
Abstract
Lipocalin-2 mediates neuro-inflammation and iron homeostasis in vascular injuries of the central nervous system (CNS) and is upregulated in extra-CNS systemic inflammation. We postulate that cerebrospinal fluid (CSF) and blood lipocalin-2 levels are associated with markers of inflammation and functional outcome in subarachnoid hemorrhage (SAH). We prospectively enrolled 67 SAH subjects, serially measured CSF and plasma lipocalin-2, matrix metallopeptidase 9 (MMP-9), interleukin-6 (IL-6) and tumor necrosis factor-alpha (TNF-α) on post-SAH days 1-5 and assessed outcome by modified Rankin Scale (mRS) every 3 months. Unfavorable outcome is defined as mRS > 2. Twenty non-SAH patients undergoing lumbar drain trial were enrolled as controls. Lipocalin-2 was detectable in the CSF and significantly higher in SAH compared to controls (p < 0.0001). Higher CSF LCN2 throughout post-SAH days 1-5 was associated with unfavorable outcome at 3 (p = 0.0031) and 6 months (p = 0.014). Specifically, higher CSF lipocalin-2 on post-SAH days 3 (p = 0.036) and 5 (p = 0.016) were associated with unfavorable 3-month outcome. CSF lipocalin-2 levels positively correlated with CSF IL-6, TNF-α and MMP-9 levels. Higher plasma lipocalin-2 levels over time were associated with worse 6-month outcome. Additional studies are required to understand the role of lipocalin-2 in SAH and to validate CSF lipocalin-2 as a potential biomarker for SAH outcome.
Keywords
Introduction
Subarachnoid hemorrhage (SAH) remains one of the most lethal and morbid forms of stroke leading to significant long-term disability and high socioeconomic impact to the society.1,2 Several recent phase-3 randomized clinical trials failed to find therapeutics that improve SAH outcome despite promising preclinical data.3–5 Emerging evidence suggest human SAH is a complex condition with pathophysiologic processes extending beyond the central nervous system (CNS). Clinically, SAH patients develop both neuroinflammation as well as acute systemic inflammatory response syndrome (SIRS) which is associated with poor outcome.6–8 The relationship between systemic and neuroinflammation in SAH is insufficiently understood. Currently, there is no validated blood or CSF biomarkers in clinical use for SAH-associated brain injury and outcome prediction.
Lipocalin-2, also known as neutrophil gelatinase-associated lipocalin, is an important mediator of neuroinflammation in vascular brain injury9–11 via several putative mechanisms. In the brain, lipocalin-2 knock out reduces tumor necrosis factor (TNF-α) and interleukin 1β (IL-1β) levels and improves outcomes in acute ischemic stroke models. 11 On the other hand, lipocalin-2 upregulation in injured brain may be part of an endogenous repair response 12 that promotes angiogenesis and neurovascular recovery. 13 In addition to neuroinflammation, brain lipocalin-2 may also mediate iron toxicity in hemorrhagic stroke models.14,15 On the systemic side of the blood brain barrier, lipocalin-2 stimulates neutrophil migration and neutrophilic cytokines production including IL-6, TNF-α and IL-1β and elevated blood lipocalin-2 is associated with in systemic inflammation.16,17 Furthermore, lipocalin-2 binds and stabilizes human matrix metallopeptidase-9 (MMP-9), 18 which regulates neutrophils in inflammatory processes19,20 and is implicated in multiple vascular brain injury mechanisms including blood brain barrier (BBB) injury. 21 Increased blood IL-6, TNF-α, and MMP-9 are associated with poor outcome6,22–24 in human SAH. Taken together, lipocalin-2 is an important candidate biomarker for both SAH-associated neuroinflammation and systemic inflammation. We hypothesize that cerebrospinal fluid (CSF) and plasma lipocalin-2 levels may be associated with inflammatory mediators and with functional outcome in a human SAH cohort.
Materials and methods
Prospective SAH and control cohorts
In this study, 67 consecutive SAH and 20 control subjects with both CSF and blood biospecimens available were derived from an existing parent cohort of 102 SAH subjects and 24 control subjects from an existing single-center observational cohort enrolled between 2009 – 2014 at the Brigham and Women’s Hospital. 25 Inclusion criteria were consecutive adult patients with spontaneous SAH within 96 hours of onset. Exclusion criteria were pregnancy, end-stage renal or liver diseases, active malignancy, any CSF infection, or human immunodeficiency virus infection as these conditions may confound blood and CSF biomarker compositions. Non-SAH patients with suspected normal pressure hydrocephalus (NPH) undergoing lumbar CSF drain trial were enrolled as controls. All subjects were enrolled following self or surrogate informed consent. This study was conducted according to the ethical standards of Brigham and Women’s hospital (BWH) Institutional Review Board (IRB) (approval number: 2007P002168). A written informed consent was obtained from all subjects or their health care surrogate.
Standardized institutional SAH protocol
All SAH subjects were treated according to standardized institutional treatment protocol in a dedicated neurological intensive care unit. All patients received enteral nimodipine. Per standardized SAH treatment protocol, patients do not receive steroids or nonsteroidal anti-inflammatory (NSAID) medications. All patients underwent digital subtraction cerebral angiography upon initial presentation and again on post-SAH days 6-8 for evaluation of angiographic vasospasm. Clinically significant vasospasm was treated with induced hypertension, euvolemia, and intra-arterial vasodilator injection and/or balloon angioplasty in refractory cases. Patients were monitored for delayed cerebral ischemia throughout acute hospital stay for up to 21 days from SAH onset.
Outcome and clinical endpoints definition
The primary endpoint of this study was 3-month SAH functional outcome measured by modified Rankin Score (mRS) validated for telephone assessment.26,27 The secondary endpoint was 6-month SAH outcome. All enrolled SAH subjects underwent prospective, standardized outcome assessment at 3- and 6-months post SAH using scripted telephone interview by trained research personnel under principal investigator supervision (SC). Unfavorable outcome was defined as mRS > 2. SAH clinical severity was measured by Hunt and Hess (HH) grade and radiographic severity was measured by modified Fisher grade. 28 Both scores were systematically adjudicated by the principal investigator (SC). Angiographic vasospasm (VSP) was defined as >50% reduction in diameter of any cerebral artery on angiogram. Delayed cerebral ischemia (DCI) was defined as areas of new hypodensity on CT scans after exclusion of peri-operative injuries or injuries prior to SAH onset. 29 Both vasospasm and DCI were adjudicated by a trained neuroradiologist (LH).
Biospecimen and biomarker methods
After informed consent, all enrolled SAH patients underwent prospective, standardized serial blood and CSF collections on post-SAH days 1, 3 and 5. Day 0 is defined as day of SAH symptom onset. Per human subject protection considerations, all samples were taken after ruptured cerebral aneurysms have been secured. Blood samples were collected via indwelling arterial or central venous catheters if they were present and otherwise via standard venopuncture. CSF was collected using sterile technique and only in subjects with external ventricular drain (EVD) present for a clinical indication. In control subjects, blood and CSF samples were collected upon study enrollment and CSF was collected through a lumbar drain placed for a clinical indication. To minimize risk of infection to human subjects, we do not aspirate directly from the EVD or lumbar drain (LD) catheter. Fresh CSF is allowed to drip out into the collection cylinder in the EVD/LD system and this CSF is then collected from the collection graduated cylinder in the EVD system using sterile technique.
Immediately following collection, all biospecimens (CSF and blood) were centrifuged at 3900 revolutions per minute for 15minutes at room temperature. Post centrifugation, the cell-free CSF supernatant and plasma/serum from blood are aliquoted into micro-aliquot tubes and immediately frozen at −80 C until analysis.
Lipocalin-2 was measured from EDTA plasma samples using Human Magnetic Luminex Assay and TNFα and IL-6 were measured using Human XL Cytokine Discovery Premixed High Performance Assay (R&D Systems, Minneapolis, MN). Biomarker analyses were performed by the Clinical Research, Investigation, and Systems Modeling of Acute Illness (CRISMA) center Core Laboratory at the University of Pittsburgh. We performed careful internal validation of biomarker assays. A standard curve was generated for each experiment. The standards (provided by the manufacturer) used to generate the standard curve were analyzed as unknowns within the experiment across all plates. Random experimental samples were analyzed across all plates to assess whether variations in the results occurred between plates. Additionally, we spiked pooled CSF samples with the standards provided by manufacturer and calculated the % recovery, which ranged from 90-99%.
Statistical analysis
Continuous variables were compared using a two-sided Student’s t or Mann-Whitney U test depending on data distribution. Paired tests were used for in-sample comparisons. The association between CSF and plasma lipocalin-2 levels over time with the primary and secondary endpoints were assessed using mixed effect models. For lipocalin-2 level comparisons at individual measurement time points, we applied Bonferroni correction to control family-wise error rate. We performed exploratory bivariate analyses to assess associations of lipocalin-2 with TNF-α, IL-6, and MMP-9 levels using Pearson’s or Spearman’s test depending on data distribution. Ordinal logistic regression models were used to explore the independent effect of lipocalin-2 levels on mRS after adjusting for HH grade, modified Fisher grade, and age, which are known important predictors of SAH outcome. Data analyses were performed using GraphPad Prism 8 and JMP Pro 13.0.
De-identified data from this study will be made available to other investigators in accordance to the National Institute of Health’s data sharing policy and in observance of all regulatory and human subject protection requirements.
Results
Patient characteristics
Table 1 summarizes baseline characteristics of the SAH (N = 67) and control (N = 20) cohorts. Compared to controls, the SAH cohort had lower mean age and higher proportion of females. Poor outcome occurred in 48% of SAH subjects at 3-month and 36% at 6-month post SAH. Thirty-seven (55%) met criteria for angiographic vasospasm. Two-thirds of patients presented with HH grade ≥ 3 and a majority of the cohort (89%) had modified Fisher grades 3–4. The SAH cohort characteristics are comparable to those reported in recent large SAH trials.4,5,30
Baseline characteristics of SAH and control subjects.
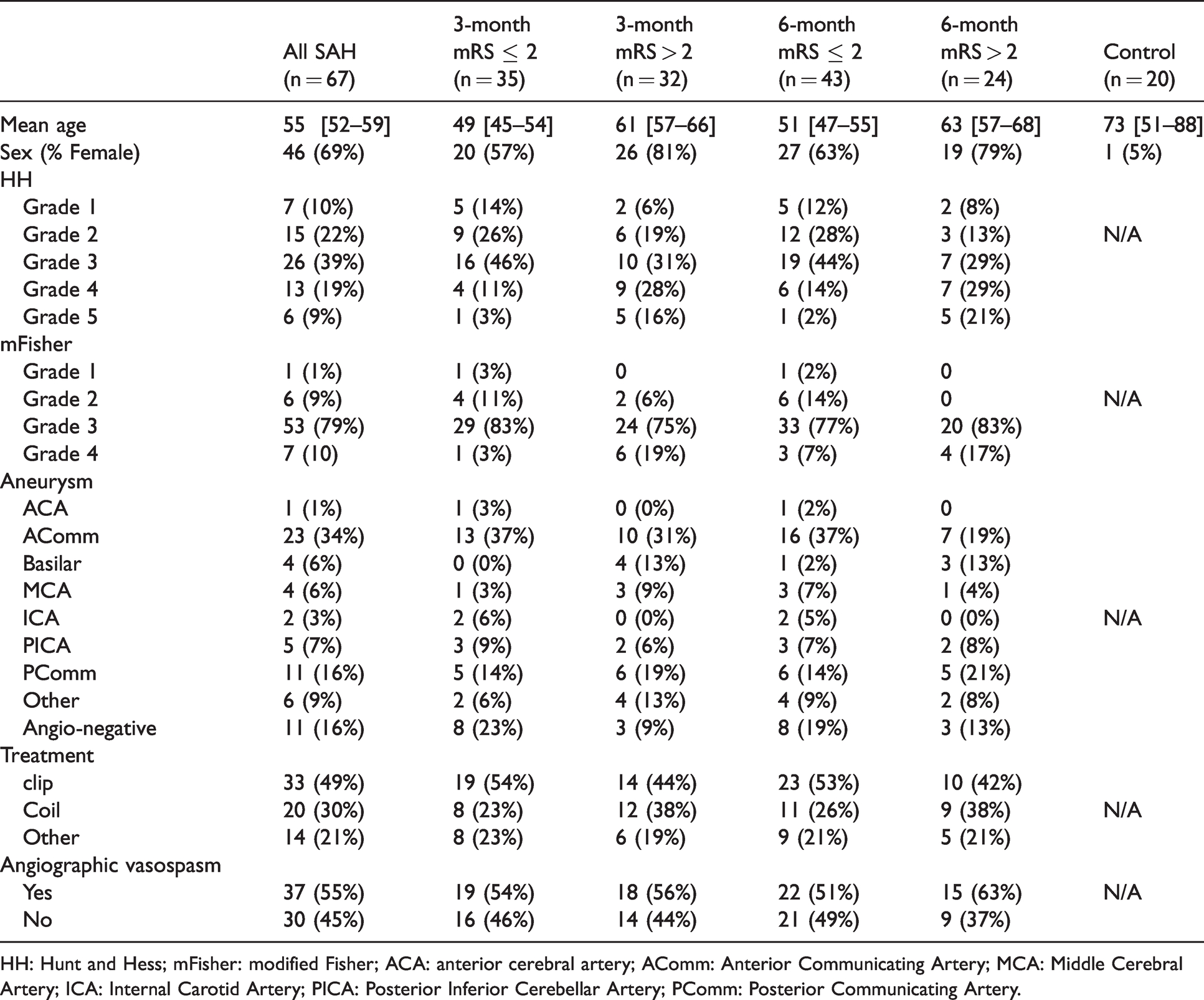
HH: Hunt and Hess; mFisher: modified Fisher; ACA: anterior cerebral artery; AComm: Anterior Communicating Artery; MCA: Middle Cerebral Artery; ICA: Internal Carotid Artery; PICA: Posterior Inferior Cerebellar Artery; PComm: Posterior Communicating Artery.
CSF lipocalin-2 and SAH outcome
CSF lipocalin-2 levels were higher at all time points after SAH compared to controls (Figure 1(a)). Within the SAH cohort, CSF lipocalin-2 levels increased over time from post-SAH day 1 to 5, and higher CSF lipocalin-2 throughout post-SAH days 1-5 was associated with unfavorable outcome both at 3-months (p = 0.0031) and at 6-months (p = 0.014) (Figure 1(a) and (b)). For individual time points comparisons, higher CSF lipocalin-2 levels on post-SAH day 3 (p = 0.036) and 5 (p = 0.016) were associated with unfavorable 3-month outcome (Figure 1(a)). CSF lipocalin-2 levels did not differ between low (HH grade 1-3) and high (HH grade 4-5) SAH clinical severity (Figure 1(c)). Similarly, CSF lipocalin-2 did not vary with SAH radiographic severity measured by modified Fisher grade (Figure 1(d)).
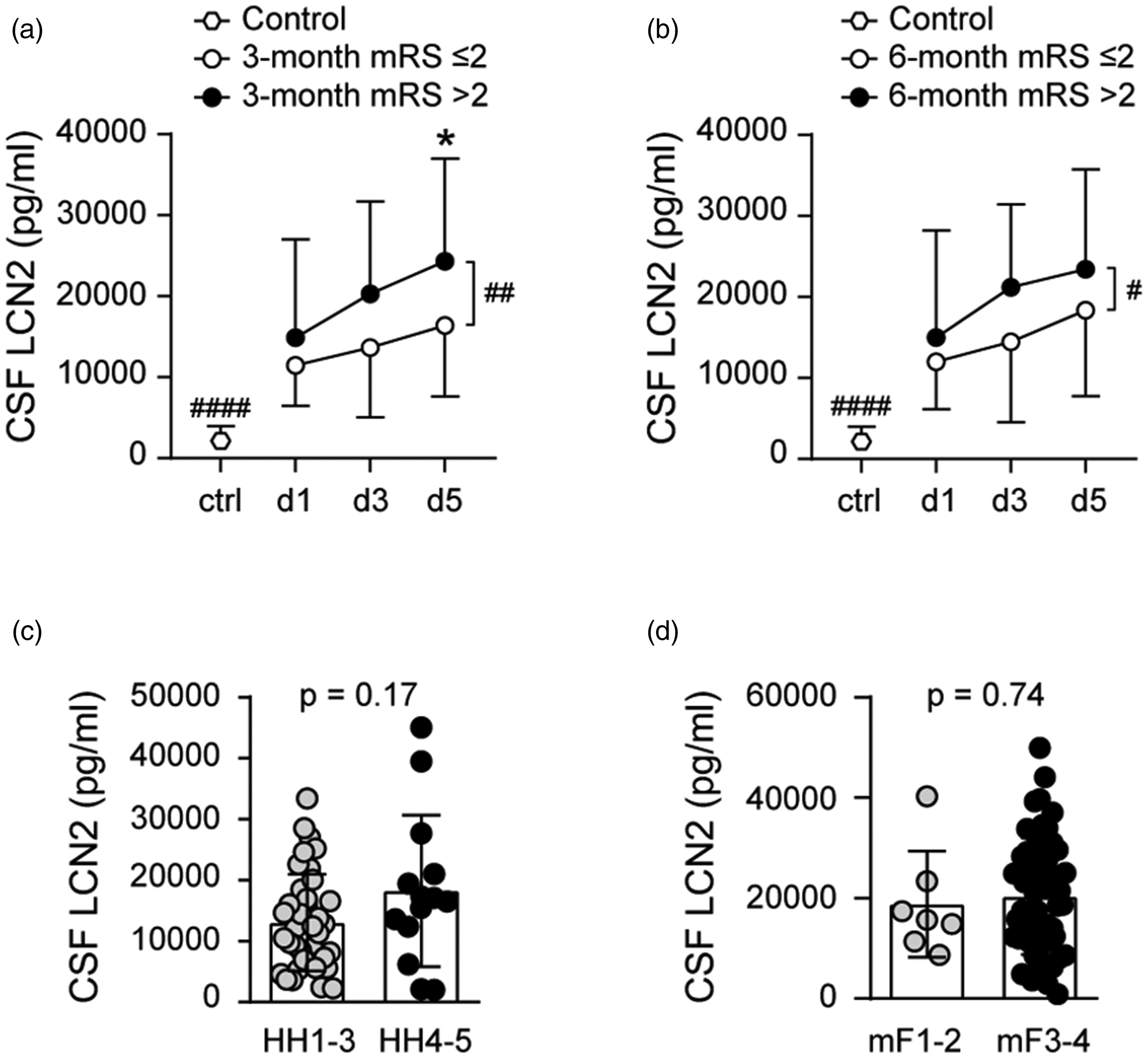
CSF lipocalin-2 (LCN2) levels and SAH functional outcomes at 3-month (a) and 6-month (b) and CSF lipocalin-2 levels relative to SAH clinical severity measured by Hunt Hess grade (c) and SAH radiological severity measured by modified Fisher grade (d) (*p < 0.017, #p < 0.05,
Univariate analyses of our data showed age as a continuous variable is significantly associated with 3- (p < 0.0001) and 6-month (p < 0.0001) SAH outcomes in this patient cohort which is consistent with published data.31,32 We used ordinal logistic regression models to explore the relationship between SAH outcome and CSF lipocalin-2 levels at specific measurement time points adjusting for effects of age, Hunt and Hess grade and modified Fisher grade as these are established prognostic factors for SAH outcome. We used ordinal logistic regression models to explore the relationship between 3-month SAH outcome and CSF lipocalin-2 levels at specific measurement time points after adjusting for all 3 covariates. Age was entered as a continuous covariate and HH and modified Fisher grades as ordinal covariates into the ordinal logistic regression where outcome measured by mRS is the dependent variable. CSF lipocalin-2 levels on post-SAH day 1 (p = 0.035) and post-SAH day 3 (p = 0.049) were associated with SAH outcome at 3 months after adjusting for all 3 covariates. CSF lipocalin-2 levels on post-SAH day 1 (p = 0.027) and post-SAH day 3 (p = 0.022) were associated with SAH outcome at 6 months after adjusting for all 3 covariates.
Plasma lipocalin-2 levels and SAH outcome
Plasma lipocalin-2 levels were lower in SAH compared to controls 29281 ± 6235 pg/ml vs 38091 ± 10904 pg/ml (p = 0.0001). Plasma lipocalin-2 levels over post-SAH days 1-5 demonstrated significant difference between favorable and unfavorable outcome groups only at 6-month (Figure 2(a) and (b)). Unlike what we observed in the CSF, plasma lipocalin-2 levels increased with both HH (p = 0.018) and with modified Fisher grade (p = 0.005) (Figure 2(c) and (d)). Mean plasma lipocalin-2 levels of SAH patients ranged from 12822 pg/ml to 50784 pg/ml while mean CSF lipocalin-2 levels ranged from 1469 pg/ml to 45052 pg/ml. CSF and plasma lipocalin-2 levels showed no correlation at any of the measured time points (Supplemental Figure 1(a) to (c)).
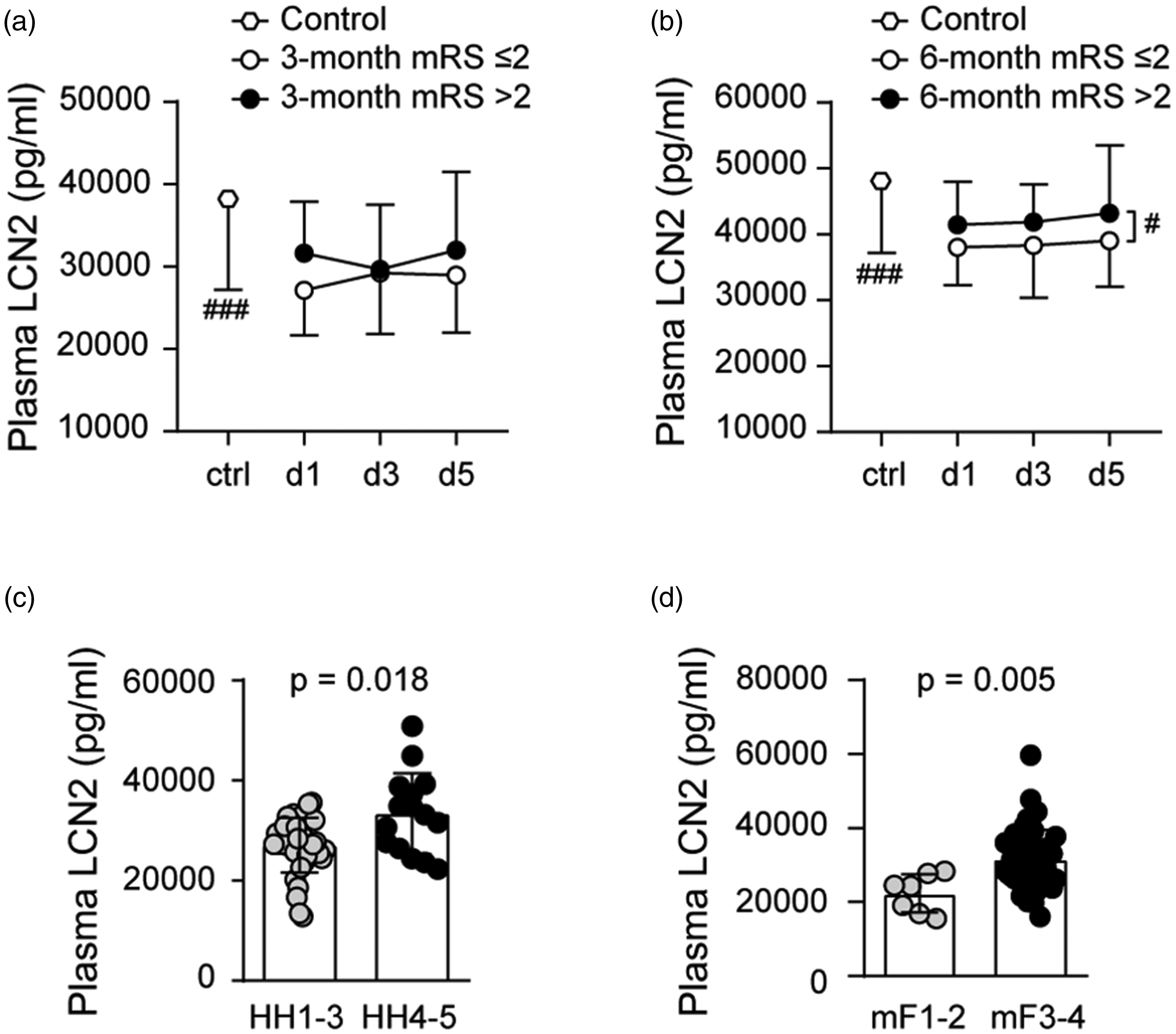
Plasma lipocalin-2 (LCN2) levels and SAH functional outcomes at 3-month (a) and 6-month (b) and plasma lipocalin-2 levels relative to SAH clinical severity measured by Hunt Hess grade (c) and radiographic severity measured by modified Fisher grade (d) (#p < 0.05,
Lipocalin-2 vs. Inflammatory mediators TNF-α, IL-6 and MMP-9
We assessed the relationship between CSF lipocalin-2 and its effects on inflammatory mediators IL-6, TNF-α, and MMP-9 in both CSF and blood. In CSF, lipocalin-2 levels were consistently and positively associated with IL-6 and TNF-α levels at all measurement time points with strongest associations on post-SAH days 3 and 5 (Figure 3(a) to (f)). Plasma lipocalin-2 did not show consistent associations with IL-6 and TNF-α.
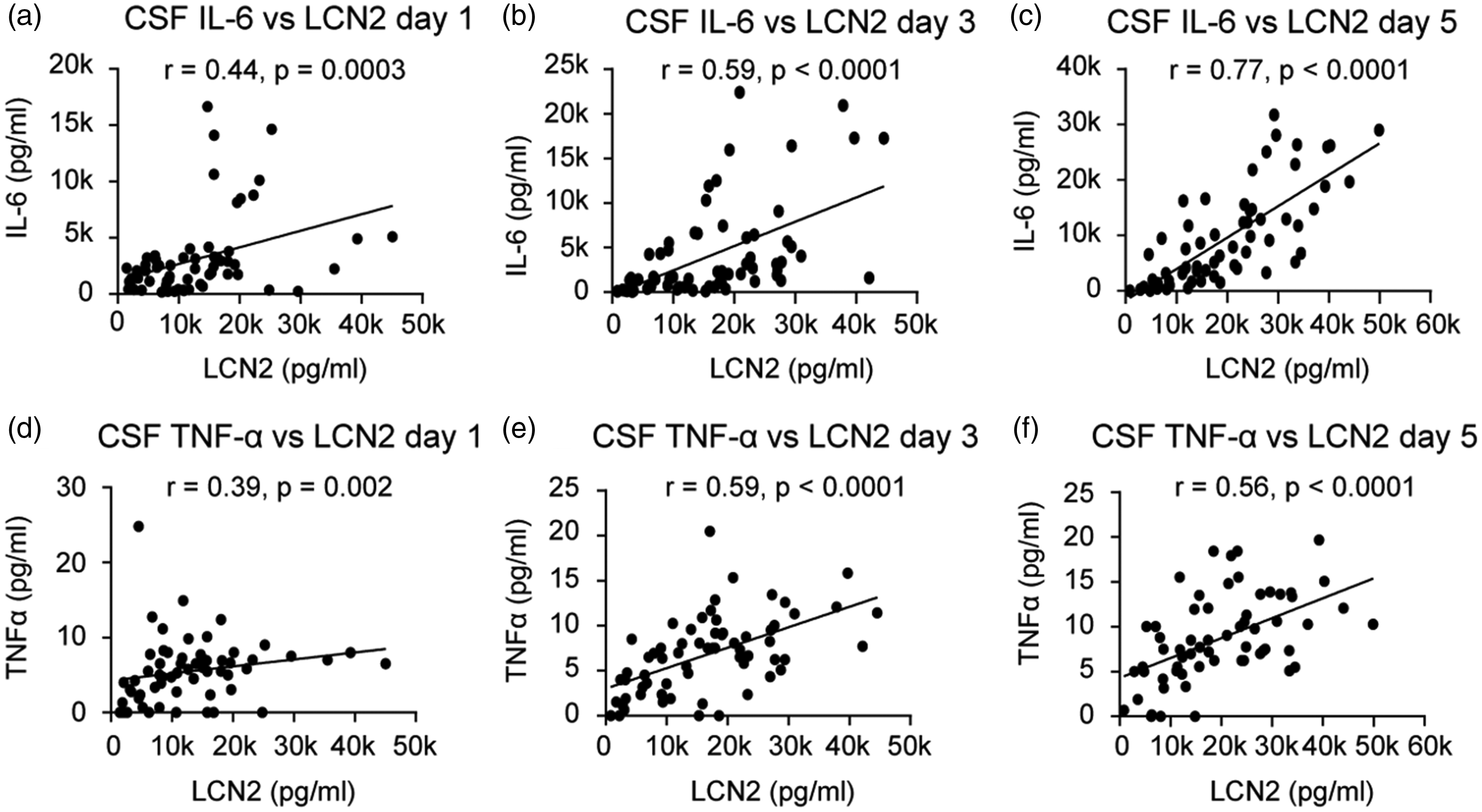
Associations between CSF lipocalin-2 (LCN2) and IL-6 on post-SAH days 1 (a), 3 (b) and 5 (c) and between CSF LCN2 and TNF-α on post-SAH days 1 (d), 3 (e) and 5 (f).
CSF lipocalin-2 did not show associations with MMP-9 except on post-SAH day 5 (r = 0.64, p = 0.0001, Spearman’s Rank Sum test) while in plasma lipocalin-2 and MMP-9 showed trend towards positive association on post-SAH day 1 (r = 0.51, p = 0.02, Spearman’s Rank Sum Test) (Figure 4).
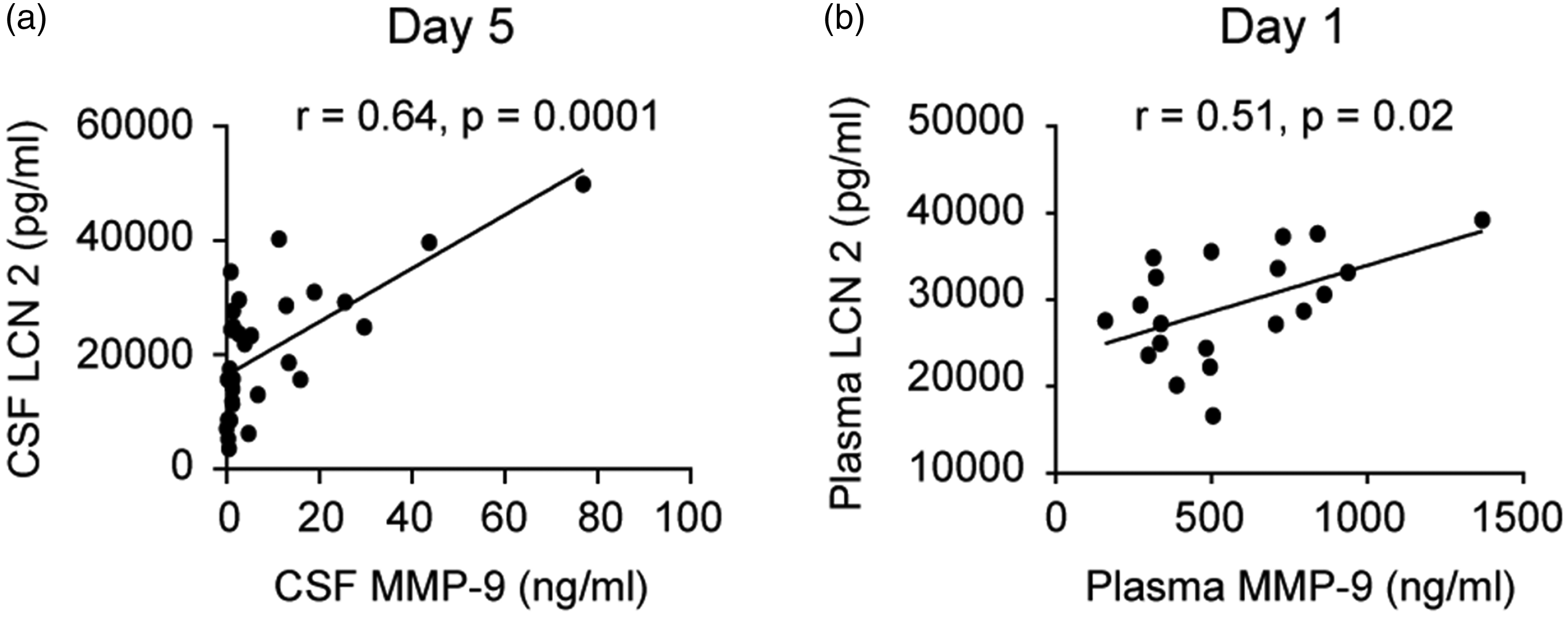
Relationships between lipocalin-2 (LCN2) and MMP-9 in CSF (a) and in blood (plasma) (b).
Effect of age and sex difference on liopocalin-2 levels
We explored whether CSF and plasma lipocalin-2 levels differed by age or sex. Higher CSF lipocalin-2 was associated with older age on post-SAH days 3 (r = 0.29, p = 0.02) and 5 (r = 0.44, p = 0.0003) (Supplemental Figure 2(a) and (b)). Plasma lipocalin-2 did not demonstrate significant changes with age except on post-SAH day 5 (r = 0.36, p = 0.008) (Supplemental Figure 2(c)). Neither CSF nor plasma lipocalin-2 levels differed by sex (Supplemental Figure 2(d) and (e)).
Effects of angiographic vasospasm and aneurysm treatment modalities on lipocalin-2 levels
We performed exploratory analyses to assess whether lipocalin-2 levels differed in patients with digital subtraction cerebral angiography confirmed aneurysm, or in patients who subsequently developed angiographic vasospasm, and whether cerebral aneurysm treatment modalities affected lipocalin-2 levels, as craniotomy may conceivably alter CSF biomarker levels. No difference of either plasma or CSF lipocalin-2 was observed between patients with digital subtraction cerebral angiography confirmed aneurysm and angionegative results (Supplemental Figure 3(a) and (b)). Meanwhile, neither plasma nor CSF lipocalin-2 were associated with subsequent angiographic cerebral vasospasm (Supplemental Figure 3(c) and (d)). Lipocalin-2 levels did not differ by cerebral aneurysm treatment modalities (Supplemental Figure 3(e) and (f)).
Discussion
In this study, we simultaneously examined lipocalin-2 and its associated inflammatory mediators on both sides of the BBB during the acute phase of SAH (days 1-5) in a prospective human cohort. We showed that in SAH, lipocalin-2 behaves very differently on the CNS versus the blood side of the BBB. CSF lipocalin-2 levels were significantly higher in SAH compared to controls, levels increased over time, and elevated CSF lipocalin-2 throughout post-SAH days 1-5 was associated with unfavorable outcome at both 3 and at 6 months. CSF lipocalin-2 did not change with clinical (HH grade) or radiographic (modified Fisher grade) SAH severity. Similar to many other biomarkers associated with SAH outcome,33–35 CSF lipocalin-2 was not associated with angiographic vasospasm. In contrast, CSF lipocalin-2 showed consistent positive associations with pro-inflammatory mediators IL-6 and TNF-α throughout post-SAH days 1-5, supporting the hypothesis that CSF lipocalin-2 may be involved in the acute neuroinflammatory processes in SAH. As open craniotomy may conceivably impact CSF biomarker levels, we specifically examined CSF lipocalin-2 levels in relation to aneurysm treatment modalities and found that craniotomy did not impact CSF lipocalin-2 levels. Taken together, these data suggest that CSF lipocalin-2 in SAH is a potential prognostic biomarker for SAH outcome and validation in a separate large cohort is warranted. Furthermore, these data suggest that CSF lipocalin-2 may reflect its important role in the underpinnings of the neuroinflammatory processes in SAH, and it or the purported associated markers IL-6, TNF-α, and/or MMP-9 could potentially serve as pharmacodynamics or response biomarkers. 36
Contrary to the CSF results, plasma lipocalin-2 did not show association with SAH outcome. We observed that plasma lipocalin-2 is lower in SAH group compared to controls. This may be related to age effect, as plasma lipocalin-2 levels increased with age (Supplemental Figure 2(c)), and the SAH group is younger than the control group. Future studies are needed to determine of plasma lipocalin-2 is indeed lower in SAH compared to a normal, age-matched control group. The observation that higher plasma lipocalin-2 levels are associated with increased SAH clinical severity (HH grade) and a greater amount of blood on CT (modified Fisher grade) may reflect a non-specific response to increased severity in disease states. The relationship between plasma lipocalin-2 and known mediators of systemic inflammation such as IL-6 and TNF-α was much less consistent compared with CSF observations. These data suggest that on the blood side of the BBB, the role of lipocalin-2 in SAH and SAH-associated systemic inflammation is less clear.
One important question is the source of CSF lipocalin-2 in SAH. In CNS injuries, lipocalin-2 can be produced on both sides of the BBB from different sources. Major CNS lipocalin-2 sources in hemorrhagic strokes include astrocytes, 15 infiltrating neutrophils, 37 and even injured white matter. 38 On the blood side, lipocalin-2 is primarily produced by activated neutrophils and increased plasma lipocalin-2 has been described in numerous acute (systemic inflammatory response syndrome and sepsis)39,40 and chronic (COPD, diabetes, and atherosclerosis)40–42 inflammatory conditions. In a condition such as SAH where the BBB integrity is breached, an important consideration is whether blood lipocalin-2 may cross the injured BBB and become an important source of CSF lipocalin-2. Our data did not find correlations in CSF and plasma lipocalin-2 to support this notion. Further, little is yet know about potential role of CSF lipocalin-2 in SAH-associated brain injury. While lipocalin-2 has been implicated as deleterious in many neurological diseases,14,43–47 emerging evidence suggest its role in brain injury may be complex and dynamic. In-vitro models suggested neuron secreted lipocalin-2 in ischemia as a “help me” signal that promoted recovery 12 and enhanced angiogenesis. 13 Lipocalin-2 release may reflect an endogenous response to the initial injury and subsequently participate in neuro-recovery. Such dichotomous roles for neuroinflammatory markers are well recognized in other types of CNS injuries. 48 Our study focused on the acute phase of SAH (post-bleed days 1-5). Comprehensive study of lipocalin-2 through a more extensive time course after SAH is needed to more fully define its potential role in human SAH and whether or not it represents a plausible therapeutic target in SAH.
Results of this study should be interpreted with the following caveats: 1) we recruited older patients with suspected NPH as controls to obtain control CSF with minimal risk to the human subject. These controls are not age- or sex-matched with the SAH cohort. Plasma lipcalin-2 results from this study must be interpreted with potential age effect in mind, and future studies with age-matched controls are needed to understand the effect of SAH on plasma lipocalin-2 levels. 2) While, to our knowledge, this is the largest prospective human study of lipocalin-2 in SAH to date, it is a single-center study with modest sample size. Future larger, multi-center studies are needed to validate CSF lipocalin-2 as potential prognostic biomarker for SAH. 3) This study did not directly assess mechanistic roles of lipocalin-2 in SAH associated brain injury. Future targeted studies are needed to further elucidate the roles of lipocalin-2 in SAH-related brain injury mechanisms and define whether lipocalin-2 or its associated inflammatory mediators could serve as pharmacodynamic/response biomarkers in therapeutic clinical trials. 4) This study did not explore lipocalin-2’s effect on cellular iron trafficking15,49 in SAH. Follow-up studies are needed to determine whether lipocalin-2 participates in iron-mediated brain injury in human SAH.
Summary/conclusions
This is the first prospective human study to date that simultaneously examined lipocalin-2 on both sides of the BBB in SAH and control cohorts. We identified distinct profiles of lipocalin-2 responses to SAH in the CSF compartment compared to blood and showed that early increases in CSF lipocalin-2 over time are associated with neuroinflammation and unfavorable SAH outcome. We further examined lipocalin-2 and its associated inflammatory mediators and generated human data that supports prior in-vitro and in-vivo findings that lipocalin-2 stimulates IL-6 and TNF-α secretion in the brain.49,50 Taken together, our results suggest that CSF lipocalin-2 is a promising prognostic and/or pharmacodynamic/response biomarker in SAH worthy of further validation in a larger sample.
Supplemental Material
sj-pdf-1-jcb-10.1177_0271678X211012110 - Supplemental material for CSF lipocalin-2 increases early in subarachnoid hemorrhage are associated with neuroinflammation and unfavorable outcome
Supplemental material, sj-pdf-1-jcb-10.1177_0271678X211012110 for CSF lipocalin-2 increases early in subarachnoid hemorrhage are associated with neuroinflammation and unfavorable outcome by Fang Yu, Aisha Saand, Changhong Xing, Jong Woo Lee, Liangge Hsu, Octavia P Palmer, Vanessa Jackson, Lu Tang, MingMing Ning, Rose Du, Patrick M Kochanek, Eng H Lo and Sherry H-Y Chou in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Dr. S. Chou has been funded by the National Institute of Neurological Disease and Stroke (1K23-NS073806, R21-NS113037), the University of Pittsburgh Physician Foundation Award, and the University of Pittsburgh Dean's Faculty Advancement Award.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Chou, S, Ning, M, and Lo, EH are co-inventors on MITOCHONDRIAL BIOMARKERS OF, AND THERAPEUTICS AQ15 FOR, CNS INJURY AND DISEASE filed in the United States Patent and Trademark Office as Application 62/3817.
Authors’ Contributions
SHYC and EHL designed research; JWL, LH, MMN, RD, CX and SHYC collected the samples; FY, AS, CX and SHYC performed research; FY, AS, LT and SHYC analyzed data; FY and SHYC wrote the paper and OPP, VJ, PMK and EHL revised the manuscript.
Supplementary material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.