
Abstract
We aim to determine the cerebrospinal fluid levels of high mobility group box 1 in subarachnoid hemorrhage patients and to investigate the involvement of the receptor for advanced glycation end products and high mobility group box 1 in the pathogenesis of post-subarachnoid hemorrhage neuronal death. The study included 40 patients (mean age, 59 ± 19 years) with Fisher's grade ≥ III aneurysmal subarachnoid hemorrhage. Cerebrospinal fluid was collected on the seventh day post-hemorrhage. Receptor for advanced glycation end products expression was examined in rat brain tissue following subarachnoid hemorrhage and in cultured neurons exposed to post-subarachnoid hemorrhage cerebrospinal fluid. Therapeutic effects of the recombinant soluble form of RAGE on subarachnoid hemorrhage models were also investigated. The results indicated that a higher level of cerebrospinal fluid high mobility group box 1 was independently associated with unfavorable outcome at three months post-subarachnoid hemorrhage (OR = 1.061, 95% CI: 1.005–1.121). Expression of RAGE increased in post-subarachnoid hemorrhage rat brain cells and in cultured neuron with stimulation of post-subarachnoid hemorrhage cerebrospinal fluid. Administration of recombinant soluble form of RAGE significantly reduced the number of positive TUNEL staining cells in subarachnoid hemorrhage rat and improved cell viability in post-subarachnoid hemorrhage cerebrospinal fluid-treated cultured neurons. Thus, the level of cerebrospinal fluid high mobility group box 1 can be a prognostic indicator for patients with Fisher's grade ≥ III aneurysmal subarachnoid hemorrhage and that treatment with soluble form of RAGE is a novel approach for subarachnoid hemorrhage.
Introduction
Aneurysmal subarachnoid hemorrhage (SAH) is a serious cerebral vascular event that is frequently associated with high morbidity and mortality.1–3 After the occurrence of SAH, blood extravasates into the subarachnoid and ventricular spaces, spreading out and coating other vessels and the brain substance. Hemolysis of red blood cells results in rapid release of hemoglobin and induces the activation of associated enzymes and proinflammatory cytokines.4–8 Theoretically, these post-hemorrhagic cascades and products may contribute to further vascular ischemia and neuronal injury.9–11
High mobility group box 1(HMGB1) is a nuclear factor that has been identified as an important immune mediator, triggering the production of various inflammatory cytokines in many diseases. 12 HMGB1 has been shown to be released in large amounts into the extracellular space immediately after an ischemic brain insult, and subsequently activates the receptor for advanced glycation end products (RAGE) as well as activating downstream signaling that includes mitogen-activated protein kinases (MAPKs), and nuclear factor kappa B pathways.13–15 Soluble form of RAGE (sRAGE) may counteract the detrimental effects of RAGE. We and others have recently established that blockage of HMGB1-RAGE signaling by exogenous administration of either HMGB1 blocking antibody or sRAGE significantly improves cell survival and reduces infarct volume in experimental stroke models.14,16
A recent study showed that the level of cerebrospinal fluid (CSF) HMGB1 increased significantly in patients with SAH. 17 However, whether circulating HMGB1 plays a role as a surrogate marker for prognosis or directly participates in the pathophysiology of post-SAH brain injury remains unclear. In the present study, we performed a comprehensive investigation involving clinical and basic aspects on the expression and role of HMGB1 and RAGE singling in SAH patients as well as experimental models of SAH. We also demonstrated the therapeutic effects of exogenous administration of recombinant sRAGE in experimental SAH models.
Materials and methods
In clinical part, the study was approved by the National Taiwan University Hospital Committee of Human Research and conducted in accordance with human ethics regulations. Written informed consent was obtained from the patients or from the next of kin of patients with decreased consciousness. In basic part, the experimental procedures were approved by the National Taiwan University Animal Care and Use Committees.
SAH patients
The study included 18 to 80-year-old patients with Fisher’s Grade ≥ III aneurysmal SAH (defined as SAH ≥ 1 mm thick or of any thickness with extension of intraventricular hemorrhage (IVH) or parenchymal) during the period between January 2009 and June 2010 in National Taiwan University Hospital. Patients with IVH in the 3rd and 4th ventricle causing obstructive hydrocephalus, arterial dissection or infectious aneurysm, massive intracerebral hemorrhage (defined as a hematoma volume > 30 ml) or uncal brain herniation, documented meningitis or brain tumor, or spinal cord tumor were excluded. Control subjects were patients with a clinical diagnosis of normal pressure hydrocephalus.
The patients received standard treatment, which was provided by an integrated team that included neurosurgeons, neuroradiologists, and neurointensivists.7,8 The management protocol consisted of resuscitation, early surgical or endovascular obliteration of the aneurysm, standard management of intracranial pressure and neurointensive care, and aggressive medical or endovascular therapy for vasospasm if it occurred. After surgery, patients were monitored in the neurointensive care unit. Head computed tomography (CT) angiography or conventional angiography was performed when vasospasm was suspected, or if neurological deterioration was noted. Delayed ischemic neurological deficit was defined as clinical deterioration (i.e. a new focal deficit, decrease in the level of consciousness, or both) and/or a new infarct on a CT scan that was not visible on the admission or immediate post-operative scans. 18 Functional outcome was evaluated using the Glasgow outcome scale (GOS) at three months after onset. GOS ≤ 3 was defined as an unfavorable functional outcome. 19
CSF sample collection, preparation, and analysis
Intrathecal CSF was obtained via lumbar puncture or lumbar drain on the seventh day after SAH based on our previous protocol.7,8 The CSF samples were immediately centrifuged at 900 × g at 4℃ for 20 min before being divided into suitable aliquots. Then all aliquots were snap-frozen at −80℃ within 30 min after the centrifuge. The concentrations of proteins, glucose, and lactate in the CSF samples were determined by using an automatic chemistry analyzer. The levels of HMGB1 were determined by using a standard ELISA Kit (IBL, Hamburg, Germany).
Rat model of SAH
Male Wistar rats were maintained in our animal facility under pathogen-free conditions with continuous access to food and water until three months old (weighing 250 to 300 g each). Rats were anesthetized with 1.5% halothane in a mixture of 30% O2 and 70% N2O. The right femoral artery was cannulated for continuous arterial pressure monitoring. Systemic arterial pressure was monitored using an RFT Biomonitor, obtained from VEB Messgeraete Werk (Zwoenitz, Germany). Body temperature was maintained at 37 ± 0.5℃ by using a heating pad. A rat SAH model was created by following a standard procedure. SAH was induced by injecting 0.3 ml of fresh autologous blood into the cisterna magna over a 5-min period with the animal in a 20° sign head-down position. Rat CSF samples were obtained from the cisterna magna. The CSF was centrifuged at 3000 r/min for 10 min. The supernatant was kept at 30℃ and then sent for the related study immediately.
Primary neuronal culture
Dissociated neuron-enriched cultures of cerebral cortex were established from 18-day Sprague-Dawley rat embryos as described. 20 Experiments were performed using seven to nine-day-old cultures. Approximately 95% of the cells in such cultures were neurons; the remaining cells were astrocytes. 21
Cell viability assay
Cell survival was evaluated with Alamar blue dye using methods similar to those described previously. 21 Dissociated cells were counted and plated in 24-well plates and exposed to treatments for a pre-defined period. The culture medium was removed and replaced with 300 L per well of 0.5% Alamar blue diluted in Locke's solution and incubated for 1–2 h at 37℃ in a 5% CO2 incubator. Levels of the Alamar blue reaction product were measured using a HTS 7000 Plus Bio Assay Reader (540-nm excitation and 590-nm emission wavelengths). Values for cultures exposed to experimental treatments were expressed as a percentage of the mean value for untreated control cultures. 22
Immunocytochemistry
Primary neurons were grown on 25-mm microscope coverslips and fixed in 4% paraformaldehyde for 20 min. After washing, the coverslips were blocked in phosphate-buffered solution (PBS) containing 10% fetal calf serum (FCS) and 0.1% Triton X-100, incubated at room temperature for 1 h with primary rabbit anti-RAGE antibody (Abcam), mouse anti-MAP2 (Millipore), in blocking buffer, followed by a 1 h incubation with Alexa Fluor 488- and 568-conjugated corresponding secondary antibodies (invitrogen) at room temperature. In vitro analysis of RAGE positive neurons was made by quantifying the amount of staining positive cells based on five randomly selected microscopic images (40 × magnification).
Immunohistochemistry
Brains were fixed in 4% formaldehyde in PBS (freshly prepared from paraformaldehyde powder) overnight at 4℃ before being transferred to sequential 20% and 30% solutions of sucrose (w/v) at 4℃ until the brains sank to the bottom of the solution. The brains were embedded in Tissue-Tek (Sakura) before sectioning (10 M sections made using a cryostat) in the coronal anatomical plane. Sections were first exposed for a minimum of 30 min to PBS containing 0.1% Triton X-100 (Amresco) and 10% normal goat serum (Sigma) to block nonspecific antibody binding, followed by incubation overnight with the TdT FragEL DNA fragmentation detection kit. Immunohistochemical analysis for 8OHdG was performed by the labeled polymer method using the EnVision kit (DAKO). After blocking endogenous peroxidase with 0.3% hydrogen peroxidase in methanol for 30 min, the slides were washed and then incubated with anti-8OHdG monoclonal antibody (diluted 1:100; NOF corporation, Japan) overnight at 4℃ and exposed to goat anti-mouse immunoglobulin conjugated to peroxidase-labeled dextran polymer for 30 min, followed by staining with 3,3′-diaminobenzidine tetrahydrochloride. Slides were then counterstained with hematoxylin and analyzed. In our experiment, the percentage of positive immunohistochemical staining cells with the aforementioned antibodies was blindly counted and expressed as an average based on three separate sections from four rats for each group.
Statistical analysis
Continuous data and categorical data were expressed as the mean ± standard deviation and percentage, respectively. Statistical analyses were performed using GraphPad Prism 4.0 software (GraphPad Software, Inc. USA) and SPSS software package version 18.0 (SPSS Inc., Chicago, IL, USA). Experimental results are expressed as mean ± SEM. Comparisons between subgroups were made with student’s t-test, logistic regression, and ANOVA, followed by Newman–Keuls post hoc analysis. Statistical significance was defined as p < 0.05.
Results
Levels of CSF HMBG1 significantly correlate with outcomes in SAH patients
Associations between clinical and cerebrospinal fluid parameters and outcome in patients with subarachnoid hemorrhage.
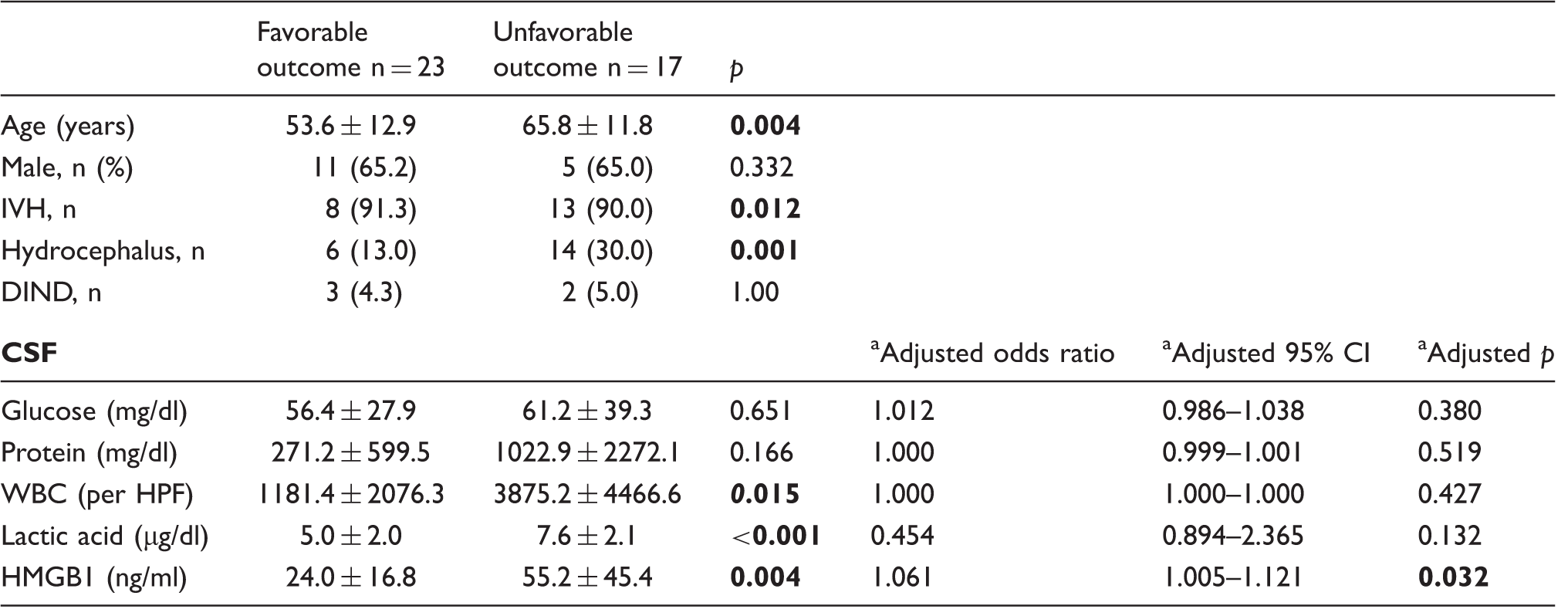
IVH: intraventricular hemorrhage; DIND: delayed ischemic neurological deficit; CSF: cerebrospinal fluid.
Adjusted with age, gender, presence of IVH, and hydrocephalus. Bold values indicate a p value less than 0.05.
Expression of RAGE increases in post-SAH rat brain cells and cultured neurons exposed to post-SAH CSF
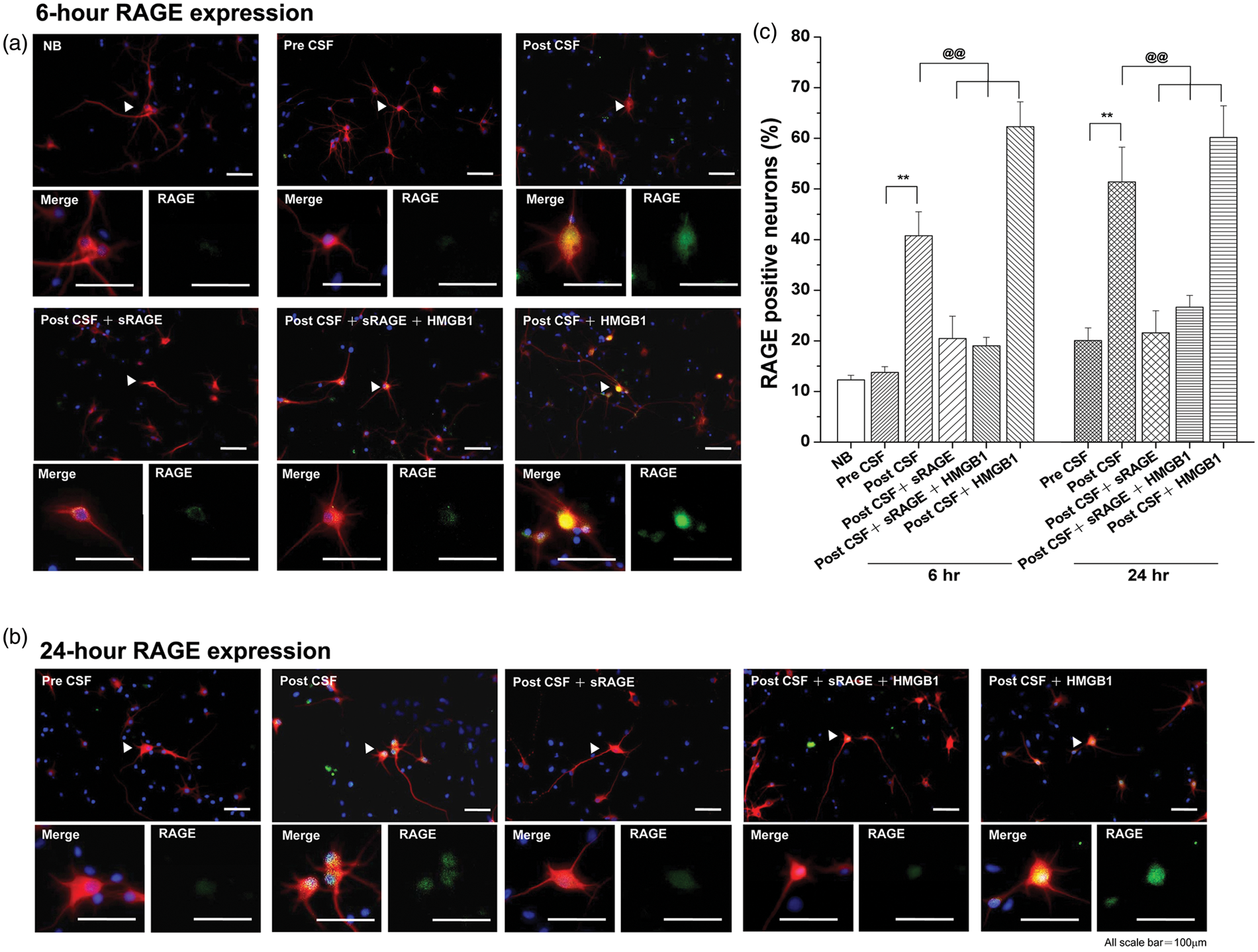
We first analyzed the level of HMGB1 in CSF obtained from rats following 24 h SAH injury (Figure 1(a)). Immunoblot assay of CSF shows increased levels of HMGB1 following SAH injury compared to pre-SAH CSF samples (Figure 1(b)). Next, we analyzed the expression pattern of RAGE in the brain following SAH. Brain cells especially in cortical layers had increased RAGE expression after 24 h of post-SAH injury (Figure 1(c)). Similarly, RAGE expression significantly increased in cultured primary neurons after exposure to CSF obtained from rats following both 6 and 24 h of SAH (CSF from rat post-SAH 24 h with a concentration of 1 µL/mL) or pre-administration of recombinant HMGB1 (50 ng/mL). Importantly, pre-treatment of recombinant sRAGE into culture medium significantly inhibited the expression levels of RAGE following either CSF or HMGB1 (Figure 2(a) to (c)).
Levels of CSF HMBG1 and expression of RAGE in brain cells increase following SAH in rats. (a) Representative images of rat brains following SAH. (b) Immunoblot assay for CSF obtained from post SAH rats showed increased levels of HMGB1 after 24 h following SAH injury. (c) Expression of RAGE in rat brain cells increased significantly 24 h after SAH injury. Expression of RAGE increases in cultured cortical neurons following post-SAH CSF treatment. RAGE expression significantly increased in terms of signal intensities and number of cells after 6 (a and c) and 24 (b and c) h exposure to CSF from rat SAH model (CSF from rat post-SAH 24 h with a concentration of 1 L/mL) (both p < 0.01 especially after 24 h stimulation). CSF-induced increase in RAGE positive neurons was ameliorated by the administration of recombinant sRAGE into culture medium and aggravated by that of the recombinant HMGB1 (a–c). **p < 0.01 versus pre-CSF cultures. @@p < 0.01 versus post-CSF cultures. Statistical comparisons were made with ANOVA, followed by Newman–Keuls post hoc analysis.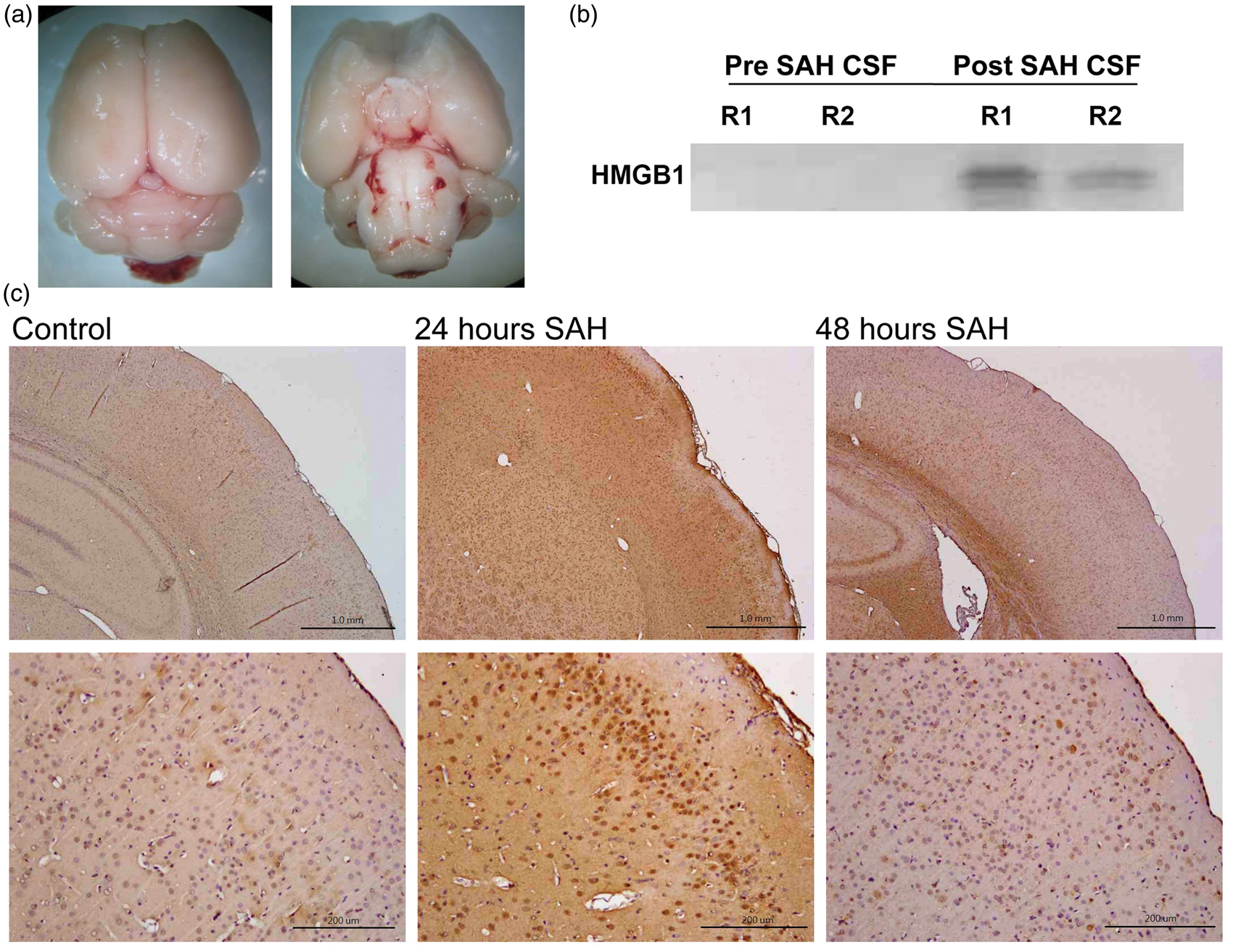
Administration of recombinant sRAGE protects cultured neurons exposed to post-SAH CSF
In order to examine the possible detrimental effect of CSF following SAH injury, we tested CSF from a patient with normal pressure hydrocephalus and two SAH patients (one with relatively low HMGB1 and the other one with high HMGB1 levels) and artificial CSF, on neuronal cultures for cell viability. Our data show that CSF from SAH patients, especially the patient who had high level of HMGB1, induced significantly higher level of neuronal death compared to other groups, especially after 24 h stimulation (Figure 3(a)).
Post-SAH CSF from patients and rats induces neuronal cell death. CSF obtained from the patient who had high level of HMGB1, induced significantly higher level of neuronal death compared to other groups (a) (p < 0.01; especially after 24 h stimulation). Exogenous administration of recombinant sRAGE (1 g/mL) to cultured primary neurons exposing to different sources of CSF resulted in significantly less number of cell death (b). By contrast, exogenous HMGB1 (50 ng/mL) significantly aggravated post-SAH CSF-induced neuronal death, and its detrimental effect was partially reversed by pre-mixing with sRAGE (b).**p < 0.01 versus ACSF control group (a) or pre-CSF group (b). @@p < 0.01 versus post-CSF cultures. &&p < 0.01 versus post-CSF + sRAGE + HMGB1 cultures. Statistical comparisons were made with ANOVA, followed by Newman–Keuls post hoc analysis.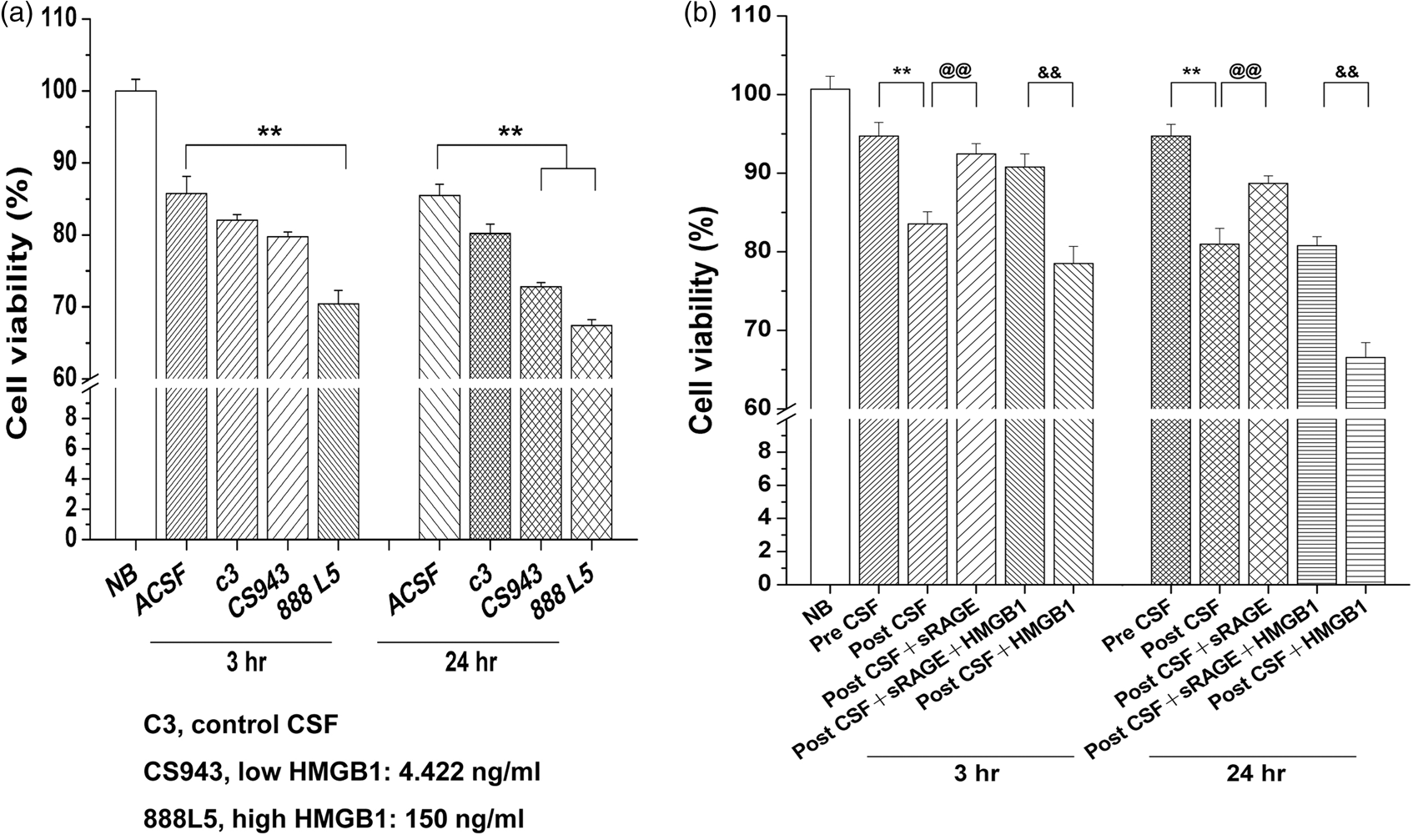
We next analyzed rat primary neuronal cell viability under different conditions to study the protective effect of sRAGE against post-SAH CSF (1 µL/mL) and HMGB1-induced neuronal death (Figure 3(b)). Our data show that cell viability of cultured neurons decreased significantly following treatment with CSF from post-SAH compared to pre-SAH rats. Importantly, neurons treated with recombinant sRAGE (50 ng/mL) were significantly less vulnerable to death caused by CSF from post-SAH rat. By contrast, exogenous HMGB1 (50 ng/mL) aggravated CSF from post-SAH rat-induced neuronal death, and its detrimental effect was partially reversed by pre-mixing with recombinant sRAGE (Figure 3(b)).
Intrathecal injection of recombinant sRAGE protects rat brain cells against SAH injury
Intrathecal administration of recombinant sRAGE (0.1 µg/rat) at the time of the SAH procedure significantly reduced the number of positive TUNEL-stained brain cells compared to controls at both 24 and 48 h after SAH injury (Figure 4(a) and (b)). Similarly, 8OHDG which is an oxidized derivative of deoxyguanosine and one of the major products of DNA oxidation also significantly reduced (Figure 4(c) and (d)).
Administration of sRAGE protects rat brain against SAH-induced cell death and DNA damage. Intrathecal injection of recombinant sRAGE (0.l g/rat) at the time of the SAH procedure significantly reduced the number of TUNEL-positive brain cells (a and b) and 8OHDG expression (c and d) compared to controls at both 24 and 48 h after SAH injury. **p < 0.01 versus control group at 24 h. ***p < 0.001 versus control group at 48 h.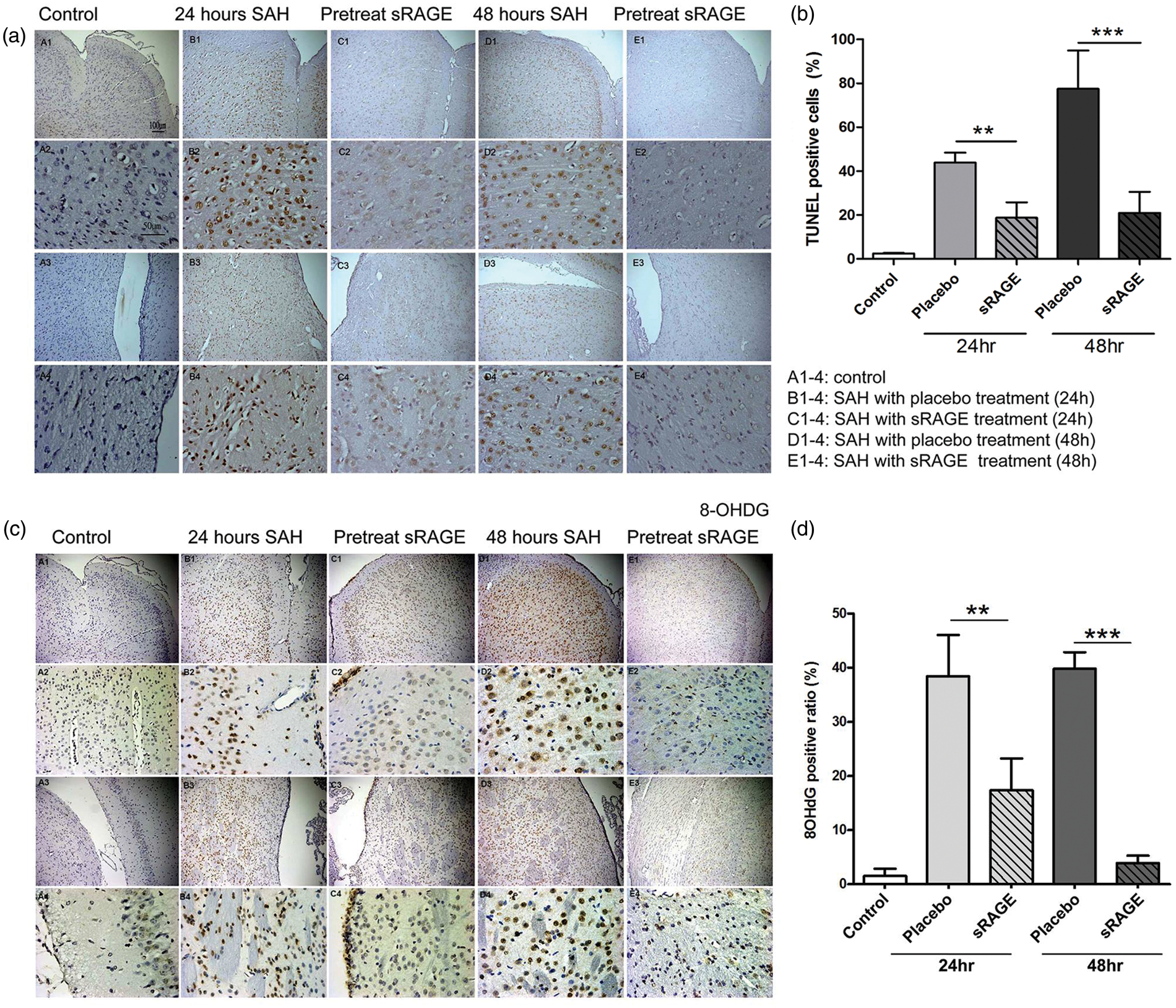
Discussion
Although vasospasm has been studied and treated with a wide range of drugs during the past decades, the outcomes of patients suffering from SAH have not obviously changed.9,23 On the other hand, the importance of early brain injury after SAH, which is possibly related to multiple factors such as direct subarachnoid blood toxicity, global ischemia, and microcirculation impairment, has been addressed in recent years.10,23–26 Previous data have proved the occurrence of apoptotic cell death in neurons and endothelial cells during the acute stage of SAH. However, the mechanisms that induce the cell death after SAH are still undefined, though inflammation, such as via the MAPKs pathway, has been mentioned as one of the critical mediators.27–29
There is growing evidence that HMGB1 and RAGE signaling and related inflammatory reactions are involved in the pathogenesis of various disorders, including cardiovascular, neurodegenerative, and immune disorders.12,20,30–33 We have recently established that plasma levels of HMGB1 as well as interactions between HMGB1 and RAGE were increased significantly after ischemic stroke onset both in human and mice. 14 We also found that administration of recombinant sRAGE effectively protects mice from ischemic reperfusion injury and protects primary neurons from energy deprivation-induced cell death via a mechanism involving reduced RAGE-associated inflammatory pathways. 14
The present study showed that CSF levels of HMGB1 increased significantly in SAH patients, and the values were positively correlated with outcome even after adjustment for clinical variables. In addition, RAGE expression increased in cultured neurons exposed to post-SAH CSF. Therefore, HMGB1 in CSF may be directly responsible for the activation of cellular RAGE and downstream pathways in acute SAH. Furthermore, the increased neuronal death observed when exposed to either CSF from SAH patients or SAH rats prove that post-SAH CSF is toxic to neurons and may responsible for delayed neuronal cell death following SAH. The current study for the first time demonstrates that external administration of recombinant sRAGE can significantly reduce the number of cell deaths based on the aforementioned in vivo and in vitro experimental models.
Increased CSF HMGB1 has also been reported in patients with neuromyolitis optica and multiple sclerosis.34–36 The levels of CSF HMGB1 are correlated with CSF cell counts, and levels of protein, interleukin 6, and glial fibrillary acidic protein in those patients.34,36 Another study also demonstrated the direct effect of CSF from multiple sclerosis patients on neuronal apoptosis, and the severity of cell viability correlated with the clinical disease activities. In addition, treatment with caspase 3 inhibitor in neurons exposed to CSF from multiple sclerosis patients effectively preserved neuronal survival, indicating the direct involvement of CSF in the apoptotic pathway. 37 In our study, positive TUNEL staining cells in SAH rats provided the evidence of cell apoptosis during the acute stage of SAH and the effect of recombinant sRAGE establishes the therapeutic potential of blocking HMGB1- and RAGE-mediated signaling in reducing SAH-induced apoptotic cells.
There are several possible limitations in our study. This study was limited with patients from single hospital and thus patient selection bias might be possible. Futher study with larger sample sizes and a muti-center study design may be considered to strengthen our findings. In the basic part, although our data clearly showed the direct effect of post-SAH CSF on neuronal apoptosis, whether other cells types such as endothelial cells or glial cells have similar mechanisms is uncertain. In the rat SAH model, we administered the intrathecal injection of recombinant sRAGE only at the time of the SAH procedure since intrathcal injection at the post-SAH time point presents a technical difficulty with regard to increased incranial pressure after SAH. Therefore, it is uncertain whether delayed treatment of recombinant sRAGE enables providing the similar protective effect in acute SAH.
Our study revealed the pivotal role of HMGB1-RAGE-mediated signaling in the pathogenesis of neuronal injury after the occurrence of SAH. Measurement of CSF levels of HMGB1 can be a prognostic indicator for patients with Fisher's grade ≥ III aneurysmal SAH. Administration of recombinant sRAGE might be a potential therapy in rescuing post-SAH neuronal death.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by (1) National Taiwan University Hospital grant (NTUH102-M2313), (2) the Taiwan National Science Council grants (101-2314-B-002-160-MY3 and 102-2314-B-002-148) and (3) Academia Sinica, Taiwan, BM103010096 and BM104010092.
Acknowledgement
The authors would like to thank the 3rd core facility at National Taiwan University Hospital for technical assistance and facility support.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
KCW and SCT designed research; KCW, SCT, JEL, YIL, YSH, and WSY performed research and data analysis; KCW, SCT, and JSJ wrote the paper. TVA, JSJ, and YKT gave the critical revision.