
Abstract
Transforming growth factor-β1 (TGF-β1) is a fibrogenic cytokine that is involved in postinjury repair and is implicated in the etiology of postsubarachnoid hemorrhage (SAH) chronic communicating hydrocephalus. TGF-β1 was measured by enzyme-linked immunosorbant assay (ELISA) in sequential samples of cerebrospinal fluid (CSF) in 11 patients with hydrocephalus after SAH; levels were seen to be biphasically elevated and sources were investigated. TGF-β1 levels were compared with albumin levels that estimated CSF blood content. Control samples from nonhemorrhagic hydrocephalics were tested similarly. Mean total TGF-β1 levels were elevated to 4400 ± 3435 (±SD) pg/mL greater than control levels of 97 ± 42 at 1 to 2 days posthemorrhage. Thereafter, levels fell to 714 ± 401 by 5 to 6 days posthemorrhage, then rose to a second peak of 1667 ± 774 at 9 to 10 days posthemorrhage, remaining significantly increased until 19 days posthemorrhage (P = 0.007). The first peak probably derived from extravasated platelets and correlated with increased albumin levels in the CSF. The second TGF-β1 peak rose greater than CSF albumin levels that had stabilized at this time, and thus was attributed to a tissue-specific response rather than a re-bleed. TGF-β1 was detected in the choroid secretory epithelium from controls, but levels were greater in SAH patients at 10 to 12 days posthemorrhage. The authors conclude that the elevated levels of TGF-β1 in CSF after SAH are derived initially from blood and later from endogenous sources such as the choroid plexus.
Keywords
Transforming growth factor-β1 (TGF-β1) is one of three mammalian TGF-β isoforms that promote epithelial and mesenchymal cell proliferation and differentiation. The mature form of TGF-β exists as a latent, biologically inactive 25 kDa protein, composed of two identical 12.5 kDa subunits. Activation of latent TGF-β allows binding to a cell-surface complex composed of receptors coupled to cytoplasmic signal transducers, which generate cellular responses (Brown et al., 1990; Wrana et al., 1994). TGF-β1 modulates extracellular matrix synthesis, the actions of other cytokines, and the immune system (Cui and Akhurst, 1996). Thus, by altering the expression of integrins bound to extracellular matrix proteins, inhibiting matrix protein degradation through protease down-regulation, and up-regulating protease inhibitor genes, TGF-β1 is a prototypic fibrogenic factor active during tissue remodeling, fibrosis, and tissue repair (Roberts et al., 1992).
In experimental animals (for example, a rat), normal adult central nervous system parenchyma contains little or no TGF-β1, but factors derived from central nervous system lesions, where the blood–brain barrier is breached, both trigger release of TGF-β1 from platelet α granules (Assoian et al., 1983) and up-regulate TGF-β1 expression in macrophages, astrocytes, and other injury responsive cells, including those of the vascular endothelium, and the meninges and choroid plexus, which secrete TGF-β1 into the cerebrospinal fluid (CSF) (Logan et al., 1992; Berry et al., 1999). Such high and sustained TGF-β1 levels induce fibrogenesis and scar formation (Logan and Berry, 1998; Berry et al., 1999) after experimental brain trauma (Logan et al., 1992, 1994a).
Transforming growth factor-β1 released into the CSF after subarachnoid hemorrhage (SAH) may similarly stimulate fibrogenesis in the subarachnoid space that is implicated in the etiology of post-SAH chronic hydrocephalus. Elsewhere (Daniel et al., 1997), the authors have demonstrated a biphasic response of TGF-β1 in humans after a single hemorrhagic event. Here, the authors continue their investigation of the TGF-β1 changes in the CSF of post-SAH human patients, identifying the source of the second, late phase increase in TGF-β1 levels by analyzing titers in serial samples of CSF from individual patients and monitoring CSF blood levels using albumin as an indicator of hemorrhage.
MATERIALS AND METHODS
Cerebrospinal fluid samples
Ethical approval for this study was obtained from the South Birmingham Health Authority Ethics Committee. Sequential CSF samples were collected from 11 patients with diagnosed SAH admitted to the Neurological Critical Care Unit at the Queen Elizabeth Hospital, Birmingham. All samples were taken through external intraventricular drains fitted for routine patient management. Approximately 5 to 8 mL CSF was collected every 1 to 2 days, centrifuged at 717 g for 10 minutes to sediment both hematogenous and other cells contaminating the sample, and the supernatant was aliquoted and stored at −70°C. Over a 2-month period, the development of acute and chronic post-SAH hydrocephalus was recorded for each patient. Chronic hydrocephalus was defined as clinical and radiologic hydrocephalus requiring treatment, demonstrable two weeks after SAH, as either the progression of acute hydrocephalus or the development of chronic hydrocephalus de novo. Day 1 was designated as the day of hemorrhage and samples were collected between 1 to 19 days post-SAH (days posthemorrhage). Clinical profiles of patients with post-SAH hydrocephalus are shown in Table 1. Control samples were collected from seven unmatched patients with nonhemorrhagic-associated hydrocephalus (Table 2) and were processed in the same way. Subarachnoid hemorrhage and control patient groups had the same male:female ratio of 0.8:1 and a mean age of 52.3 and 58.5, respectively.
Characteristics of the 11 patients (P1–P11) with SAH
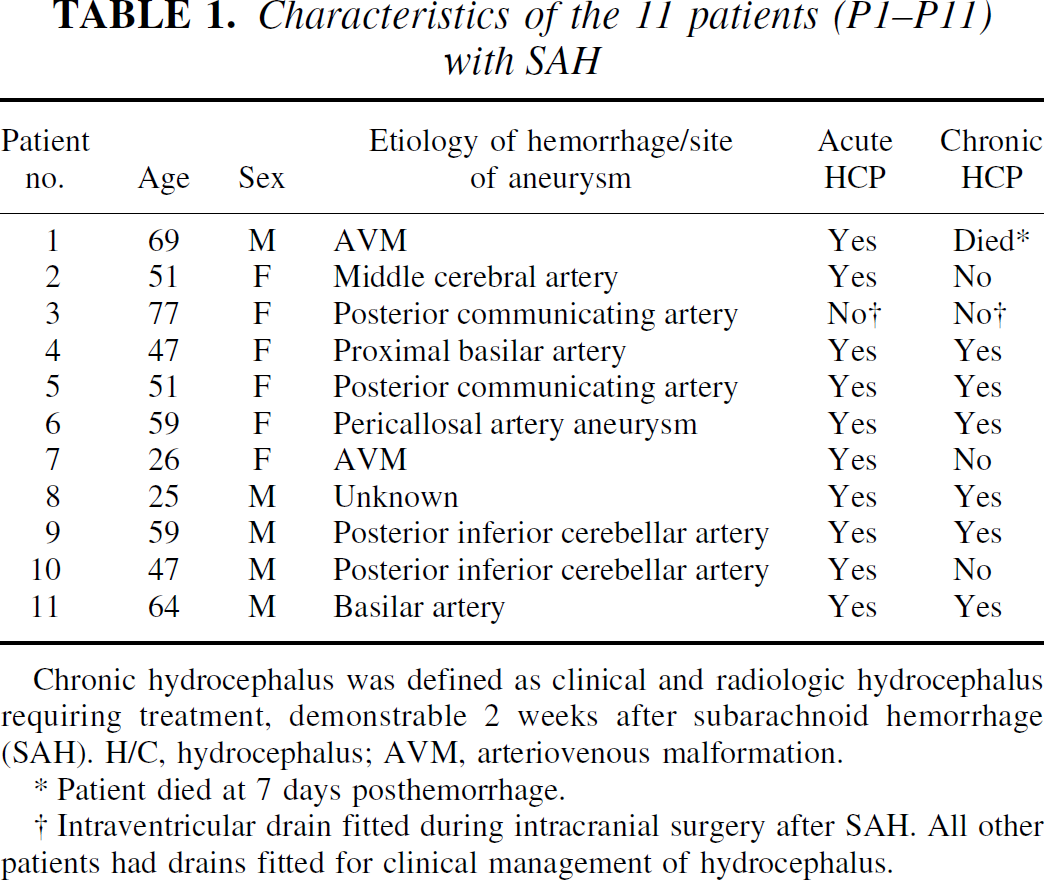
Chronic hydrocephalus was defined as clinical and radiologic hydrocephalus requiring treatment, demonstrable 2 weeks after subarachnoid hemorrhage (SAH). H/C, hydrocephalus; AVM, arteriovenous malformation.
Patient died at 7 days posthemorrhage.
Intraventricular drain fitted during intracranial surgery after SAH. All other patients had drains fitted for clinical management of hydrocephalus.
Characteristics of 7 control patients
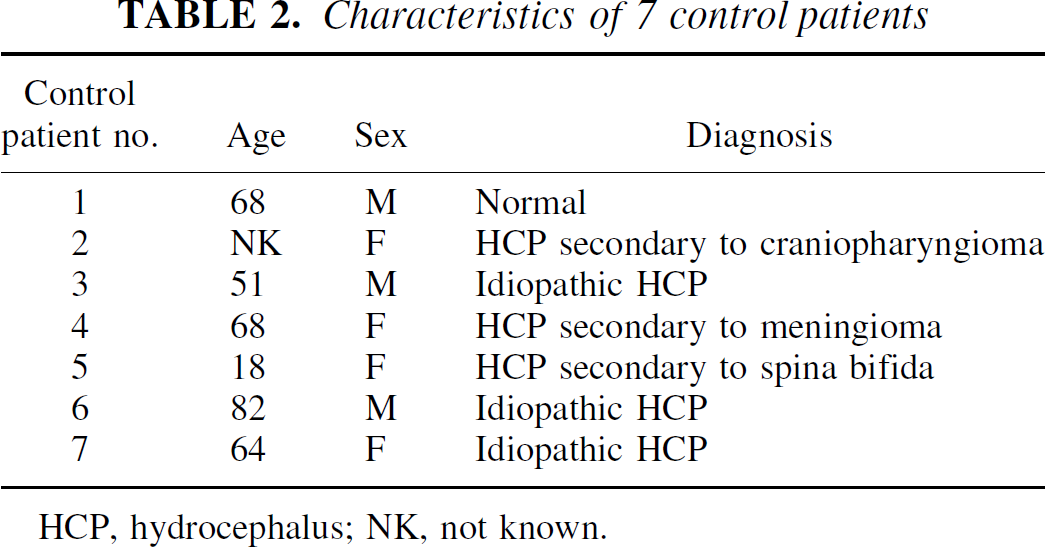
HCP, hydrocephalus; NK, not known.
Choroid plexus samples
Paraffin wax sections (donated by Peter Barber, neuropathologist at the Queen Elizabeth Hospital, Birmingham) of normal choroid plexus from biopsies of suspected choroid plexus papillomas (used as non-SAH controls), together with choroid plexus from autopsy of SAH patients at 10 to 12 days posthemorrhage, were processed for immunohistochemistry as detailed below.
ELISA
Concentrations of TGF-β1 in each CSF sample were measured using the Promega TGFβ1 Emax ImmunoAssay System (Promega, Southampton, U.K.), an antibody sandwich enzyme-linked immunosorbant assay (ELISA), using antibodies that recognize only the biologically active molecule. Thus, all non-manipulated CSF samples provided values of biologically active TGF-β1 and acidified samples, in which latent TGF-β1 was activated, gave values of total TGF-β1 (1.5 μL 1N HCl per 50 μL CSF, incubated at room temperature for 15 minutes, followed by neutralization with 1 μL 1N NaOH per 50 μL CSF). Different dilutions of CSF in Promega TGF-β sample buffer were assayed to obtain an optical density for the range of 0.2 to 0.4 for which the standard curve was most accurate.
Albumin assay
Albumin levels in each sample were measured using the bromocresol green dye reagent (Sigma, Poole, U.K.) that binds with human albumin, causing the maximum absorbance of the solution to shift to 628 nm. The resultant optical density at 638 nm is proportional to the amount of albumin in the sample. The albumin in the samples was assayed by mixing 200 μL of undiluted CSF to 1000 μL of the dye reagent in a cuvette, and immediately measuring the absorbance at a wavelength of 628 nm. A standard curve was prepared using dilutions of human albumin solution (Sigma) in phosphate-buffered saline.
Immunohistochemistry
Transforming growth factor-β1 was detected in sections of human choroid plexus by diaminobenzidine immunohistochemistry using the avidin-biotin amplification method (Vectastain Elite ABC kit; Vector Laboratories, Peterborough, U.K.) as described previously (Logan et al., 1994a). The primary antibody used was anti-TGF-β1 chicken-anti-human IgY antibody, at a 1/500 dilution (R&D Systems, Abingdon, U.K.), and specificity of staining was tested using sections prepared by the same method, with either a 1/500 dilution of nonimmune IgY solution (Sigma), or a 1/500 dilution of anti-TGF-β1 antibody preabsorbed with excess TGF-β1 peptide (R&D Systems, Abingdon, U.K.).
Statistical analysis
Because of the small number of CSF samples collected at each day posthemorrhage to examine the temporal changes in both TGF-β1 and albumin levels, sample data were pooled into groups every 2 days posthemorrhage until 14 days posthemorrhage. All samples collected at 15 to 19 days posthemorrhage comprised the 1 remaining group. When multiple samples were collected per patient at each time interval, the mean value of the measurements per patient was used for analysis. The significance of the differences in TGF-β1 and albumin levels between time intervals was established by a one-way analysis of variance and by Student's t-test. Because the data obtained in these studies was discontinuous, an analysis of covariance was used to examine the association of TGF-β1 with time after allowing for albumin level changes post-SAH. Probability values of P ≤ 0.05 were considered statistically significant.
RESULTS
TGF-β1 levels in the cerebrospinal fluid
Levels of total TGF-β1 in the CSF from control patients varied between 32 and 269 pg/mL ± SD (mean 97 ± 42). Active TGF-β1 levels were less than 32 pg/mL. Total TGF-β1 levels recorded during the post-SAH measurement period ranged between 229 and 9828 pg/mL (mean 1763 ± 1094), and the active TGF-β1 levels recorded were between 32 and 363 pg/mL (mean 88 ± 71). The temporal distribution of total TGF-β1 for each time period was biphasic, with a primary peak at 1 to 2 days posthemorrhage, followed by a secondary peak at 9 to 10 days posthemorrhage (Fig. 1). Total TGF-β1 levels were statistically significantly greater (analysis of variance) at each time point in SAH patients compared with control levels (for example, 1 to 2 days posthemorrhage, P = 0.01; 5 to 6 days posthemorrhage, P = 0.002; 9 to 10 days posthemorrhage, P = 0.0003; 15 to 19 days posthemorrhage, P = 0.007). The elevation of active TGF-β1 in the CSF post-SAH, compared with that of control patients throughout the observation period, was also highly significant (P < 0.001 for each time interval).
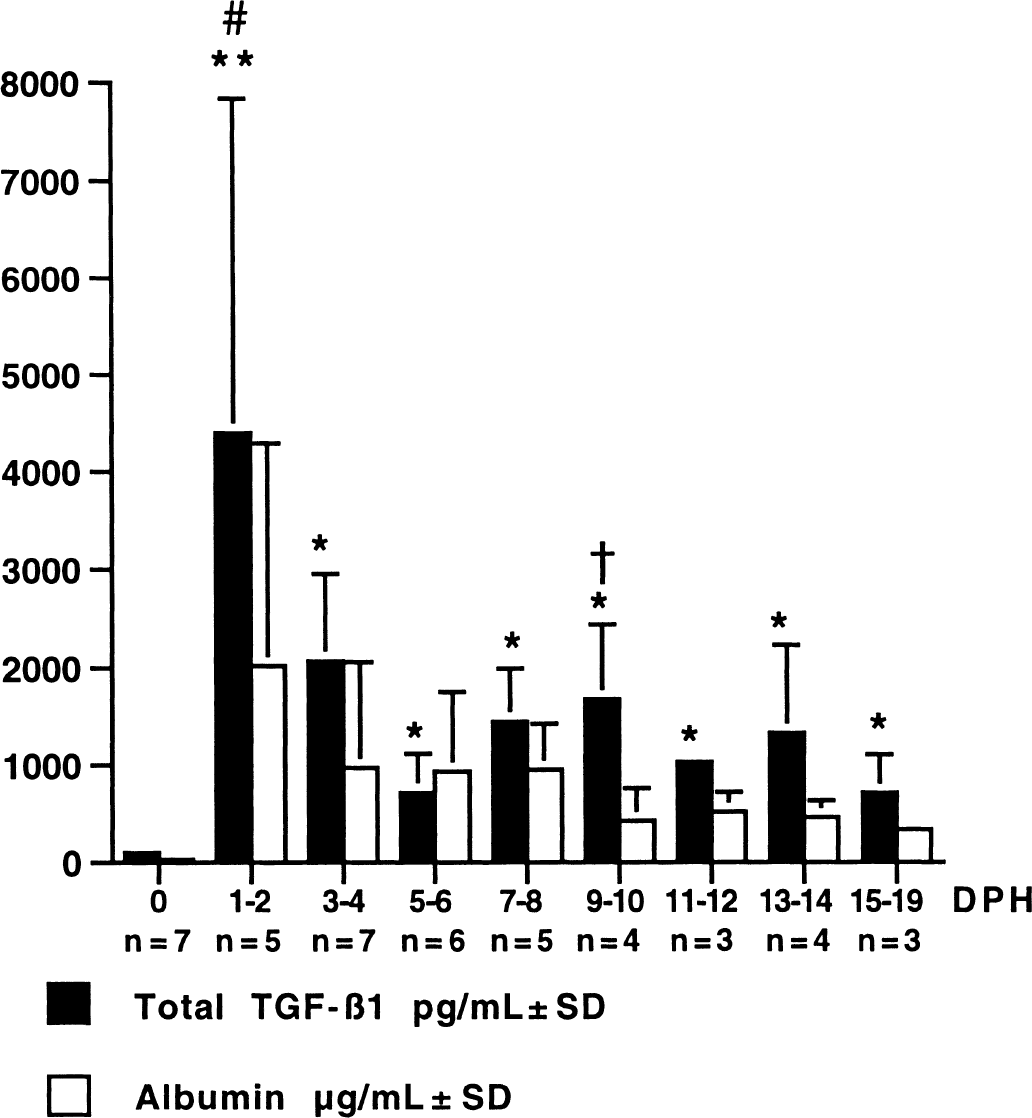
Temporal variation in mean total transforming growth factor-β1 (TGF-β1) and albumin levels in the cerebrospinal fluid for all subarachnoid hemorrhage patients. Error bars represent standard deviation. At all time points, total TGF-β1 levels were significantly greater than in controls (0 days posthemorrhage [DPH]) (*P < 0.002, **P < 0.01). Statistically significant (analysis of variance) differences between TGF-β1 levels at 1 to 2 days posthemorrhage and 9 to 10 days posthemorrhage compared with levels at 5 to 6 days posthemorrhage are also indicated (#P < 0.04, †P < 0.03, respectively). n = number of patients.
To further evaluate the biphasic response of total TGF-β1, both primary (1 to 2 days posthemorrhage) and secondary (9 to 10 days posthemorrhage) peak data were compared with the posthemorrhagic trough data at 5 to 6 days posthemorrhage and were shown to be significantly greater in both cases (P = 0.04 and P = 0.03, respectively). Increased albumin levels were seen in the CSF after SAH, peaking at 1 to 2 days posthemorrhage, and levels declined continuously toward control values (Fig. 1), suggesting that no further hemorrhagic events had occurred and that the secondary peak in total TGF-β1 was not blood derived. Correlation between albumin and total TGF-β1 in the posthemorrhagic period (1 to 19 days posthemorrhage) was further analyzed by an analysis of covariance. This analysis revealed an underlying association of total TGF-β1 with albumin post-SAH (F1,23 = 4.43, P = 0.046), but also demonstrated that even allowing for this variation there was a significant variation in total TGF-β1 with time post-SAH (F1,23 — 2.8, P = 0.029) that was independent of albumin. This finding indicated that, after the initial hemorrhagic period, the levels of total TGF-β1 were not related to those of albumin and, therefore, were probably not blood derived.
Choroid plexus immunohistochemistry
Immunohistochemistry of human choroid plexus localized TGF-β1 to this site (Fig. 2). Antibody preabsorbed with both TGF-β1 peptide and IgY established the specificity of the antibody (Fig. 2A to 2B). Nonhemorrhagic choroid plexus (controls) showed low levels of immunoreactivity in the cytoplasm of the secretory cuboidal epithelium (Fig. 2C to 2D). Sections of choroid plexus from SAH patients taken at 10 to 12 days posthemorrhage had obvious dense concentrations of TGF-β1 in the epithelial cytoplasm and occasional nuclei (Fig. 2E to 2F).
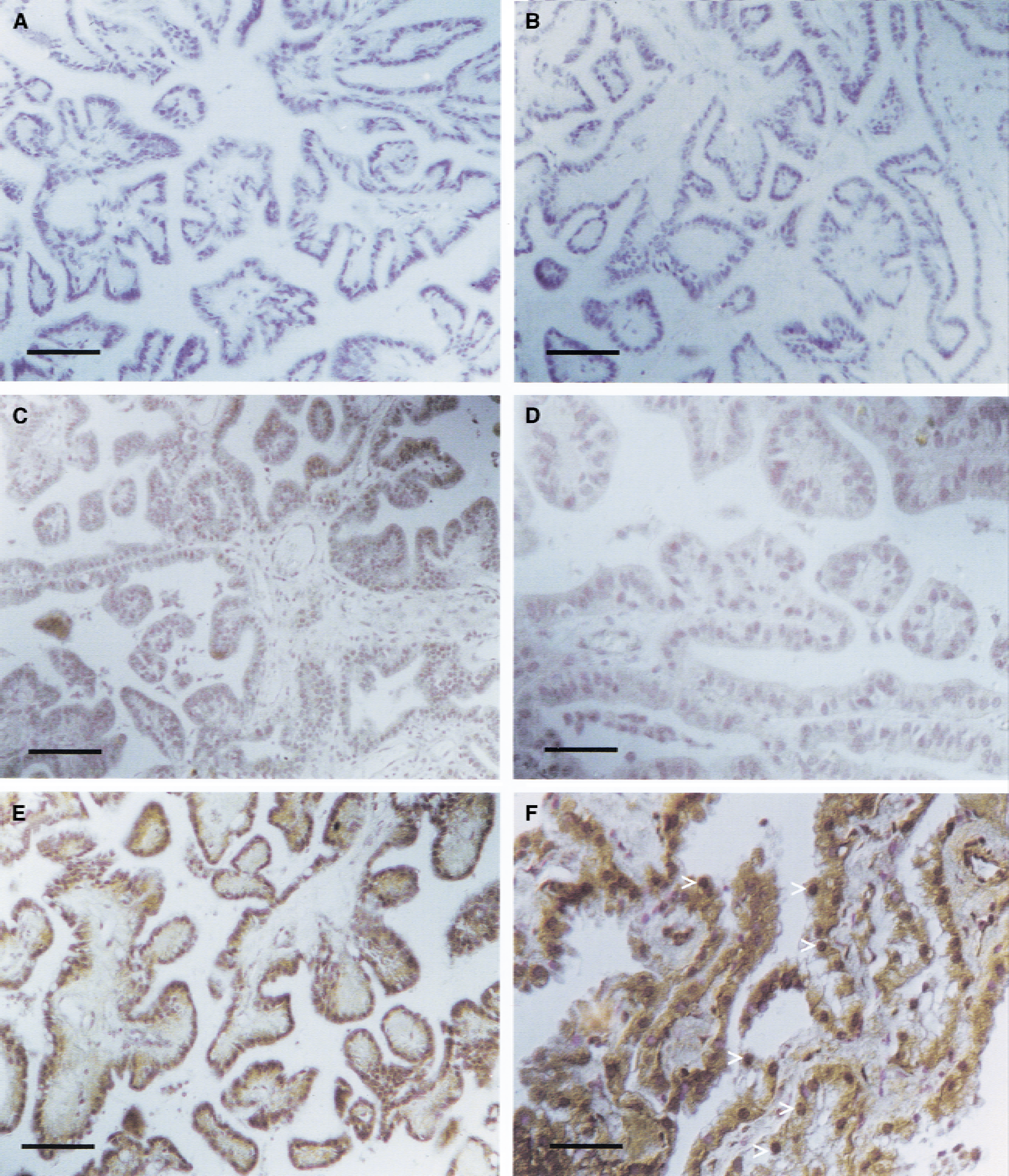
Staining of choroid plexus sections to localize immunoreactive TGF-β1 (brown deposit).
DISCUSSION
The authors measured TGF-β1 levels in sequential CSF samples taken from SAH patients to confirm the biphasic response of TGF-β1 in the CSF after a bleed observed elsewhere by the authors (Daniel et al., 1997) and to investigate its origin. By establishing that after the initial hemorrhage variation in albumin levels was not correlated with that of total TGF-β1 in the CSF, the authors have demonstrated that the second TGF-β1 peak observed was probably not derived from a re-bleed, but rather from injury-responsive cells in the brain. TGF-β1 immunostaining of human choroid plexus showed immunoreactivity in the secretory epithelium, with dramatically more intense staining detected in sections taken from patients at 10 to 12 days post hemorrhage compared with controls. This observation suggests that the choroid plexus may be one source of the second TGF-β1 peak.
The level of active TGF-β1 in the CSF of each of the control patients was <32 pg/mL, the limit of detection for the assay. In post-SAH CSF, active TGF-β1 levels were consistently much less than total TGF-β1 (mean levels of active TGF-β1 in all samples throughout the post-SAH observational period were 88 ± 71 pg/mL (±SD) compared with 1763 ± 1094 for total TGF-β1). This was to be expected as TGF-β1 is secreted in a latent form, with activation occurring only at the target site, thereby preventing inappropriate widespread bioactivity (Sato and Rifkin, 1989; Brown et al., 1990). After SAH, activation of latent TGF-β1 is promoted locally in the microenvironment of the subarachnoid space by proteases such as plasmin, local acidification after tissue damage (Jullien et al., 1989), and interaction with other cytokines. Such activation could account for the small but significant (P < 0.001) rise in levels of active TGF-β1 in the CSF of all patients after SAH.
Relatively low levels of total TGF-β1 are normally found in the CSF in humans (Mogi et al., 1995; Krupinski et al., 1996; Samuels et al., 1989; Johnson et al., 1992) and rats (Logan et al., 1992; Unsicker et al., 1991). The current study confirms this, with levels of 97 ± 42 pg/mL (±SD) recorded in the CSF from control patients. In normal rats, TGF-β1 is synthesized and secreted by the choroid plexus epithelium and leptomeningeal cells, the few cell groups that can express TGF-β1 in the undamaged central nervous system (Unsicker et al., 1991; Logan et al., 1992), but thus far only the leptomeninges have been identified as a source of production in humans (Johnson et al., 1992). This study suggests that the choroid plexus epithelium also produces TGF-β1 in humans.
There was a 45-fold increase of basal levels of total TGF-β1 immediately after SAH, which averaged 4400 ± 3435 pg/mL during the first 2 days posthemorrhage (P = 0.01). The source of TGF-β1 during this time was almost certainly platelets (Assoian et al., 1983), plasma (Huang et al., 1988; Wakefield et al., 1988), and hematogenous monocyte macrophages (Wahl et al., 1991) from blood extravasated into the subarachnoid space. Others reported levels of total TGF-β1 in the CSF of between 1427 and 1930 pg/mL post-SAH (Daniel et al., 1997; Kitazawa and Tada, 1994), but the levels in the current patient set appear higher. This probably reflects not only the different ELISA systems and time intervals used in the various studies, but also the fact that all of these patients were in a critical care unit after sustaining severe hemorrhages. After the acute peak in CSF TGF-β1 titers, the levels declined, although they remained significantly (P = 0.002) greater than control levels (714 ± 401 versus 97 ± 42 pg/mL at 5 to 6 days posthemorrhage). This initial decrease in TGF-β1 can probably be attributed to either degradation, removal by phagocytic cells, CSF reabsorption, sequestration by the extracellular matrix, or a combination of these. Because unbound active TGF-β1 in the plasma has a short half-life of 2 to 3 minutes and latent TGF-β1 of approximately 100 minutes (Wakefield et al., 1990), the authors would expect that TGF-β1 derived from the hemorrhagic event to be cleared within days. However, significantly raised TGF-β1 levels remained until at least 19 days posthemorrhage (P < 0.002 throughout the post-SAH period), with levels rising to a second peak of 1667 ± 774 pg/mL (±SD) at 9 to 10 days posthemorrhage, P = 0.0003), indicating an additional, probably endogenous, source of TGF-β1.
The cells of the choroid plexus and leptomeninges are likely candidates for the endogenous source of TGF-β1, as they are known to synthesize and secrete TGF-β1 into the CSF in the rat in response to trauma (Logan et al., 1992, 1994a, b ; Unsicker et al., 1991; Johnson et al., 1992). Furthermore, TGF-β1 up-regulates its own production at sites of synthesis (Cui and Akhurst, 1996), including the choroid plexus of the rat (Logan et al., 1994a). The authors' immunohistochemical analysis of TGF-β1 in the human choroid plexus indicates increased TGF-β1 levels after SAH, and implies a reactive up-regulation of TGF-β1 production in the secretory epithelium of choroid plexus after SAH as a probable source of the second TGF-β1 peak. Alternative sources of post-SAH TGF-β1 titers may include increased selective permeability of the blood–brain barrier to TGF-β1 after SAH (Germano et al., 1992), release of TGF-β1 from platelets in a dissolving blood clot in the subarachnoid space (Grainger et al., 1995), and/or release of sequestered TGF-β1 from extracellular matrix, but these would not be sufficient either to produce the high levels of TGF-β1 nor to explain the consistent temporal changes in the TGF-β1 levels observed.
Albumin in the CSF was used as a marker for the hemorrhage, and, at 1 to 2 days posthemorrhage, levels increased coincidentally with total TGF-β1 to more than 80 times the normal CSF levels (from 25 ± 10 to 2014 ± 1699 μg/mL (±SD) albumin). Although albumin is the main CSF protein, it is neither metabolized nor secreted intrathecally, and its concentration in whole blood is approximately 1000 times that of normal CSF, hence its use as a blood marker (Tietz, 1994). Albumin leakage through the blood–CSF barrier (that is, a re-bleed) would dramatically increase levels in the CSF. As no further rise in CSF albumin was detected, statistically coincident with TGF-β1 at 9 to 10 days posthemorrhage, re-bleeds could not account for the second peak in TGF-β1 levels. The analysis of covariance supported this contention by demonstrating that the changes in total TGF-β1 in the CSF post-SAH were not significantly correlated with the observed changes in albumin. In fact, albumin levels in the CSF of SAH patients fell steadily after 1 to 2 days posthemorrhage, probably because of the removal of the blood from the subarachnoid space, possibly through the arachnoid granulations (Weller, 1995; Upton and Weller, 1985; Alksne and Lovings, 1972).
In conclusion, the authors believe that this study, which describes brain-specific responses in the TGF-β1 axis after SAH, provides further evidence that TGF-β1 is a fibrogenic factor involved in the etiology of post-SAH pathology. With continued research in this area, anti-fibrotic strategies may be developed to prevent complications of SAH, including chronic communicating hydrocephalus.
Footnotes
Acknowledgments:
The authors thank Peter Barber of the Queen Elizabeth Hospital, Birmingham, U.K., for help in making clinical samples available and Dr. Roger Holder, University of Birmingham, for advice on the statistical analysis.