
Abstract
Lipocalin-type prostaglandin (PG) D synthase (L-PGDS) is the second major protein in human cerebrospinal fluid (CSF) and belongs to the lipocalin superfamily composed of various secretory lipophilic ligand transporter proteins. However, the endogenous ligand of L-PGDS has not yet been elucidated. In this study, we purified L-PGDS from the CSF of aneurysmal subarachnoid hemorrhage (SAH) patients. Lipocalin-type PG D synthase showed absorbance spectra with major peaks at 280 and 392 nm and a minor peak at around 660 nm. The absorbance at 392 nm of L-PGDS increased from 1 to 9 days and almost disappeared at 2 months after SAH, whereas the L-PGDS activity decreased from 1 to 7 days and recovered to normal at 2 months after SAH. These results indicate that some chromophore had accumulated in the CSF after SAH and bound to L-PGDS, thus inactivating it. Matrix assisted laser desorption ionization time-of-flight mass spectrometry of L-PGDS after digestion of it with endoproteinase Lys-C revealed that L-PGDS had covalently bound biliverdin, a by-product of heme breakdown. These results suggest that L-PGDS acted as a scavenger of biliverdin, which is a molecule not found in normal CSF. This is the first report of identification of a pathophysiologically important endogenous ligand for this lipocalin superfamily protein in humans.
Keywords
INTRODUCTION
Lipocalin-type prostaglandin (PG) D synthase (L-PGDS, EC 5.3.99.2) is one of the most abundant proteins in human cerebrospinal fluid (CSF); and it is expressed in various parts of the mammalian central nervous system (CNS), such as the choroid plexus, leptomeninges, and oligodendrocytes. 1 Lipocalin-type PG D synthase is the enzyme responsible for the biosynthesis of prostaglandin D2 (PGD2) from PGH2, a common precursor of various prostanoids. 2 In the CNS, PGD2 induces sleep and regulates body temperature, nociception, and the release of luteinizing hormone. 3 Homology searches in the data base of protein primary structure showed that L-PGDS is a member of the lipocalin family, which is composed of various secretory lipid-transporting proteins such as retinoic acid-binding protein, odorant-binding protein, and tear lipocalin.4–6 The sequence similarity is considerably low among the members of the lipocalin family, which are known to share very diverse identity and a highly conserved structure. The lipocalins are thought to serve as storage or transport proteins of physiologically important lipophilic ligands. A series of our studies showed that L-PGDS could bind a large variety of lipophilic ligands such as heme metabolites, retinoids, thyroids, steroids, flavonoids, gangliosides, and amyloid-β peptides in vitro.7–11 Most recently, we found that human L-PGDS has a protective effect against reactive oxygen species-induced neuronal cell death by scavenging the active oxygen via a thiol of Cys65 in the L-PGDS molecule. 12 In addition, it is reported that L-PGDS contributes to the balance between carbohydrate and lipid utilization in vivo. 13 Therefore, L-PGDS is a unique multifunctional protein in the lipocalin family of proteins, acting as a PGD2 synthase, a reactive oxygen species scavenger, a regulator of glucose utilization, and an extracellular transporter for several small lipophilic ligands. However, the endogenous and/or exogenous ligand for L-PGDS in vivo has not been found at all.
The members of the lipocalin family share a conserved barrel of eight antiparallel strands as their central folding motif and a large cup-shaped hydrophobic cavity within the β-barrel. 14 This β-barrel commonly encloses a ligand-binding site composed of both an internal cavity and an external loop scaffold. We also reported that the molecular structure of recombinant mouse L-PGDS, as determined by nuclear magnetic resonance spectroscopy and X-ray crystallography, exhibited the typical lipocalin fold consisting of an 8-stranded antiparallel β-barrel and a long α-helix associated with the outer surface of the barrel, with the interior of the barrel forming a hydrophobic cavity.15–17
In terms of clinical importance, many reports have been suggested that the concentration of L-PGDS in the CSF is closely associated with various neurologic diseases. The L-PGDS concentration is increased in the CSF of patients with spinal canal stenosis, 18 whereas it is significantly lower in patients with schizophrenia, 19 brain tumors, 20 bacterial meningitis, 18 or normal pressure hydrocephalus. 21 However, the CSF concentration of L-PGDS is unchanged in patients with multiple sclerosis, Human immunodeficiency virus infection/acquired immunodeficiency syndrome-related neuropathies, viral meningitis, or fibromyalgia. 22 Lipocalin-type PG D synthase was also proposed to be a diagnostic marker for N-glycosylation defects in the brain. 23 These reports suggest that the L-PGDS level in CSF is a useful clinical biomarker to diagnose neurologic diseases. In addition, we found that the L-PGDS level in the CSF of patients with aneurysmal subarachnoid hemorrhage (SAH) is transiently increased to become approximately twofold over normal with a peak at day 5. 24 However, the pathophysiological role of this accumulated L-PGDS in the CSF of patients with SAH remains unclear.
In this study, we investigated the role of L-PGDS accumulated in the CSF of SAH patients. Based on the results of spectroscopic analyses, we found that biliverdin, a metabolite of heme, covalently bound to L-PGDS in the CSF of patients with aneurysmal SAH. Our results suggest that L-PGDS acts as a scavenger of biliverdin, and thus has an important role for the quick clearance of unwanted lipophilic molecules that break into the CSF in humans after an aneurysmal SAH.
MATERIALS AND METHODS
Reagents
Biliverdin and bilirubin were purchased from MP Biomedicals (Santa Ana, CA, USA). High-performance liquid chromatography (HPLC)-grade acetonitrile, trifluoroacetic acid (TFA), dithiothreitol, trichloroacetic acid, and guanidine hydrochloride (GdnHCI) were purchased from Wako (Osaka, Japan). All other chemicals were of analytical grade.
Sampling of Human Cerebrospinal Fluid after Subarachnoid Hemorrhage
Thirteen patients with aneurysmal SAH, who were consecutively admitted to the Department of Neurosurgery, Nagoya City University Hospital, were treated with intravascular embolization of the aneurysmal dome by using detachable coils within 24 hours after the initial SAH attack. After the treatment of aneurysms, a silicon drainage tube was placed in the lumbar CSF space for continuous draining of the subarachnoid clots and intermittent sampling of the CSF from 1 to 17 days at about every other day and then at 2 months after SAH. A patient had symptomatic vasospasm, resulted in brain death due to severe cerebral ischemia before 9 days after SAH. Thus, we collected the CSF of remaining 12 patients and measured the concentration of L-PGDS in CSF by using a sandwich enzyme-linked immunosorbent assay system. 25 Informed consent for lumbar puncture, which procedure was a part of the diagnostic workup, and for use of the sampled CSF in this study was obtained from all patients. This study protocol was governed by the guidelines of national government based on the Helsinki Declaration revised in 1983. Studies with human samples were performed under the approval by the Ethics Review Committee of Graduate School of Medical Sciences, Nagoya City University (Approved No. 799).
Purification of Lipocalin-Type PG D Synthase in Cerebrospinal Fluid after Subarachnoid Hemorrhage
Cerebrospinal fluid-lipocalin-type PG D synthase was purified from the CSF from 1 to 17 days and at 2 months after SAH of three patients who were affected with posthemorrhagic chronic hydrocephalus after SAH by immunoaffinity chromatography with a monoclonal antibody 1B7-conjugated column as reported previously. 25 An appropriate concentration of CSF, diluted in phosphate-buffered saline (PBS, pH 7.4), was filtered before being applied to a 1 B7-conjugated column and circulated twice at room temperature by a peristaltic pump. The column was washed sequentially with 2 mol/L NaCI, 0.05% Triton X-100 in PBS, and PBS. Cerebrospinal fluid–lipocalin-type PG D synthase was eluted with 100 mmol/L sodium citrate, pH 3.0. Each fraction was immediately adjusted to pH 7.0 by 2 mol/L Tris, pH 11. To avoid contamination by albumin, we further purified the CSF-L-PGDS to apparent homogeneity by using Superdex75 (GE Healthcare Biosciences, Piscataway, NJ, USA).
Enzyme Assays
The activities of CSF-L-PGDS (final concentration of 5 μg/mL) obtained from the CSF of SAH patients and patients who developed communicating hydrocephalus after SAH were measured by incubation at 25°C for 1 minute with [1-14C]PGH2 (final concentration of 5 μmol/L) in 50 μL of 0.1 mol/L Tris-HCI (pH 8.0) in the presence of 1 mg/mL immunoglobulin G and 1 mmol/L dithiothreitol. The reaction was stopped by the addition of 250 μL of diethyl ether:methanol:citric acid 1 mol/L (30:4:1), and aliquots were then subjected to thin-layer chromatography at − 20°C. [1-14C]PGH2 was prepared from [1-14C]arachidonic acid (2.20 GBq/mmol, PerkinElmer Life Sciences, Boston, MA, USA) as described previously. 2
Expression and Purification of Recombinant Human Lipocalin-Type PG D Synthase
Both recombinant human Δ1-22-C65A-substituted L-PGDS mutant (C65A-L-PGDS), in which the N-terminal signal peptide sequence had been removed and the Cys65 as an active-site residue had been replaced with alanine, and Δ1-22-C89A/C167A/C186A-substituted L-PGDS mutant (C89A/C167A/C186A-L-PGDS), in which the Cys65 was retained, were expressed as a glutathione S-transferase fusion protein in Escherichia coli BL21 (DE3, TOYOBO, Tokyo, Japan), as described previously. 8 Each fusion protein was bound to glutathione sepharose 4B (GE Healthcare Biosciences) and incubated with thrombin (Sigma-Aldrich, St Louis, MO, USA, 100 units/100 μL) to release the L-PGDS. Each recombinant protein was further purified by using size-exclusion chromatography as described previously, 8 and dialyzed against PBS. The concentrations of these proteins were spectrophotometrically determined by using the molar absorption coefficient at 280 nm, i.e., ε280 = 26, 100 L/mol per cm for the C65A-L-PGDS and ε280 = 25,700 L/mol per cm for the C89A/C167A/C186A-L-PGDS. In this study, the C89A/C167A/C186A-L-PGDS was employed instead of the wild-type protein, because the amount of the wild type is limited owing to the incorrect intra and intermolecular disulfide bonds and protein aggregation.
Circular Dichroism Measurements
The CD spectra of CSF-L-PGDS and the complex of C89A/C167A/C186A-L-PGDS and biliverdin were measured with a spectropolarimeter model J-820 (Jasco, Tokyo, Japan). The temperature of the solution in the cuvette was controlled at 4.0 ± 0.5°C by circulating water. The path length of the quartz cuvette was 10 mm for near ultraviolet (UV) to visible range CD measurements. The protein concentrations of CSF-L-PGDS were 40 μg/mL in PBS. Biliverdin was dissolved in dimethyl sulfoxide to give a 2 mmol/L stock solution. The desired amounts of biliverdin were added to 40 μg/mL of C89A/C167A/C186A-L-PGDS in PBS. The concentration of biliverdin was determined spectroscopically, i.e., ε382 in dimethyl sulfoxide for biliverdin = 49,400 L/mol per cm. 11
Biliverdin Treatment of Recombinant Human Lipocalin-Type PG D Synthase and Size-Exclusion Chromatography of Biliverdin/Lipocalin-Type PG D Synthase Complex
Biliverdin at 400 μmol/L was added to 400 μmol/L C65A-L-PGDS or C89A/C167A/C186A-L-PGDS in PBS. After incubation at 37°C for 0, 0.5, 1, 3, 6, 9, 12, or 24 hours, the samples were denatured by 6 mol/L GdnHCI and analyzed by size-exclusion chromatography performed by using a SMART system (Superdex75 PC 3.2/30, GE Healthcare Bioscience). A C89A/C167A/C186A-L-PGDS/biliverdin complex was eluted with 6 mol/L GdnHCI in PBS, and the elutions were monitored by UV absorption at 280 and 377 nm.
Identification of Biliverdin-modification Site of Recombinant Lipocalin-Type PG D Synthase
C89A/C167A/C186A-L-PGDS/biliverdin complex was incubated at 100°C for 3 minutes with 2 mmol/L dithiothreitol. The sample was digested with endoproteinase Lys-C (Boehringer Mannheim, Mannheim, Germany) at 37°C for 24 hours at an enzyme:substrate ratio of 1:20 (w/w). Peptide samples were purified by using a reversed-phase HPLC system (Agilent 1100 series HPLC system, Agilent, Santa Clara, USA). A Protein-R column (C18, Waters, Milford, MA, USA) was used for separation at 35°C. The mobile phase consisted of water containing 0.1% TFA (solution A) and acetonitrile containing 0.1% TFA (solution B). The gradient programmer was used according to the following procedure: 0 to 5 minutes hold at 5% B; 5 to 25 minutes linear increase in B from 5% to 50%; 25 to 30 minutes hold at 95% B; with a flow rate of 0.5 mL/minute. The UV spectra were recorded from 190 to 400 nm, and the monitor wavelengths were set at 280 and 377 nm. The peptides purified by HPLC were analyzed by matrix assisted laser desorption ionization time-of-flight (MALDI/TOF) mass spectrometry (MS) and tandem MS (MS/MS). They were demineralized and concentrated by applying the C4 Zip-tip (Millipore, Tokyo, Japan) according to the manufacturer's instruction. Subsequently, the solutions were bound by subjecting the solutions to Zip-tip treatment, washed with water, and eluted with a solution of 80% acetonitrile. The eluates were mixed with a saturated solution of α-cyano-4-hydroxycinnamic acid (Sigma-Aldrich) containing 60% acetonitrile and dried on stainless steel targets at room temperature and pressure. Analyses were performed by using an Ultraflex MALDI TOF/TOF (Bruker Daltonics, Billerica, MA, USA) with a nitrogen laser of 377 nm. All analyses were carried out in the positive-ion mode.
Identification of Modification Site of Cerebrospinal Fluid–Lipocalin-Type PG D Synthase
Lipocalin-type PG D synthase260 μg) was reduced with 10 mmol/L dithiothreitol followed by carboxamidomethylation. After dialysis with 5 mmol/L Tris-HCI (pH 8.0), CSF–L-PGDS was digested with endoproteinase Lys-C at 37°C for 24 hours by using an enzyme:substrate ratio of 1:20 (w/w). The digested CSF-L-PGDS fragments were analyzed by MALDI/TOF MS and MS/MS. The procedure was the same as used for the binding-site analysis of recombinant human L-PGDS.
Cell Culture and Cytotoxicity Assay
SH-SY5Y cells, which are human neuroblastoma cells, were cultured in DMEM/F12 (Life Technologies Corporation, Carlsbad, CA, USA) containing 10% fetal bovine serum (Life Technologies Corporation) in the humidified atmosphere of 5% CO2 and 95% air at 37°C.
Bilirubin or biliverdin of 10 mmol/L dissolved in dimethyl sulfoxide was added to BSA solution to obtain bilirubin/BSA and biliverdin/BSA molar ratios of 0.5. All handling of bilirubin and biliverdin was performed in dim light. SH-SY5Y cells were cultured in 96-well tissue culture plates at a density of 6 × 103 cells per well. After 48 hours of cultivation, the appropriate amounts of biliverdin or bilirubin in the presence or absence of L-PGDS were added to the culture medium. The cells were further cultured for 24 hours, and then lactate dehydrogenase (LDH) activity was measured by using Cytotoxicity Detection KitPLUS (LDH, Roche Diagnostics, Mannheim, Germany) according to the manufacturer's instruction. Absorbance was measured at 492 nm by using a microplate reader, Model 680 (Bio-Rad Laboratories, Hercules, CA, USA).
Biliverdin Reductase Assays and High-Performance Liquid Chromatography Analysis
C89A/C167A/C186A-L-PGDS at 40 μmol/L was reacted with 40 μmol/L biliverdin for 24 hours at 37°C in 5% dimethyl sulfoxide/PBS. The reaction sample was dialyzed against 0.1 mol/L potassium phosphate buffer (pH 8.5). The final concentration of biliverdin-bound L-PGDS was adjusted to 20 μmol/L with 0.2 mg/mL BSA. Then, human biliverdin reductase A (Sigma-Aldrich) of 1 unit/mL was added to 20 μmol/L biliverdin-bound L-PGDS or 20 μmol/L biliverdin according to the manufacturer's instruction. The reductive reaction was initiated by the addition of 100 μmol/L NADPH (Nacalai Tesque, Kyoto, Japan) to each sample, which was then incubated for 1 hour at 37°C and subsequently diluted twofold with 0.1% TFA in water. As controls, 20 μmol/L biliverdin, 20 μmol/L bilirubin, and 20 μmol/L biliverdin-bound L-PGDS in a 0.1 mol/L potassium phosphate buffer containing 0.2 mg/mL BSA (pH 8.5) were diluted twofold with 0.1% TFA in water. The concentration of bilirubin was determined by using the ε454 value of 59,000 L/mol per cm. 11
Reverse-phase HPLC was performed by using a Waters Alliance 2795 Chromatography Separations Module (Waters) equipped with a Protein-R column operated at 37°C A linear gradient was applied from 33% to 73% solvent B for 30 minutes at a flow rate of 0.5 mL/minute (solvent A: 0.1% TFA in water; solvent B: 0.1% TFA in acetonitrile). Detection was done by monitoring UV absorbance at 382 and 454 nm.
Statistical Analysis
Data were expressed as the means ± s.d. The statistical significance was analyzed with one-way repeated measures analysis of variance followed by Bonferroni's post hoc test for the changes in L-PGDS concentration in CSF of SAH patients, and was analyzed with one-way analysis of variance followed by Dunnett's post hoc test for the changes in absorbance at 392 nm and enzyme activities of CSF-L-PGDS. P <0.05 was considered to be significant. In cytotoxicity assay, data were expressed as the means ± s.e.m. The statistical significance between the control and the experimental group was assessed by Student's t-test. P <0.05 was considered to be significant.
RESULTS
Concentration of L-PGDS in CSF after SAH
The concentration of L-PGDS in the CSF obtained from 12 patients with SAH was determined by using a sandwich enzyme-linked immunosorbent assay system. 25 As shown in Figure 1, the L-PGDS level in the CSF of all patients transiently increased with a peak at 5 days after SAH (22.8 ± 9.0 μg/mL). After this peak, the level gradually decreased; and by 17 days after SAH it had dropped (to 11.9 ± 3.5 μg/mL) to a level comparable to that at 1 day after SAH (10.7 ± 2.3 μg/mL). These results show that the L-PGDS concentration in the CSF increased in patients after SAH.
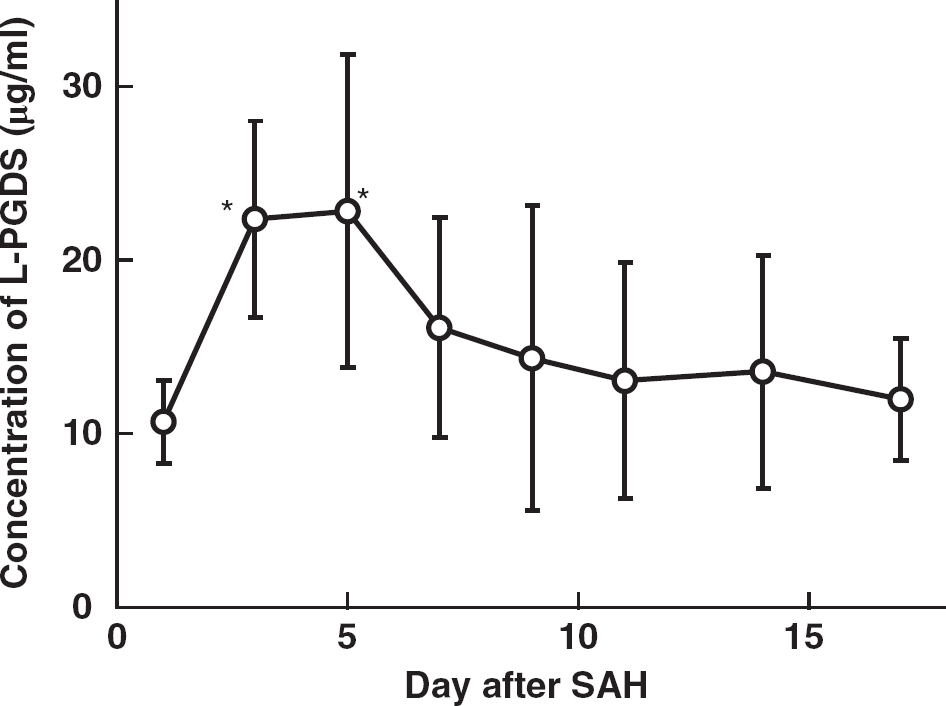
Changes in lipocalin-type prostaglandin D synthase (L-PGDS) concentration in cerebrospinal fluid (CSF) of subarachnoid hemorrhage (SAH) patients. Lipocalin-type PG D synthase concentration was determined by an enzyme-linked immunosorbent assay using two kinds of mouse monoclonal antibodies, 1B7 and 10A3, against human L-PGDS, as previously described. 25 Lipocalin-Type PG D Synthase concentrations at 3 and 5 days after SAH are significantly higher than that concentration at 1 day after SAH. Data are expressed as the means ± s.d. of 10 to 12 independent experiments. Significance of differences was determined by one-way repeated measures analysis of variance followed by Bonferroni's post hoc test, *P <0.05 versus at 1 day after SAH.
Purification of Lipocalin-Type PG D Synthase from Cerebrospinal Fluid in Patients with Subarachnoid Hemorrhage
Human L-PGDS was purified from the SAH patients' CSF by use of mAb 1B7-conjugated immunoaffinity column chromatography. 25 SDS-PAGE of L-PGDS purified from CSF of SAH patients (CSF-L-PGDS) at 5, 7 days, and 2 months after SAH gave a single but broad band at a position around Mr = 27,000 by silver staining (Supplementary Figure 1).
Ultraviolet-Visible Spectra and Enzyme Activity of Cerebrospinal Fluid-Lipocalin-Type PG D Synthase
We also purified L-PGDS from CSF in patients at 1, 3, 5, 7, 9, 11, 13, and 15 days after SAH. Supplementary Figure 2 shows a photograph of CSF-L-PGDS at 7 days and 2 months after SAH. The color of the CSF-L-PGDS solution at 7 days after SAH was light green, whereas that at 2 months after SAH was colorless. Cerebrospinal fluid-lipocalin-type PG D synthase at 7 days after SAH showed two major absorption peaks, at the wavelengths of 279 and 392 nm, and a minor peak at around 650 nm. These absorption peaks of CSF-L-PGDS showed a transient increase with time after the SAH event (Figure 2A), indicating that some chromophores had bound to the CSF-L-PGDS. The enzyme activity and the absorbance at 392 nm of CSF-L-PGDS were measured and plotted against time after SAH (Figure 2B). The enzyme activity of CSF-L-PGDS (at 1 day; 707 ± 16 nmol/minute per mg protein) decreased after SAH and reached a minimum at 7 days (240 ± 140 nmol/minute per mg protein), and then the activity gradually increased (at 15 days; 578 ± 88 nmol/minute per mg protein). By 2 months after SAH, the enzyme activity recovered to become 740 ± 126 nmol/minute per mg protein. In contrast, the absorbance at 392 nm increased after SAH and reached a peak at 9 days (0.60 ± 0.03 AU/mg protein), which was ˜2.3-fold higher than that at 1 day (0.16 ± 0.01 AU/mg protein). By 15 days after SAH, the absorbance had decreased to 0.20 ± 0.02 AU/mg protein, comparable to that at 1 day post SAH. At 2 months after SAH, the absorbance was even lower, being 0.11 ± 0.03 AU/mg protein, which was well below that at 1 day.
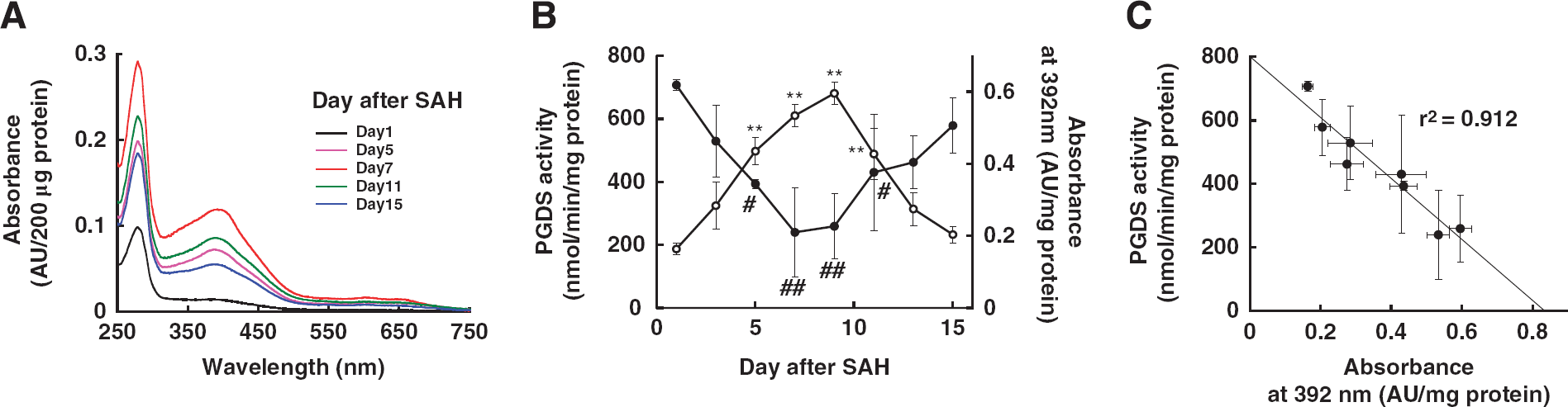
Changes in absorption spectra and enzyme activities of cerebrospinal fluid-lipocalin-type prostaglandin D synthase (CSF-L-PGDS). (
To demonstrate the correlation between the enzyme activity and the absorption of CSF-L-PGDS, we plotted the change in PGDS activity against the absorption of CSF-L-PGDS at 392 nm (Figure 2C), and thereby obtained a negative linear correlation (r2 = 0.912). These results, taken together, indicate that some chromophore(s) derived from blood constituents had inactivated L-PGDS by binding around the active center of the Cys65 residue of the enzyme.
Circular Dichroism Analyses of Cerebrospinal Fluid-Lipocalin-Type PG D Synthase
To investigate what chromophore(s) had bound to CSF-L-PGDS, we assessed CSF-L-PGDS by making CD spectrum measurements in the near UV and visible ranges. Supplementary Figure 3A shows the CD spectra of CSF-L-PGDS at 3, 7, and 13 days after SAH, from which the CD spectrum of non-colored CSF-L-PGDS at 2 months after SAH had been subtracted. The CD spectra exhibited a single major negative Cotton effect at the wavelength around 370 nm, demonstrating the existence of some chromophore showing absorption anisotropy by binding to L-PGDS. The intensity of the negative CD Cotton effect at 370 nm increased at 7 days as compared with that at 3 days, but significantly decreased at 13 days after SAH. The CD spectrum of non-colored CSF-L-PGDS at 2 months after SAH did not show any Cotton effect around 370 nm (data not shown).
(
Considering all these lines of evidence described above, we suspected that biliverdin might be a possible candidate for the chromophore that bound to CSF-L-PGDS. Thus, we measured the UV-visible absorption spectra and the CD spectra of biliverdin bound to a human recombinant L-PGDS mutant, C89A/C167A/C186A-substituted L-PGDS (C89A/C167A/C186A-L-PGDS), in which the Cys65 as an active-site residue was retained. The complex of biliverdin and C89A/C167A/C186A-L-PGDS showed three major absorption peaks at the wavelengths of 277, 382, and 650 nm (data not shown). The complex also exhibited a single negative CD Cotton peak at 383 nm, and the intensities of the negative CD Cotton effect increased in a molar ratio-dependent manner (Supplementary Figure 3B). These results, taken together, demonstrated that these spectra obtained from the biliverdin/L-PGDS complex were close to those obtained from the colored CSF-L-PGDS after SAH.
Covalent Binding of Biliverdin to Recombinant Human Lipocalin-Type PG D Synthase
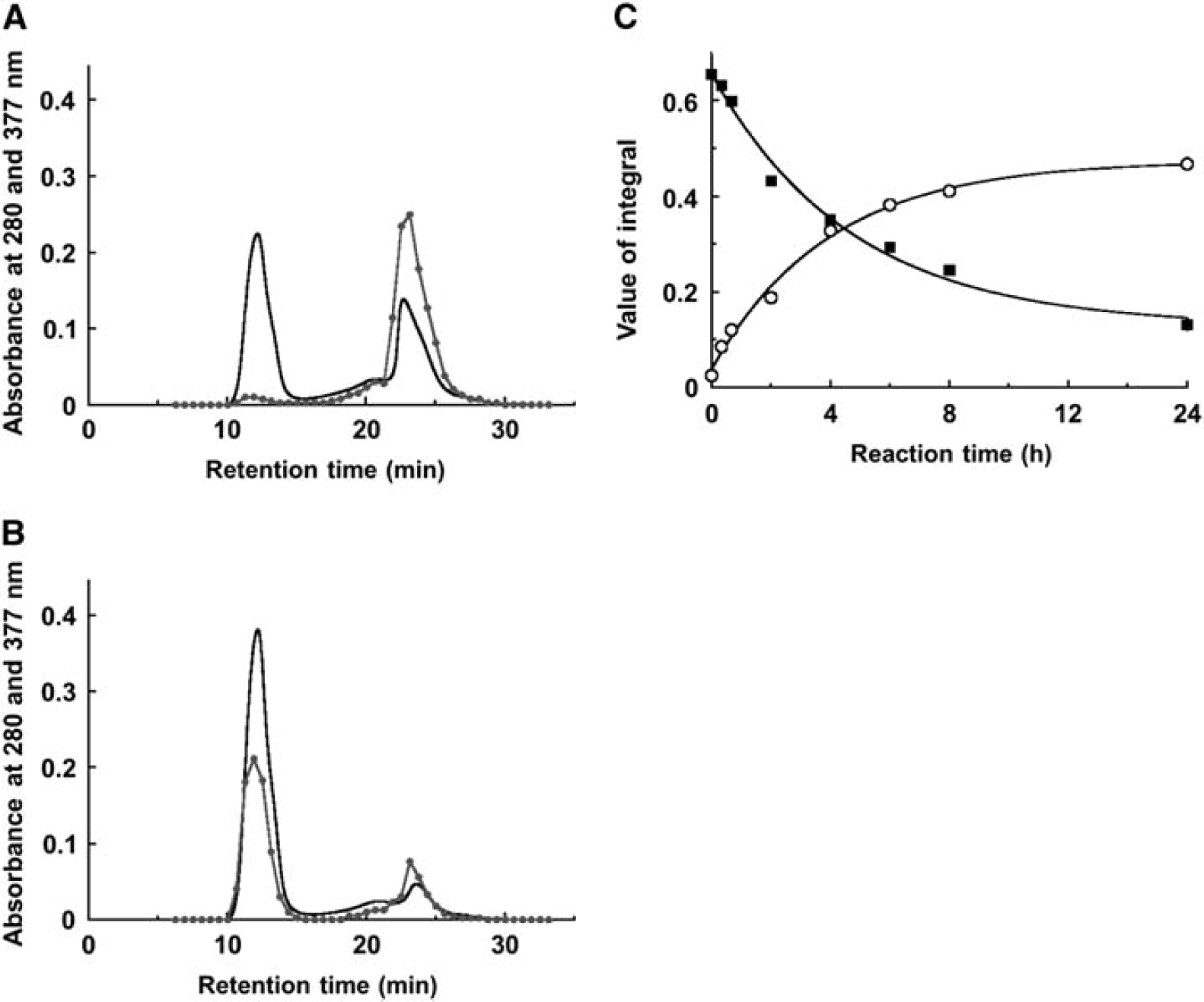
As biliverdin is regarded to be a candidate of chromophores bound to L-PGDS after SAH, we prepared complexes of biliverdin and two kinds of human recombinant L-PGDS mutants, i.e., the C65A-substituted L-PGDS mutant (C65A-L-PGDS), in which the Cys65 as an active-site residue was replaced with alanine, and C89A/C167A/C186A-L-PGDS. After the incubation of biliverdin (400 μmol/L) with 400 μmol/L C65A- or C89A/C167A/C186A-L-PGDS at 37°C for 0 to 24 hours in PBS, the samples were denatured by 6 mol/L GdnHCI and analyzed by size-exclusion chromatography in the presence of 6 mol/L GdnHCI. In all incubation times, C65A-L-PGDS, indicated by an absorption peak at 280 nm, was eluted at 13 minutes; whereas biliverdin, indicated by an absorption peak at 377 nm, was eluted at 24 minutes in almost equal amount (data not shown). However, in the case of C89A/C167A/C186A-L-PGDS, L-PGDS and biliverdin were also eluted at 13 and 24 minutes, respectively, after an incubation for 0 hour (Figure 3A). However, the elution peak at 13 minutes derived from the absorption at 377 nm was increased in a time-dependent manner, whereas the peak at 24 minutes was decreased (data not shown). The peak at 24 minutes became minimum after incubation for 24 hours (Figure 3B). The integral of the elution curve obtained at 13 and 24 minutes derived from the absorption at 377 nm was plotted against the reaction time (Figure 3C). The changes in the amount of free biliverdin and those in that of biliverdin-bound C89A/C167A/C186A-L-PGDS exhibited a mirror image. These results, taken together, demonstrate that biliverdin covalently binds to C89A/C167A/C186A-L-PGDS, but not to C65A-L-PGDS. Thus, the Cys65 residue, in the active center of the enzyme, was the site targeted for the covalent binding by biliverdin.
Determination of Biliverdin-Binding Site in Human Recombinant Lipocalin-Type PG D Synthase
Biliverdin at 400 μmol/L and C89A/C167A/C186A-L-PGDS also at 400 μmol/L were incubated for 24 hours at 37°C in PBS. Then, the sample was digested with endoproteinase Lys-C; and the digested fragments were separated by use of an HPLC system as biliverdin-modified peptides. Figure 4A shows the HPLC elution profile of the digested fragments by monitoring absorption at 377 nm, a particular absorbance wavelength of biliverdin. Four major peaks were obtained, having elution times of 16 (P1), 17 (P2), 18 (P3), and 21 minutes. Each peak was fractionated and analyzed by MALDI/TOF MS. First, free biliverdin was determined to reside in the peak at 21 minutes by MS/MS analysis (data not shown). Next, in peak P3, a mass/charge (m/z) of 1305.69 was detected, which is the sum of the mass size of an endoproteinase Lys-C-digested peptide fragment (Mr = 722.937) including the Cys65 residue and that of a single molecule of biliverdin (Mr = 582.626, Figure 4B). Figures 4C and 4D show the MS/MS spectrum of the [M + H]+ ion at m/z 1305 from a peptide in P3 and the chemical structure as revealed by MS/MS analysis, respectively. In the MS/MS analysis, the peptide 60Ala-Ala-Leu-Ser-Met-Cys-Lys66 (60AALSMCK66) was identified from N-terminal (b-ions such as b2, b3, and b4) and C-terminal (y-ions such as y2, y3, y4, and y6) fragment ions. The masses of C-terminal y-ions such as y2 (m/z 831.474), y3 (m/z 962.805), y4 (m/z 1049.920), and y6 (m/z 1234.492), and thiol residue (BV + S, m/z 616.231) were observed to increase by 583 kDa corresponding to a single molecule of biliverdin. In addition, we observed the masses of the fragment ions such as the fragment including the pyrrole C- and D-rings of biliverdin (BV-1), the fragment including pyrrole B-, C-, and D-rings of biliverdin (BV-2), and the fragment corresponding to the peptide modified by a part of the A-ring of biliverdin (BV-3). From these results, we confirmed that the nucleophilic conjugate addition of the SH group of Cys65 to the electronically deficient vinyl group of the modified pyrrole A-ring was generated by the influence of the electron-withdrawing carbonyl group of the D-ring in the conjugate system of biliverdin; that is, the SH group of Cys65 bound to the terminal position of the vinyl group attached to the A-ring of biliverdin through a Michael-type addition (Figure 4E).
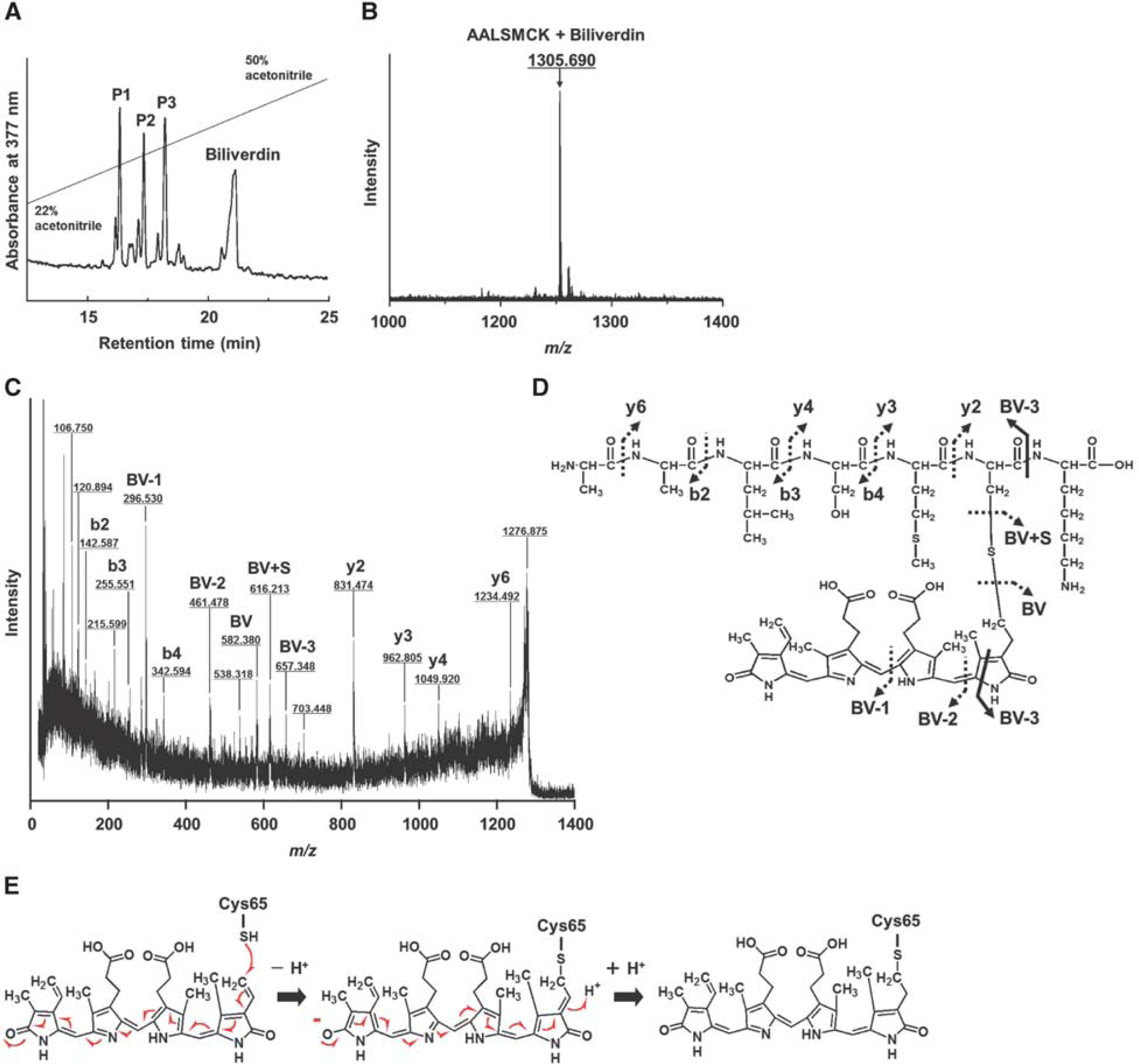
(
However, in P1, we detected two peaks, one at m/z 1322.47 and the other at m/z 1356.53. As judged from the results of MS/MS analysis, the former was considered to have been derived from a biliverdin-modified oxidized peptide formed by the oxidation of methionine to methionine-sulfoxide (data not shown). Although the peak at m/z 1356.53 was increasing by 51 kDa to m/z 1306 confirmed to be AALSMCK + biliverdin, we could not determine what molecule had been bound. In addition, in P2, we also detected two peaks, at m/z 1306.42 and m/z 1321.44. The peak at m/z 1306.42 was confirmed to be AALSMCK + biliverdin, and a peak at m/z 1321.44 was a biliverdin-modified oxidized peptide, which was the same fragment detected in PI (data not shown).
Identification of Chromophore Modification Site in Cerebrospinal Fluid-Lipocalin-Type PG D Synthase
Cerebrospinal fluid-lipocalin-type PG D synthase purified from CSF at 7 days after SAH was digested with endoproteinase Lys-C. The digested fragments were directly analyzed by MALDI/TOF MS and MS/MS without any separation on HPLC because of the limited volume of digested sample. As shown in Figure 5A, a minor peak at m/z 1305.592 and a major peak at m/z 1356.658 were detected, which represented the biliverdin-modified peptide including Cys65 and the peptide with an additional mass size of 51, respectively. Although we could not carry out MS/MS analysis of the sample at m/z 1305.592 because of its low intensity, the MS/MS spectrum and the chemical structure were obtained from the sample at m/z 1356.658 (Figures 5B and 5C). By MS/MS analysis, the peptide of 60AALSMCK66 was identified from the full-length fragment ion (m/z 722.454), N-terminal b4-ion and C-terminal (y-ions such as y2, y3, and y4) fragment ions. The masses of C-terminal fragment ions of y-ions such as y2 (m/z 831.328), y3 (m/z 962.493), y4 (m/z 1048.597), and thiol residue (BV + S, m/z 616.510) were observed to increase by 583 kDa corresponding to a single molecule of biliverdin. In addition, we observed the masses of the fragment ions of biliverdin such as BV′-1 (m/z 286.898), BV′-2 (m/z 297.630), and BV′-3 (m/z 462.514). Another prominent signal in the MALDI/TOF MS was most likely related to a peptide with a remaining ‘broken’ chromophore. These results confirmed that biliverdin bound to CSF-L-PGDS in the same way that it did to human recombinant L-PGDS. The results of proteolysis and mass spectrometry, taken together, clearly show that biliverdin was covalently attached to the Cys65 residue of L-PGDS.
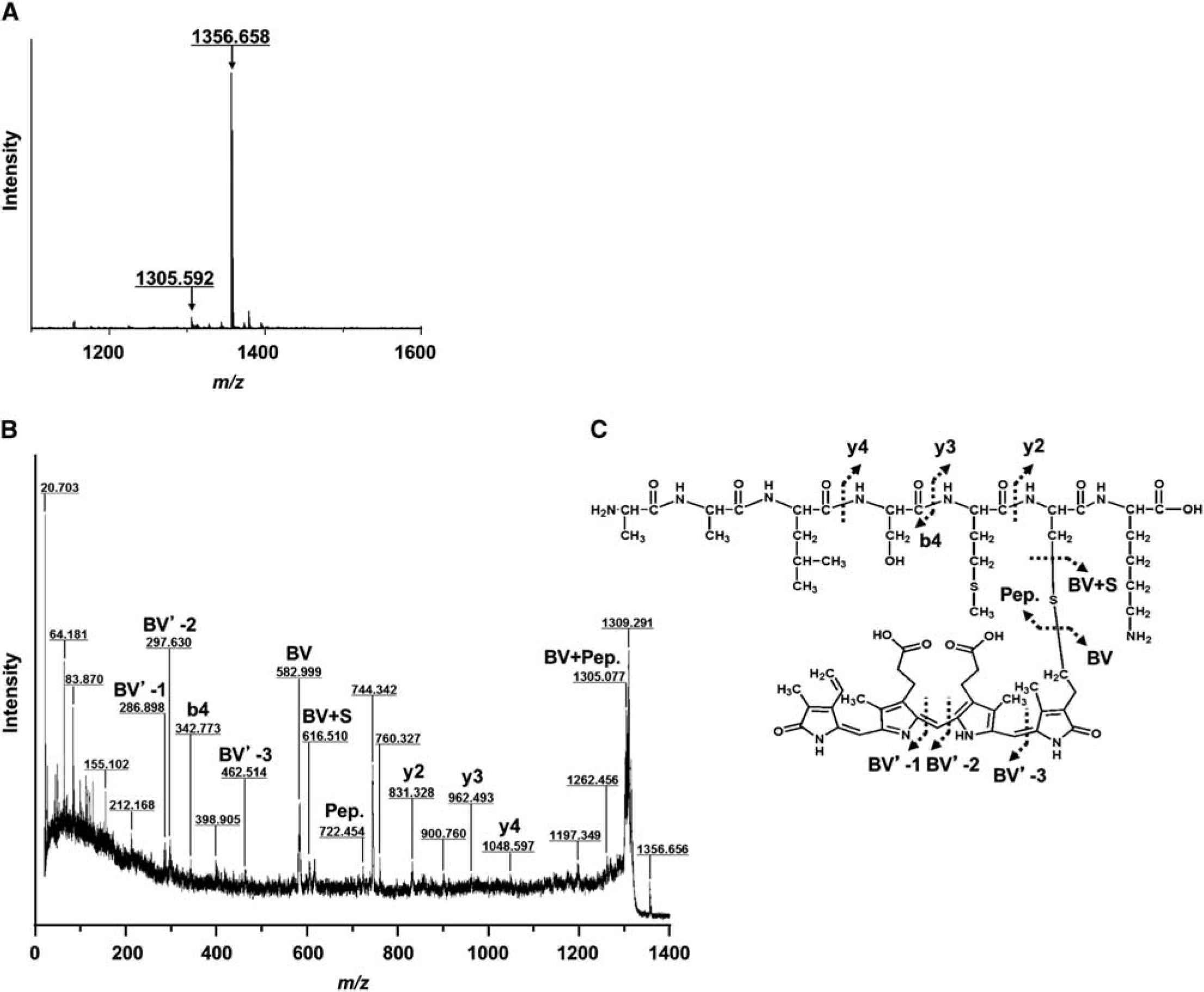
Identification of the biliverdin-modification site of cerebrospinal fluid-lipocalin-type prostaglandin D synthase (CSF-L-PGDS). (
Protective Effects of Human Lipocalin-Type PG D Synthase against Bilirubin-induced Neuronal Cell Death
The cytotoxicity of heme metabolites such as biliverdin and bilirubin toward human neuronal SH-SY5Y cells was investigated by performing a LDH assay. The results showed that biliverdin up to 100 μmol/L did not show any cytotoxicity toward SH-SY5Y cells (Supplementary Figure 4). However, bilirubin at 20 to 100 μmol/L increased the LDH activity in a concentration-dependent manner, thus showing bilirubin-induced cytotoxicity toward human neuronal cells (Figure 6A). The 50% lethal dose value was determined to be 25.2 ± 1.0 μmol/L by using MicroCal Origin 6.0 software.
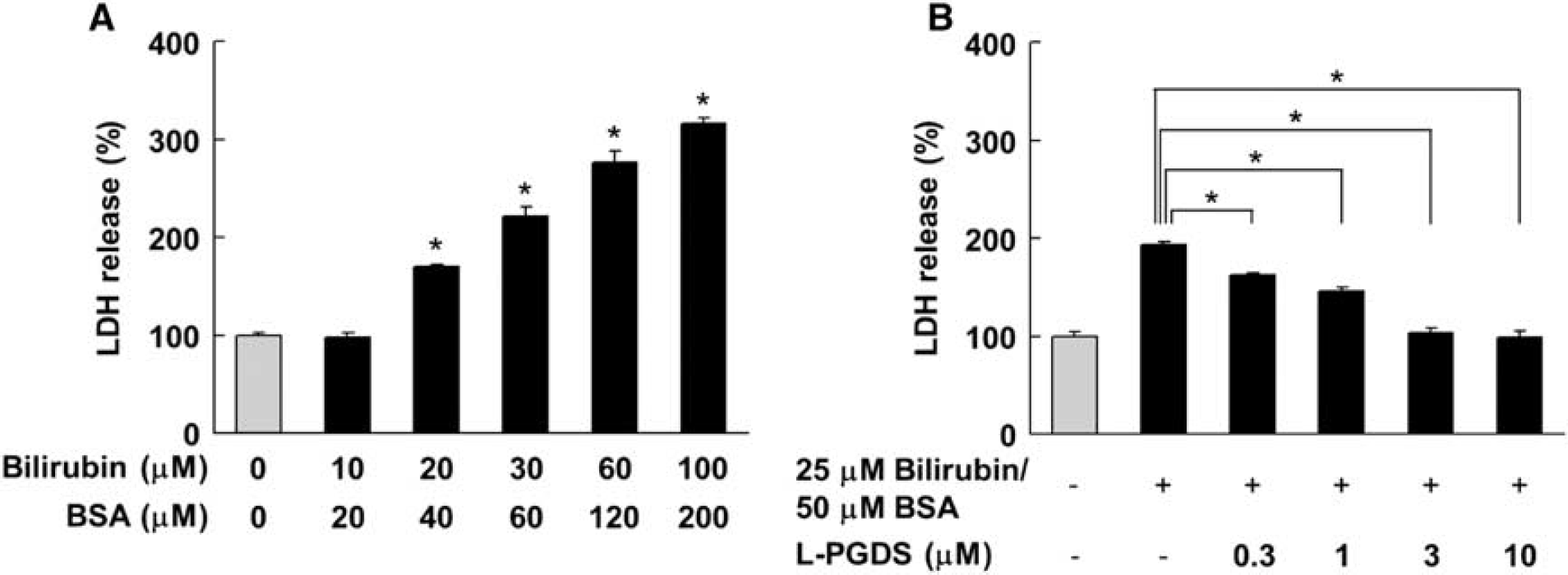
Protective effect of human lipocalin-type prostaglandin D synthase (L-PGDS) against bilirubin-induced neuronal cell death. (
As we observed the bilirubin-induced cell death of SH-SY5Y cells, we next investigated the effect of human L-PGDS on bilirubin-induced neuronal cell death. As shown in Figure 6B, the LDH activity of 25 μmol/L bilirubin-treated cells was increased ˜200% as compared with that in the untreated cells. However, C89A/C167A/C186A-L-PGDS protected against bilirubin-induced cell death in a concentration-dependent manner. In the presence of 3 μmol/L C89A/C167A/C186A-L-PGDS, the cytotoxicity induced by 25 μmol/L bilirubin was almost negligible. These results, taken together, demonstrate that L-PGDS had the ability to protect against bilirubin-induced neuronal cell death.
Effect of Human Biliverdin Reductase A on Biliverdin-Bound Lipocalin-Type PG D Synthase
As bilirubin showed cytotoxicity toward neuronal cell, we investigated whether bilirubin could be produced from biliverdin-bound L-PGDS by human biliverdin reductase A. Biliverdin, bilirubin, and biliverdin reductase A-treated biliverdin were loaded onto a C18 reverse-phase column, and the elution profiles were monitored at 382 and 454 nm (Figure 7). Biliverdin eluted at 4.7 minutes (Figure 7A), whereas bilirubin eluted at 28.1 minutes (Figure 7C). The reaction mixture elution profile of biliverdin reduction is shown in Figure 7B. The retention time of the bilirubin control and that of the biliverdin reduction product were identical. However, the reaction mixture elution profiles of biliverdin-bound L-PGDS with and without biliverdin reductase A are shown in Figures 7D and 7E, respectively. Both the control and the biliverdin reductase A-treated mixture eluted at around 6.2 to 7.2 minutes, and there was no peak at 28.1 minutes at 454 nm monitoring, showing that bilirubin had not been produced from biliverdin-bound L-PGDS in the presence of biliverdin reductase A.
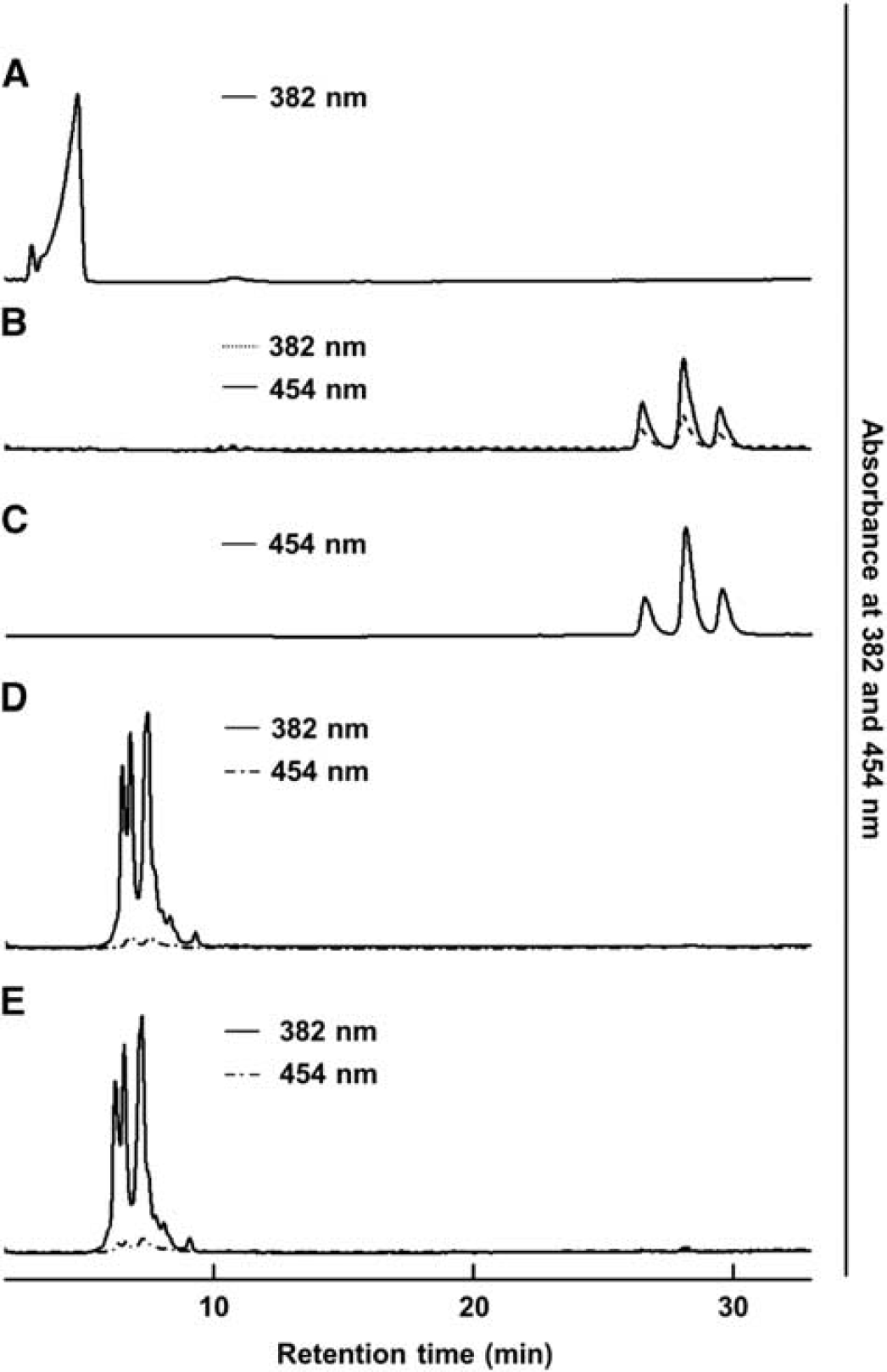
Separation of bilirubin, biliverdin and biliverdin-bound lipocalin-type prostaglandin D synthase (L-PGDS) by high-performance liquid chromatography (HPLC) on a C18 reverse-phase column. (
DISCUSSION
In this study, we identified biliverdin to be the endogenous ligand covalently bound to the thiol of the Cys65 of L-PGDS, which is increased in level in the CSF of patients with aneurysmal SAH. We could mimic the covalent binding of biliverdin to recombinant human L-PGDS through a Michael-type addition to the thiol of the Cys65. In addition, we demonstrated that L-PGDS also reduced the cytotoxicity toward neuronal cells induced by bilirubin, a metabolite of biliverdin, and that bilirubin could not be produced from biliverdin-bound L-PGDS. This is the first report to identify biliverdin as an endogenous ligand of human L-PGDS functioning as a transporter protein.
So far, a series of our studies revealed that mouse and rat recombinant L-PGDS can bind a large variety of lipophilic small ligands.7–11 We defined such a feature as the ‘broad ligand selectivity’ of L-PGDS, 7 and we proposed the structural flexibility of the L-PGDS molecule from the results showing compact packing on binding ligand, as revealed by small-angle X-ray scattering measurements. 17 Most recently, we showed that human recombinant C65A/C167A-L-PGDS, which preserves a disulfide bond between Cys89 and Cys186 without an enzymatic active center of Cys65, also can bind a variety of lipophilic small ligands with high-binding affinities. 11 Especially, it can bind heme metabolites such as hemin, biliverdin, and bilirubin with extremely high-binding affinities (KD = 3 to 18 nmol/L) compared with its binding of other lipophilic small ligands such as retinoids, thyroids hormones, steroids hormones, and flavonoids (KD >300 nmol/L) in vitro. Thus, heme metabolites are considered to be specific ligands of human L-PGDS. In addition, we reported that L-PGDS in the intrathecal space is shifted to the blood within ˜6 hours and quickly eliminated through the urine with a terminal half-life of 0.77 hours. 26 Thus, L-PGDS acts as a scavenger of biliverdin in the CSF of patients with SAH and eliminates it from the body.
Subarachnoid hemorrhage, which is often caused by the rupture of cerebral aneurysms or arteriovenous malformations, is well known to provoke sudden death at the onset of an attack. Even if the patients survive from the initial attack, the chronic cerebral vasospasms after SAH have been considered as a major cause of mortality. The acute vasospasm may occur within 4 to 24 hours and is probably due to direct contractile stimulation of vascular smooth muscle cells by circulating vasoactive substances such as serotonin and PGF2α. 27 A delayed vasospasm is then induced from 2 to 14 days after bleeding. However, the postulated mechanism for this delayed vasospasm, which is clinically more serious, is complex and is far from completely understood. A whole array of possible chemical and inflammatory mediators have been implicated in its pathogenesis, including eicosanoids, interleukins, immune complexes, and blood degradation products such as oxyhemoglobin. 28 Decreased levels of vasodilators such as PGI2, and the release of potent vasoconstrictors such as endothelin and bilirubin have been demonstrated in animal models of SAH.29–31 Biliverdin is known as the degradation product of heme catalyzed by heme oxygenase-1, whose expression level is upregulated by SAH;32,33 and biliverdin is subsequently reduced by biliverdin reductase to form bilirubin. The concentrations of bilirubin in the CSF are closely related to the time course of cerebral vasospasms. Clark's group intensely studied the metabolites of heme and suggested that bilirubin oxidation products are involved in the delayed cerebral vasospasms of patients with SAH 34 and in the pathologic vasoconstriction associated with hemorrhage. 35 Bilirubin oxidation products are considered to have a key role in SAH-induced vasospasm. In clinical practice, external drainage of bloody and xanthochromic CSF is a standard remedy for preventing vasospasm. Therefore, blocking bilirubin formation, inactivating bilirubin or bilirubin oxidation products, or removing all of the blood clot before vasospasm are potential treatment strategies. In addition, several studies have reported that bilirubin induces apoptosis both in primary cultures of fetal cortical and cerebellar rodent neurons36,37 and in cultures of fetal rodent astrocytes. 38 In the present study, we showed the toxicity of bilirubin toward human neuroblastoma cells and demonstrated the protective effect of L-PGDS against bilirubin-induced neuronal cell death, an effect mediated by the scavenging of bilirubin. We also showed that biliverdin-bound L-PGDS was not affected by biliverdin reductase, such that bilirubin could not be produced from biliverdin-bound L-PGDS. Thus, our results indicate that L-PGDS, the second most abundant protein in human CSF, acted as a scavenger of biliverdin, whose degradation products are involved in SAH-induced vasospasm and neuronal cell death.
However, oxidative stress has also been shown to be involved in the pathogenesis of cell injury after SAH; and it has been associated with reactive oxygen species production in the brain. In fact, histochemical analysis showed an enhanced production of superoxide anion in the subarachnoid space after SAH. 39 Other evidence also suggests that oxidative stress is one of the factors contributing to posthemorrhagic vasospasm. 40 In a previous study, we demonstrated the protective effect of human L-PGDS against reactive oxygen species-induced neuronal cell death by scavenging the active oxygen via the thiol of the Cys65 of L-PGDS. 12 Therefore, the active oxygen also possesses the ability to bind to this thiol. In this study, we monitored the chromophore having an absorbance at 392 nm and determined it to be biliverdin. However, we can easily predict the production of oxidized L-PGDS by the active oxygen generated in the subarachnoid space after SAH. Thus, to clarify in detail the scavenger effects of L-PGDS in the CSF of patients with SAH, it will be necessary to examine the reaction time for the binding of biliverdin and active oxygen to L-PGDS and to determine the quantities of biliverdin-bound L-PGDS and oxidized L-PGDS.
In conclusion, we investigated the role of L-PGDS, which accumulates in the CSF after SAH. Based on the spectroscopic analysis, we found that the endogenous ligand bound to L-PGDS in the CSF of patients with aneurysmal SAH was biliverdin. Our results indicate that L-PGDS acted as a scavenger of biliverdin, the metabolite of hemoglobin that accumulates in the CSF of SAH patients. L-PGDS could thus be therapeutically expected to counteract pathologic states such as cerebral vasospasm after SAH and SAH-induced neuronal cell death.
Footnotes
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
We thank Drs S Tanimori, M Hisada, H Naoki, and S Kume for their helpful discussions, and Drs T Kusumoto and CT Beuckmann, and Mr M Nakatsuji for technical assistance. We are also grateful to Drs K Seiki and H Oda for measurements of L-PGDS concentrations in CSF samples and to the Center for Medical Research and Education (Graduate School of Medicine, Osaka University) for the MALDI/TOF MS analysis.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.