAO in wt and MCP-1−/− mice. In accordance with previous observations (Brown et al., 1998), neutrophils were abundant within the lesion. There was no obvious difference in the extent of neutrophil recruitment in the wt versus the MCP-1−/− mice (data not shown).
Cytokines in MCP-1− /- mice after permanent middle cerebral artery occlusion
The effects on astrocytes and monocytes (see earlier herein) in MCP-1−/− mice occurred late after pMCAO and could thus not be implicated in infarct growth. To further explore the protective mechanism in MCP-1−/− mice, we analyzed IL-1β and IL-6, which are known to be regulated by MCP-1 (Jiang et al., 1992). In previous experiments, we found an approximate 5-fold increase of Il-1β and 20-fold increase of IL-6 in ischemic tissue at 24 hours after pMCAO in C57BL/6 mice, respectively (A.K.M. and P.M.H., unpublished observations). Because infarct growth is restricted to the first hours after pMCAO, we decided in this study to analyze brains sampled 6 hours after MCAO. In wild-type animals, cortical IL-1β protein levels were increased approximately threefold in the ischemic hemisphere as compared to the contralateral hemisphere (8.5 ± 4.1 pg/mg versus 3.5 ± 1.1 pg/mg, n = 15, P < 0.001). Although less pronounced, also in MCP-1−/− mice cortical IL-1β protein levels were higher in the ischemic hemisphere (5.6 ± 1.8 pg/mg versus 3.2 ± 2.2 pg/mg, n = 10, P < 0.01). When compared with wild-type mice, the IL-1β levels in the ischemic cortex of MCP-1−/− mice were reduced significantly by 34% (P = 0.044) (Fig. 6A).
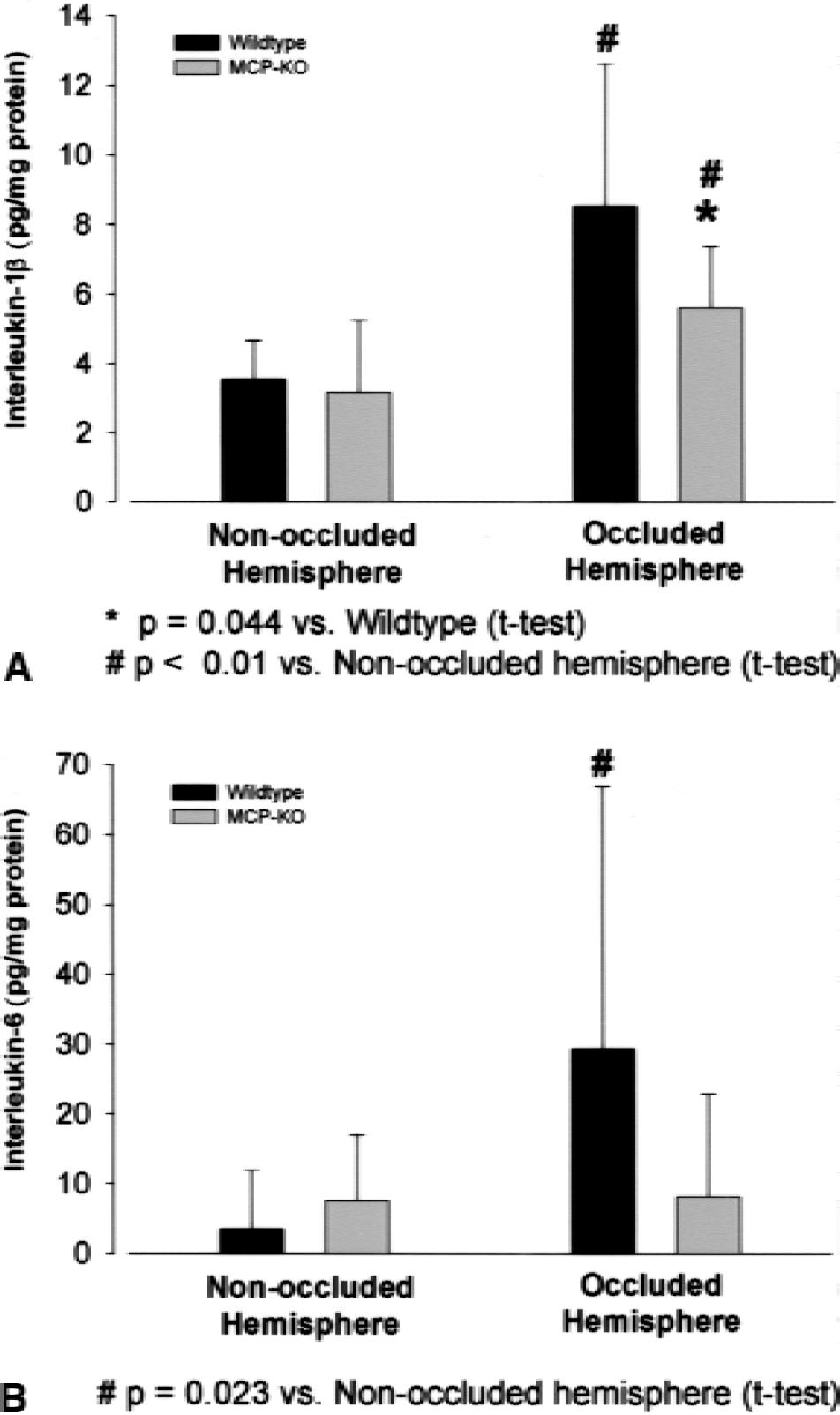
Interleukin-1β and interleukin-6 levels were decreased in MCP-1−/− mice 6 hours after middle cerebral artery occlusion. Interleukin-1β
Interleukin-6 was significantly elevated approximately 10-fold in the ischemic cortex of wild-type animals compared to the nonischemic cortex (29.3 ± 37.6 pg/mg versus 3.4 ± 8.4 pg/mg, P = 0.023), although interindividual variability was high (Fig. 6B). In contrast, in MCP-1−/− mice, Il-6 levels were not significantly different in the ischemic and the nonischemic cortex (8.2 ± 14.7 pg/mg, versus 7.5 ± 9.4 pg/mg).
DISCUSSION
Infarct size in MCP-1−/− mice
In the current study, a mouse strain deficient in MCP-1 (Lu et al., 1998) was subjected to pMCAO—an experimental stroke model. The finding that MCP-1−/− mice developed smaller infarcts after pMCAO provided the first direct evidence for a pathogenetic role of MCP-1 in infarct growth after focal brain ischemia.
Several lines of evidence indicate that smaller infarct volumes in MCP-1−/− mice were caused by the lack of MCP-1 during and after ischemia and not related to nonspecific developmental, anatomic, or general physiological differences in the mouse strains. (1) MCP-1−/− mice develop normally, do not display any malformations, have normal physiologic parameters, have the same lifespan as wild-type mice, and are fertile (Lu et al., 1998). In addition, the hematologic profiles are similar to wild-type mice (Gu et al., 2000). Staining of brain specimens from nonoperated animals revealed apparently normal cytoarchitecture, morphologies and numbers of neurons, astrocytes, and resident microglia. We did not observe any obvious abnormalities in brain architecture and anatomy of intracranial vessels. (2) Blood flow parameters are critical denominators of infarct growth and final volume. Such parameters can differ among mouse strains, which is especially important for knockout mice when compared to a wild-type strain, and may be the underlying reason for observed differences in infarct volume. We, therefore, used perfusion MR imaging to evaluate MCP-1−/− and wt mice. These measurements did not unravel any differences in basal cerebral blood flow, CBV, and blood–brain barrier permeability between wt and MCP-1−/− mice. In addition, a similar ischemic territory was found after pMCAO, indicating that the ischemic impact was similar in wt and MCP-1−/− mice. (3) MCP-1 is not expressed in the brain under normal conditions (Wang et al., 1995; Kim et al., 1995), making it unlikely that compensatory mechanisms take place in MCP-1−/− brains under normal conditions. (4) MCP-1 is well known to become upregulated early on after pMCAO, i.e., in the range of hours after the insult (Gourmala et al. 1997; Che et al., 2001). At this time, infarct volume is still expanding after pMCAO in mice (Hata et al., 2000) and, therefore, the reported expression patterns after focal ischemia are compatible with a causal role in infarct growth.
In line with the findings in MCP-1−/− mice in this study, it has previously been reported that recombinant MCP-1 injected concomitantly with N-methyl-
Mechanism of infarct reduction in MCP-1−/− mice
Several activities of MCP-1 could be involved in infarct progression after focal ischemia.
MCP-1 is involved in monocytic recruitment in several inflammatory settings, including the brain (Bell et al., 1996; Lu et al., 1998). Monocytes (phagocytes) are the most abundant leukocytes entering the brain in permanent focal ischemia (Stoll et al., 1998) commencing several days after the lesion onwards (Schroeter et al., 1994). In the present study, accumulation of phagocytes in infarcted tissue developed from 4 days after pMCAO onwards. This process was attenuated in MCP-1−/− mice suggesting that MCP-1 could be involved in phagocyte recruitment to the site of the lesion also after brain injury. In relation to the time course of infarct growth, which is completed 1 day after pMCAO (Hata et al., 2000), accumulation of phagocytes, however, is a late event. This suggests that it is a secondary response to ischemic injury and not causally involved in the manifestation of tissue damage. Thus, the attenuation of phagocyte recruitment in MCP-1−/− mice was probably not the underlying reason for the smaller infarct volumes, and it cannot be excluded that it was secondary to reduced brain damage in MCP-1−/− mice. Because blood-borne phagocytes and fully activated brain microglia are morphologically and phenotypically indistinguishable (for review see Kreutzberg, 1996) it remains unclear whether recruitment of both cell types were differentially affected in MCP-1−/− mice. Depletion of blood macrophages previously demonstrated that in permanent focal ischemia phagocytes were initially (3 days) mainly derived from resident microglia and only later (6 days) recruited from the bloodstream (Schroeter et al., 1997). The effect of MCP-1 deficiency on phagocyte accumulation was most pronounced 2 weeks after pMCAO, which suggests that mainly recruitment from the bloodstream was affected.
Although astrocytes do not express the cognate MCP-1 receptors CCR2 and CCR9, they respond to MCP-1 (Heesen et al., 1996) and have binding sites for MCP-1 (Andjelkovic et al., 1999). Their response after ischemia, therefore, could be altered in MCP-1−/− mice. Indeed, glial scar formation at the border of the infarct was attenuated in MCP-1−/− mice. However, because this effect was observed 2 weeks after pMCAO, it cannot explain smaller primary infarcts in MCP-1−/− mice and also may be secondary to reduced brain damage in MCP-1−/− mice.
MCP-1 induces the expression of IL-1β and IL-6 in human monocytes (Jiang et al., 1992). For IL-1β numerous studies provided evidence of its role as an important mediator of brain damage after pMCAO (for review see Rothwell and Luheshi, 2000). IL-6 is a pleiotropic cytokine and elevated cerebrospinal fluid and plasma levels were observed in human patients who have had strokes (Tarkowski et al., 1995), and in experimental focal ischemia (Clark et al., 1999; Legos et al., 2000). The precise role of IL-6 in stroke, however, is unclear at present (Campbell et al., 1993; Loddik et al., 1998). In our study, both IL-1β and IL-6 were expressed in the ischemic cortex 6 hours after pMCAO in wild-type mice, which is in line with previous reports for rat pMCAO (Clark et al., 1999; Davies et al., 1999; Legos et al., 2000). In MCP-1−/− mice, IL-1β levels were significantly reduced. Levels of IL-6 were also reduced, but this finding was statistically not significant because of large interindividual variability. The significant reduction of IL-1β in MCP-1−/− mice is of particular interest because it was observed at a time point after pMCAO, when infarct growth is still ongoing. At such early time points, IL-1β is produced by brain resident microglia (Davies et al., 1999). Of note, the initial morphologic activation of brain resident microglia at 6 hours and 24 hours after pMCAO was not affected in MCP-1−/− mice. Thus, although microglia appear to be “activated” by histologic criteria in MCP-1−/− mice, they may not produce proinflammatory cytokines to the same extent as in wt mice.
In conclusion, we propose that the neuroprotective effect of MCP-1 deficiency could be related to alterations of early ischemia-induced production of proinflammatory mediators, rather than changes in phagocyte recruitment and astrocytic hypertrophy. The changes in the ischemia-induced cytokine response in MCP-1−/− mice clearly deserve further investigation, and should include time course studies, and genomic/proteomic analytical procedures to cover the whole cytokine and chemokine spectrum.
Subacute cellular responses in regions of remote neuronal injury after permanent middle cerebral artery occlusion
Chronic middle cerebral artery infarcts in human patients who have had a stroke result in thalamic neurodegeneration (Ogawa et al., 1997) and microglia activation (Pappata et al., 2000). We have previously reported that also after pMCAO in mice, prominent neuronal death occurred in the thalamus with a delay of several weeks that was preceded by microglia activation (Rupalla et al., 1998). Immunohistologic evaluation of thalamic areas in our new study confirmed microglial activation, and also showed astrocytic hypertrophy at 2 weeks after pMCAO. The astrocytic response was significantly attenuated in MCP-1−/− mice. A slight attenuation of microglia activation was also observed at this time point. Aspiration lesions of the cortex led to early expression of MCP-1 and microglial activation in the lateral geniculate nucleus of the thalamus (Muessel et al., 2000a), which was attenuated in MCP-1−/− mice (Muessel et al., 2000b). Lack of MCP-1 could thus be involved directly in the observed attenuated thalamic alterations in our study. Because thalamic changes depend on the size of the primary cortical lesion, the attenuation of astrocytic responses in the thalamus could also reflect the smaller primary infarcts in MCP-1−/− mice.
Therapeutic implications
This study suggests that MCP-1 represents an important regulatory chemokine in the brain after injury and that it cannot be circumvented by parallel signaling pathways or compensatory processes. MCP-1 signals via G-protein-coupled receptors that are suitable targets for small-molecular-weight inhibitors. Therefore, the presented results indicate that targeting MCP-1 signaling could be a new acute treatment approach for stroke.
Footnotes
Acknowledgments:
The authors thank Diana Baumann, Michelle Botham, Pascale Brebbia, Francesco D'Amato, Thomas Hafner, and Willi Theilkäs for excellent technical assistance.