
Abstract
Cathepsins and caspases are two families of proteases that play pivotal roles in ischemic cell death. This study investigated the existence of a cross-talk between cathepsin B and proinflammatory caspases in stroke-induced cell death, as recently suggested by in vitro data. Cortical ischemic damage was induced in mice by distal and permanent occlusion of the middle cerebral artery. Cytoplasmic activation of cathepsin B was observed from the early stages of infarction, and displayed an activation pattern parallel to the activation pattern of caspase-1 and −11. Immunohistochemistry revealed the colocalization of cathepsin B with each caspase in cells of the infarct core. The apical position of cathepsin B in both caspase-activation cascades was confirmed by pretreatment of the animals with the cathepsin B inhibitor CA-074, which also potently protected cortical structures from ischemic damage, indicating involvement of the proteases in the lesion process. The results show that cathepsin B release is an early event following occlusion of cerebral arteries, which eventually triggers the activation of proinflammatory caspases in the absence of reperfusion. This new pathway may play a critical role in brain infarction by promoting inflammatory responses, and/or by amplifying the apoptotic process.
Cathepsins are acidic endopeptidases, which belong to the papain superfamily of proteases (Turk et al., 1997). Under physiologic conditions, their activity is restricted to the lysosomal compartment, where they participate to protein turnover by degrading unneeded proteins into amino acids (Kirschke et al., 1980). Various pathologic stresses, such as global cerebral ischemia (Nitatori et al., 1995;Uchiyama, 2001), induce the release of cathepsins into the cytoplasm, where they extensively perform their proteolytic function and can promote direct cell degradation (Boya et al, 2003). The inhibition of cathepsins leads to significant neuroprotection in experimental stroke, which also confirms their involvement in the neurodegenerative process (Seyfried et al., 2001;Yamashima et al., 1998). Although cathepsins have been mainly associated with autophagy and necrosis, recent evidence in nonneuronal cells points to a critical involvement in apoptosis, through alterations of mitochondrial homeostasis and activation of proapoptotic members of the Bcl-2 family (Boya et al., 2003;Dietrich et al., 2004;Guicciardi et al., 2000;Stoka et al., 2001).
Caspases are central players of apoptosis (Friedlander, 2003). They are activated in response to external or intrinsic stimulus in self-amplified cascades by proteolysis of their inactive zymogen. Caspases cleave a number of substrates—estimates suggest more than 200 (Brockstedt et al., 1998)—thereby contributing to the major morphologic and biochemical changes of apoptosis. The activation pattern of caspases has been extensively analyzed in both global and focal cerebral ischemia (Onténiente et al., 2003), and their participation in ischemic damage has been confirmed by inhibition studies (Benchoua et al., 2001;Endres et al., 1998;Hara et al., 1997;Himi et al., 1998; Plesnila and Moskowitz, 2000), or through the use of animals with genetic mutations (Friedlander et al., 1998;Robertson et al., 2000).
Although caspases and calpains have been associated with nonconverging intracellular death pathways, in vitro studies have recently opened the possibility of a crosstalk between both families of cysteine proteases in pathologic settings. Cathepsin B, the main lysosomal protease of the brain parenchyma, possesses a caspase-processing activity, which mainly appears on caspase-11 (Schotte et al., 1998;Vancompernolle et al., 1998) and −1 (Hentze et al., 2003;Vancompernolle et al., 1998). Caspase-1 and −11 play important roles in brain ischemia by promoting both apoptotic and inflammatory processes (Ferrer and Planas, 2003;Kang et al., 2000;Rothwell, 2003), with caspase-1, or interleukin-1β-converting enzyme (ICE), being responsible for the synthesis of the functional form of the cytokine (Ceretti et al., 1992). Activation of procaspase-1 is triggered in vitro by caspase-11 (Wang et al., 1996, 1998), which can also independently trigger or amplify the apoptotic program by direct activation of procaspase-3 in neural cell types (Shibata et al., 2000) and in mice subjected to middle cerebral artery occlusion (MCAO) (Kang et al., 2000). In addition to its role in pro—interleukin-1 maturation, caspase-1 is also involved in the maturation of interleukin-18 in monocytic cells (Hentze et al., 2003) and, as with caspase-11, is a direct activator or procaspase-3 (Enari et al., 1996). These results suggest that pathologic lysosomal degradation can trigger specific cascades that involve the activation of proinflammatory caspases.
The aim of the present study was to investigate the activation pattern of cathepsin B and caspase-11 after focal ischemia induced in mice by permanent occlusion of the middle cerebral artery (pMCAO), and to analyze the possibility of a link between lysosomal stress-induced intracellular leakage of cathepsin B and the activation of proinflammatory caspases in vivo. We show that cathepsin B, caspase-11, and caspase-1 are activated by focal cerebral ischemia with similar spatiotemporal patterns, and co-localize in neuronlike cells in the damaged cortex. We found that treatment with a specific cathepsin B inhibitor significantly reduced infarct volumes, and that cathepsin B is a causative factor of the activation of both caspases. This reveals the existence of one or more lysosomal—proinflammatory caspase pathways in the early steps of the degenerative process that leads to brain infarction.
MATERIALS AND METHODS
Surgical procedure
Studies were performed on adult C57Bl/6 male mice (25 to 30 g; Janvier, Le Genest-St-Isle, France) in accordance with the recommendations of the European community (86/609/EEC) for care and use of laboratory animals. Permanent focal cerebral ischemia was performed by electrocoagulation and section of the left middle cerebral artery according to the methods described by Tamura et al. (1981) under halothane anesthesia (4% for induction, 1.5% for maintenance). Sham-operated animals were subjected to the same surgical procedure without pMCAO. Animals were killed by decapitation 30 minutes, 1 hour, or 6 hours after occlusion for biochemical studies, or by intracardiac perfusion after 30 minutes for immunohistochemical studies.
Infarct volumes
Mice were killed 24 hours after MCAO and brains were cut into coronal 2-mm slices (Mouse Brain Slicer Coronal Matrix; Harvard Apparatus, Holliston, MA, U.S.A.), and sections were stained with 2% TTC (2,3,5-triphenyltetrazolium chloride). Infarcted areas were measured with the KS400 software (Zeiss, Oberkocken, Germany) by subtracting the healthy areas of the lesioned cortex from that of the contralateral side. The total infarct volume was calculated after integrating areas with the distance between each level (Guégan et al., 1998).
Immunohistochemistry
Immunofluorescence experiments were performed on 10-μm-thick brain sections obtained after perfusion of the animals with 4% PFA in 0.1 mol/L PBS, and paraffin embedding of the brains. After removal of paraffin in xylene, sections were re-hydrated and incubated overnight at 4°C in 0.1 mol/L PBS containing 0.3% Triton X100 (PBS-T) with rabbit polyclonal anti—cathepsin B (1/20°; Oncogene, San Diego, CA, U.S.A.), anti—caspase-1 antibodies (1/500° in PBS-T), or anti—caspase-11 antibodies (1/10° in PBS-T; gift from J. Yuan, Boston, MA, U.S.A.;Wang et al., 1996). Sections were washed in PBS-T and incubated with biotinylated goat antirabbit antibodies (1/300° in PBS-T; Vector Laboratories, Burlingame, CA, U.S.A.) for 1 hour at room temperature, then in streptavidinbiotin-peroxidase complex (Vector Elite, Vector Laboratories) 1/400 for 30 minutes. Antigens were revealed using the Tyramide System Amplification (NEN, Boston, MA, U.S.A.) according to the manufacturer's instructions with cyanin-3 or FITC fluorophores. Bisbenzimide (0.5 μL/mL, Sigma, St. Louis, MO, U.S.A.) was added to the last incubation medium to visualize nuclei. For double labeling, sections were processed for cathepsin B staining and were then washed in acidic glycine buffer (0.1 mol/L, pH 3.34) for 10 minutes to detach antigen—antibodies bonds (Nakane, 1968). This procedure does not detach former precipitates obtained with the amplification system and allows for double-fluorescent analysis. Sections were then reprobed with anti—caspase-1 or anti—caspase-11 antibodies, and were processed for immunodetection as described earlier. Images were collected on a Zeiss Axioplan II microscope equipped with a Coolsnap digital camera, or on a Zeiss LSM confocal microscope. Artefactual immunostaining was controlled for by omission of primary antibodies (control sections).
Cathepsin B activity assay
A cytoplasmic-enriched protein fraction was prepared from the cortical middle cerebral artery territory as described elsewhere (Benchoua et al., 2002). Cathepsin B catalytic activity was measured on the peptide sequence carbobenzoxy-L-arginyl-arginine (z-Arg-Arg), a specific substrate that does not cross-react with other cathepsins (Knight, 1980), linked to 7-amino-4-methyl-coumarin (z-R-R-AMC, Calbiochem, San Diego, CA, U.S.A.). Proteins (30 μg) were diluted in 95 μL of a reaction buffer composed of (in micromoles per liter) sodium acetate 400 (pH 5.5) and L-cysteine 8. The reaction was started by addition of 5 μL of a 1-mmol/L stock solution of z-R-R-AMC and was incubated for 1 hour at 37°C. Free AMC release, which indicates cleavage of the substrate, was measured on a LS50 fluorimeter (Perkin-Elmer Life Sciences, Norwalk, CT, U.S.A.) with excitation and emission wavelengths of 380 nm and 460 nm, respectively. Fluorescence units were converted into micromoles of AMC release per hour and per milligram protein using a standard curve of free AMC (Calbiochem).
Caspase activity assay
Caspase activity was measured on cytoplasmic-enriched tissue extracts. Thirty micrograms of protein was diluted to a final volume of 90 μL in caspase assay buffer (in micromoles per liter: HEPES 50, pH 7.4; NaCl 100, EDTA 1, DTT 10). The reaction was started by the addition of 10 μL of a 2-mmol/L stock solution of 7-amino-4-trifluoromethyl-coumaryl (AFC)-coupled substrates (ac-YVAD-AFC for caspase-1–like activity, ac-LEVD-AFC for caspase-4/-11–like activity, and ac-DEVD-AFC for caspase-3–like activity). Free AFC was measured (excitation wavelength 400 nm, emission wavelength 500 nm) after 2 hours of incubation at 37°C. Fluorescence units were converted into picomoles of AFC released per hour and per milligram protein using a standard curve of free AFC (Sigma).
Western blot analysis
Twenty micrograms of protein from cytoplasmic-enriched extracts was resolved on a 15% SDS-PAGE gel and transferred onto polyvinylidene difluoride membranes. Membranes were blocked in 5% nonfat dry milk in TBS containing 0.1% Tween 20 (T-TBS) for 1 hour at room temperature, and were probed with rat monoclonal anti—caspase-11 antibodies (1/10°), or with goat polyclonal anti—caspase-1 antibodies (1/500°; M-19, Santa-Cruz Biotechnology, Santa Cruz, CA, U.S.A.), in T-TBS with 5% nonfat dry milk, overnight at 4°C. After 1 hour of incubation with the appropriate HRP-coupled secondary antibodies at room temperature, antigens were revealed using enhanced chemiluminescence reaction (ECL+, Amersham Pharmacia Biotech, Les Ulis, France). Quantification of signal intensity was performed using the ImageQuant software (Amersham Biosciences, Freiburg, Germany) and was normalized to α-tubulin contents or Coomassie brilliant blue staining.
Cathepsin B inhibition
For pharmacologic studies, mice received 20 μg of a specific cathepsin B inhibitor, the epoxysuccinyl peptide CA-074 (N-(L-3-trans-propyl-carbamoyloxirane-2-carbonyl)-L-isoleucyl-L-proline, Calbiochem) 30 minutes before pMCAO. CA-074 was injected stereotaxically into the right lateral ventricle in 1 μL of sterile saline for a 5-minute period. Controls were performed with intracerebroventricular injections of vehicle alone. Mice were killed 30 minutes and 1 hour after the induction of ischemia for biochemical analyses (activity assays and Western blots) and 24 hours after MCAO for TTC studies.
Statistical analysis
Data are expressed as mean and standard error. One-way analysis of variance (ANOVA) was used to compare intergroup differences for protein contents or protease activities, followed with the Fischer PLSD test.
RESULTS
Early release and activation of cathepsin B into the cytoplasm of ischemic neurons
According to its known localization in lysosomes, cathepsin B appeared in sham-operated animals as a discrete punctate labeling observed in randomly distributed cortical cells (Figs. 1A and 1B). The strongest labeling was observed in layer V. Thirty minutes after pMCAO, cathepsin B immunoreactivity had increased in layers III to V in the lesioned cortex (Fig. 1C), and displayed a diffuse intracellular distribution pattern that evoked cytoplasmic redistribution of the protease (Fig. 1D). No labeling was observed on control sections (Fig. 1H).
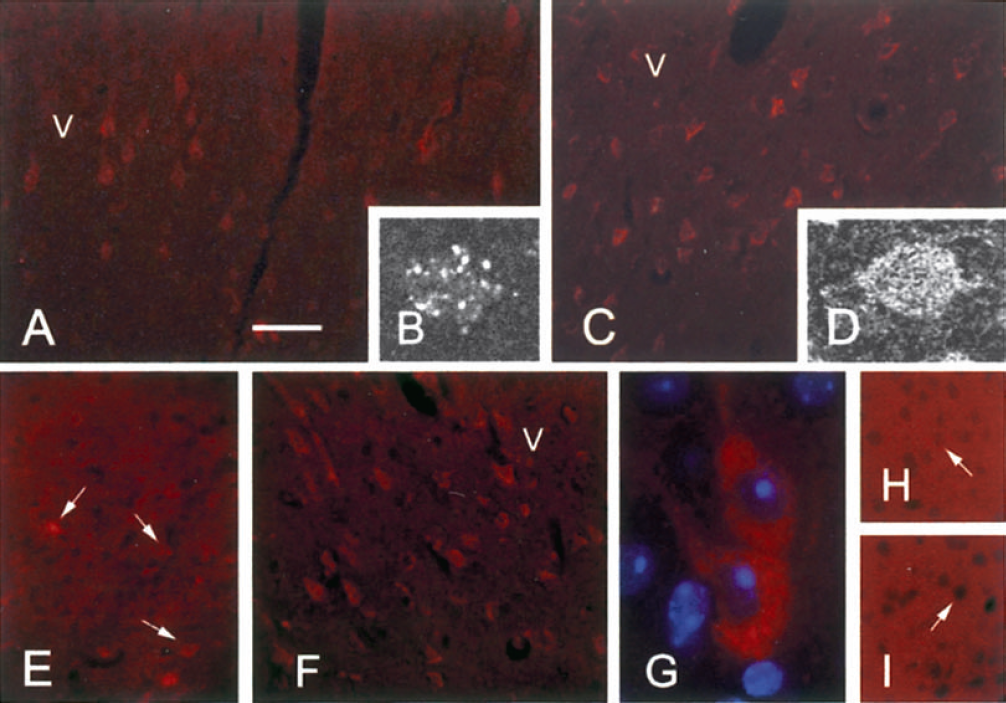
Immunohistochemical labeling of cathepsin B (
The extralysosomal redistribution was confirmed by the kinetics of cathepsin B activity (Table 1). The cleavage of z-R-R-AMC significantly increased from 30 minutes after occlusion (+25%) in cytoplasm-enriched fractions from the cortical territory ipsilateral to the occluded middle cerebral artery. High levels of cytoplasmic activity were maintained at 1 hour (+44%) and at 6 hours (+20%) (Table 1).
Time course of cathepsin B activity and caspase processing after MCAO
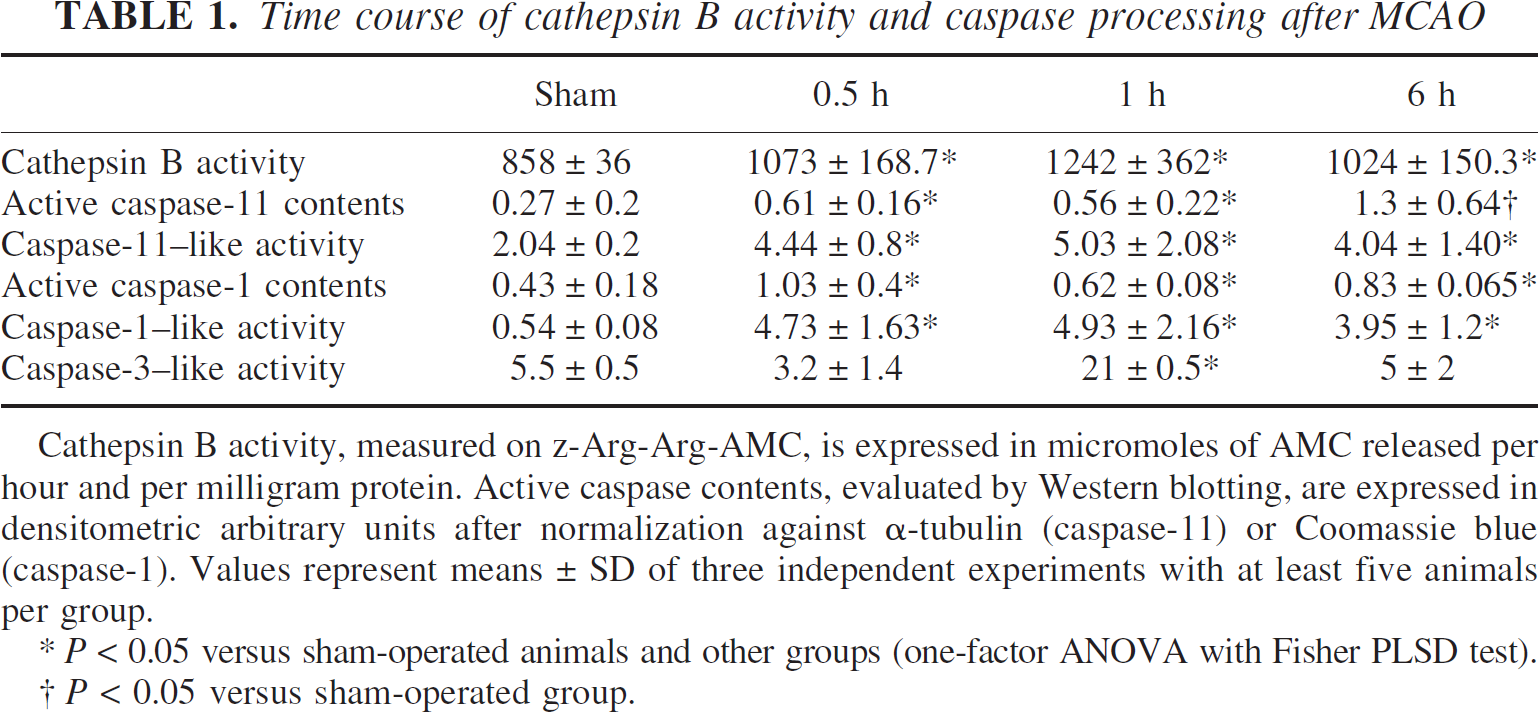
Cathepsin B activity, measured on z-Arg-Arg-AMC, is expressed in micromoles of AMC released per hour and per milligram protein. Active caspase contents, evaluated by Western blotting, are expressed in densitometric arbitrary units after normalization against α-tubulin (caspase-11) or Coomassie blue (caspase-1). Values represent means ± SD of three independent experiments with at least five animals per group.
P < 0.05 versus sham-operated animals and other groups (one-factor ANOVA with Fisher PLSD test).
P < 0.05 versus sham-operated group.
Activation of caspase-11 in pMCAO
No staining with anti—caspase-11 antibodies was observed in sections from sham-operated animals or in the cortex contralateral to the lesion (Fig. 1E). Labeling appeared in cells of the infarct core 30 minutes after occlusion (Fig. 1F), distributed throughout the cell body, and in proximal dendrites of strongly labeled cells, with a reticulate pattern (Fig. 1G). No labeling was observed on control sections (Fig. 1I).
With ac-LEVD-AFC cleavage assays, a strong increase in caspase-11–like activity was detected 30 minutes after pMCAO that was maintained for up to 6 hours (Table 1). Considering that the LEVD sequence is recognized by other caspases, the activation of caspase-11 was also evaluated by Western blot analysis of its large active fragment (p25) and full-length form (48 kd). Low levels of the p25 form were detected in sham-operated animals (Table 1 and Fig. 2A). Active caspase-11 levels were significantly increased 30 minutes after pMCAO, and were kept up to 6 hours after occlusion. However, no further increase was observed at 6 hours, indicating that the strong activity observed on the LEVD substrate at that time was due to other caspases. Amounts of the zymogen were also rapidly increased by ischemia, and were kept at high levels afterwards (Fig. 2A).
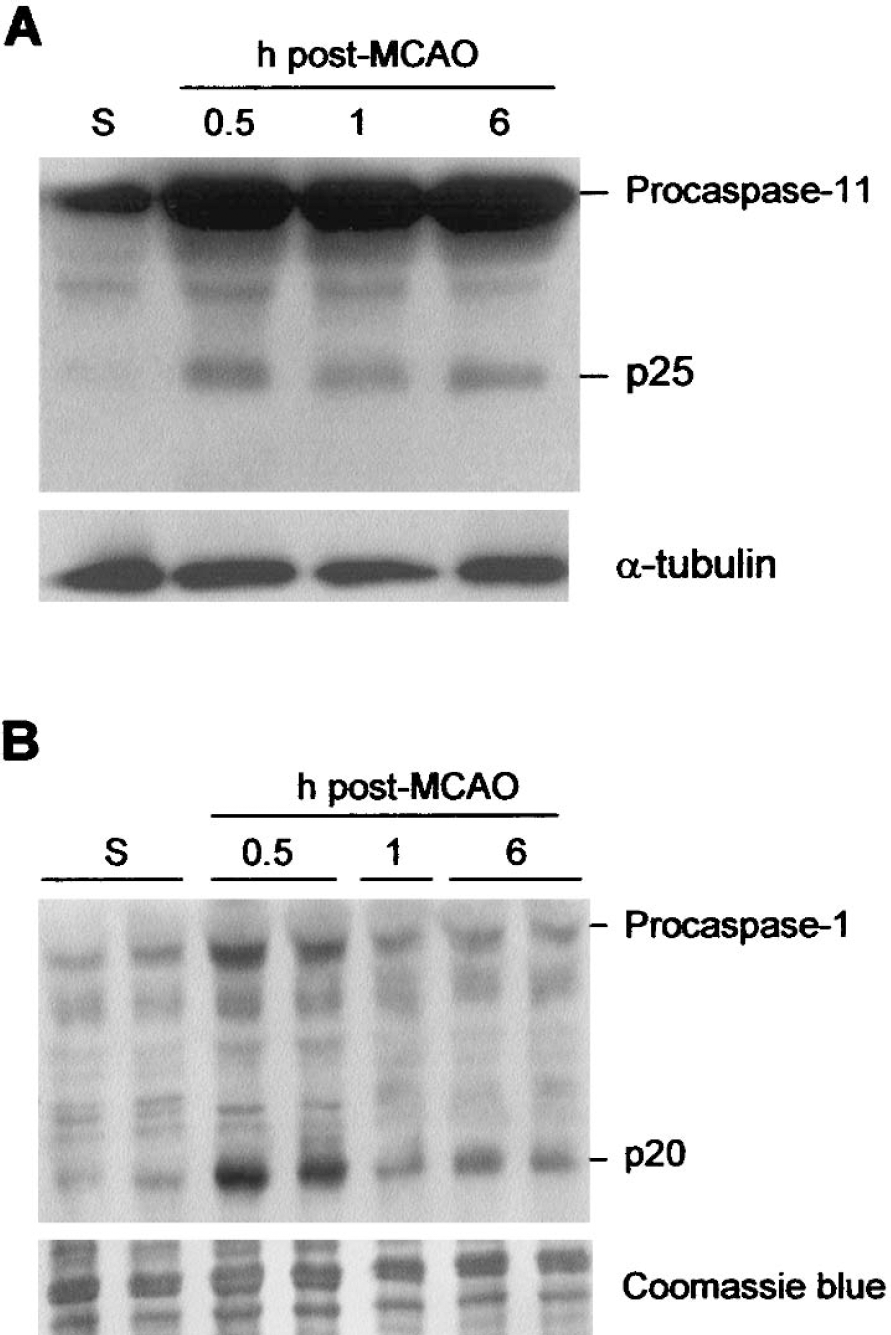
Caspase-11 and caspase-1 processing after MCAO. The time course of ischemia-induced procaspase-11 (
Activation of caspase-1
We next analyzed the activation of the putative target of caspase-11, caspase-1. Caspase-1–like activity, measured on ac-YVAD-AFC, conspicuously increased from 30 minutes after occlusion, and levels remained high for up to 6 hours (Table 1). Western blotting for caspase-1 confirmed the activity assay data, showing a significant increase in caspase-1 active p20 subunit 30 minutes after occlusion (Table 1, Fig. 2B), followed by a slight decrease 1 hour and 6 hours after pMCAO when compared with levels observed at 30 minutes. Upregulation of the caspase-1 proform was also detected at 30 minutes, but amounts decreased afterwards (Fig. 2B). As described previously, activation of the downstream caspase-3 was detected at 1 hour after occlusion (Table 1).
Colocalization of cathepsin B with caspase-1 or caspase-11 in the infarct core
Confocal microscopy of tissue sections double-labeled with cathepsin B and one of the caspases 30 minutes after occlusion revealed the colocalization of cathepsin B with caspase-11 (Fig. 3A–Fig. 3C) or caspase-1 (Fig. 3D–Fig. 3F). Because of the lack of specific antibodies, this included pro- and active forms of caspases.
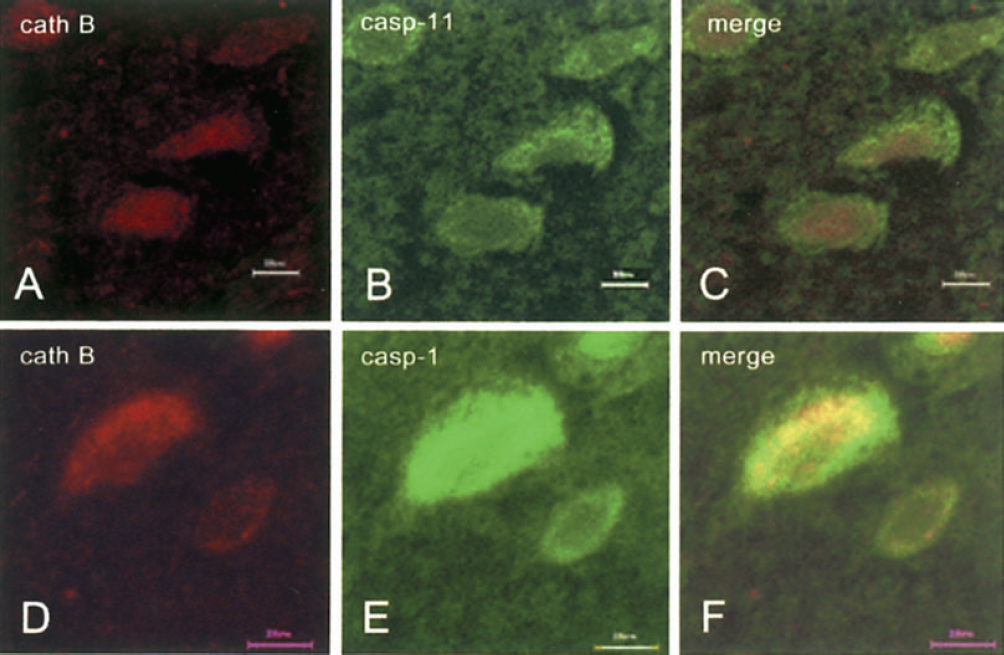
Colocalization of cathepsin B with proinflammatory caspases 30 minutes after MCAO in cortical layer V. (
Pharmacologic inhibition of cathepsin B protects ischemic lesions and results in defective caspase activation after pMCAO
Pharmacologic experiments were performed to examine whether cathepsin B acts upstream of caspase-11 and/or caspase-1. At the doses used, the inhibitor CA-074 shows no cross-reaction with other cathepsins in vivo (Towatari et al., 1991). CA-074 had no effect on basal caspase activation levels (Fig. 4B). As indicated by the dramatic decrease in z-R-R-AMC cleavage, pretreatment of the animals with CA-074 blocked the activation of cathepsin B observed 30 minutes after pMCAO (Fig. 4A), and inhibited ischemia-induced increases in caspase-11–, caspase-1–, and caspase-3–like activity (Fig. 4B). CA-074 significantly decreased infarct volumes (Fig. 5), confirming the involvement of these proteases in pMCAO-induced lesions.
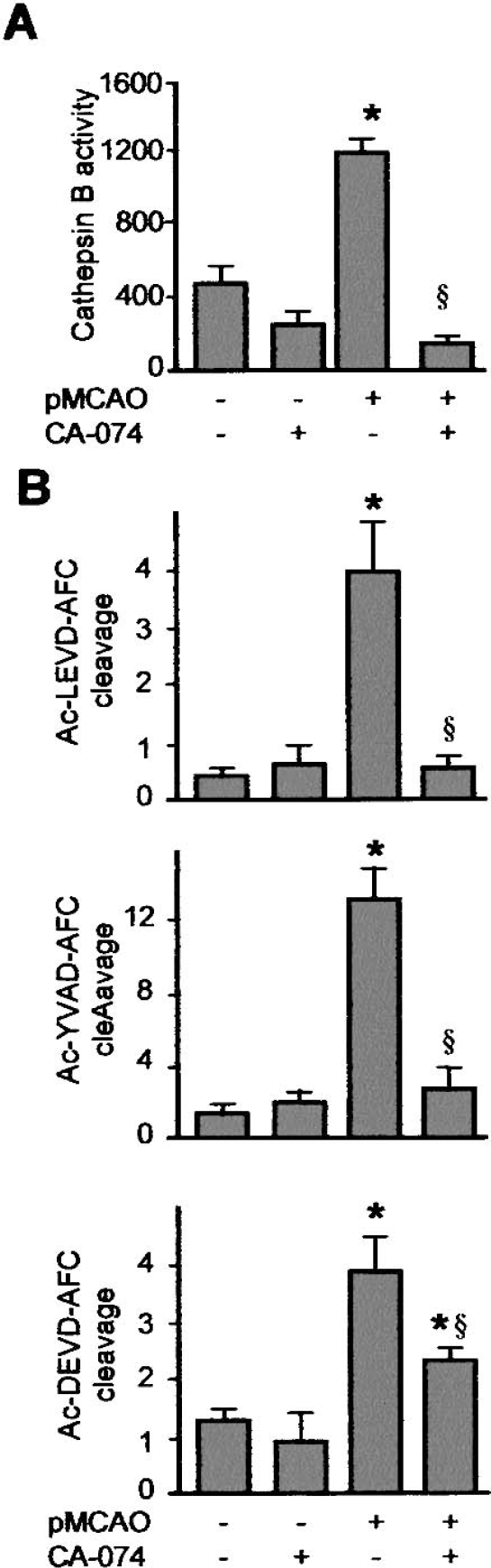
Inhibition of cathepsin B leads to decreased caspase-11 and caspase-1 activity after MCAO. Cathepsin B inhibition was performed 30 minutes before occlusion of the artery with CA-074. (
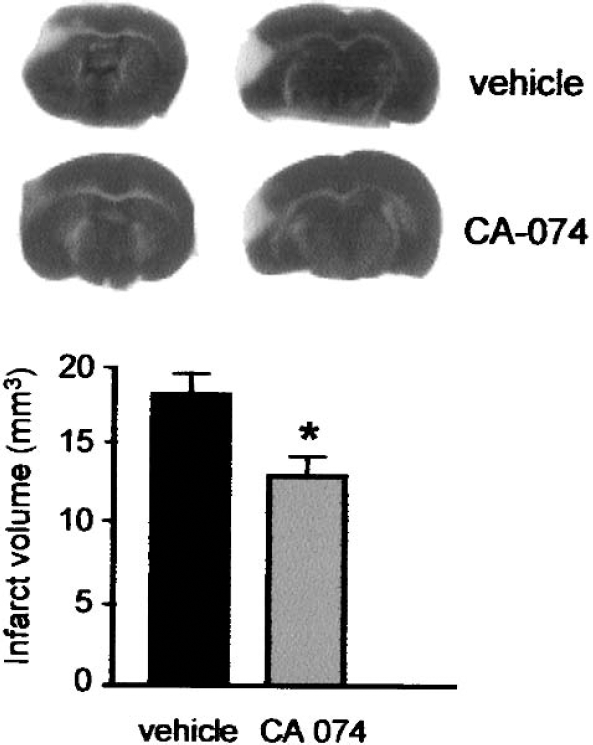
Representative TTC staining showing the neuroprotective effect exerted by CA-074 in pMCAO. *P < 0.05 versus non-treated (vehicle) group (n = 6; one-factor ANOVA with Fisher PLSD test).
Western blot analysis of the same cortical extracts confirmed a defective maturation of both caspase-1 and caspase-11 in ischemized cortices from animals treated with the inhibitor (Fig. 6). Administration of CA-074 abolished ischemia-induced processing of both caspases (Fig. 6A). Decreased amounts of active caspase-1 were not reflected by increases in procaspase contents (Fig. 6B).
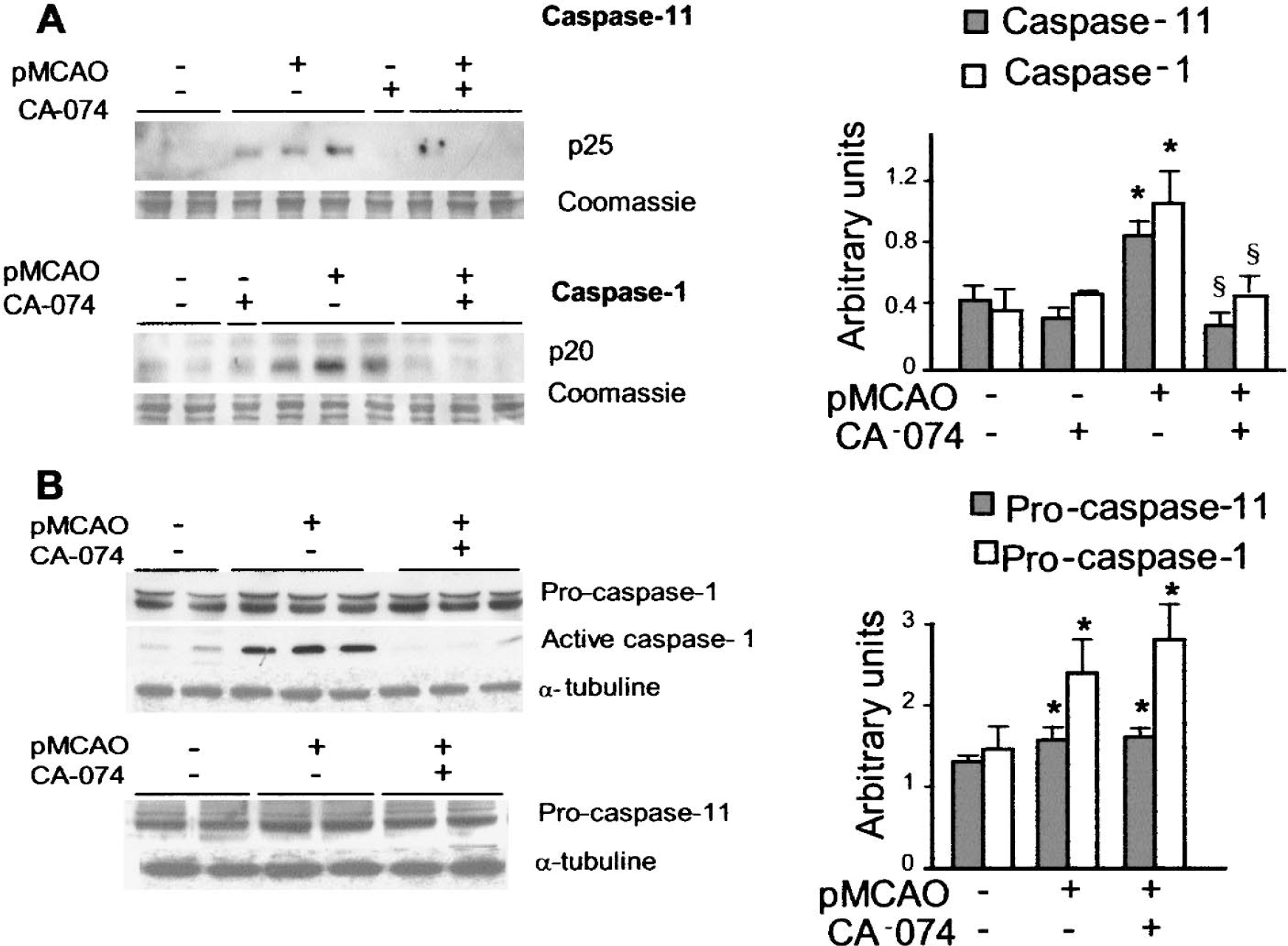
Effects of cathepsin B inhibition on caspase-11 and caspase-1 protein contents. (
DISCUSSION
This study investigated the relationships between two families of proteases involved in ischemic cell death, cathepsins and caspases. Our results show that cathepsin B, caspase-11, and caspase-1 follow parallel activation patterns in the early stages of cortical infarction after pMCAO, and that cathepsin B acts upstream of the two caspase cascades. Inhibition of cathepsin B activity offered protection from pMCAO lesions, defining a new lesional cascade that links lysosomal stress and caspase activation in cerebral focal ischemia.
By direct degrading of cell components, cathepsins have been originally associated with “passive” cell death forms, such as autophagia (Uchiyama, 2001) or necrosis (Tsukada et al., 2001;Yamashima, 2000). Autophagia and necrosis were also considered parallel to, and independent of, the “active,” energy-requiring, apoptotic machinery, of which caspases are central players. This dichotomy has been challenged by recent experiments that provided evidence of a direct involvement of cathepsins in apoptosis. Cathepsin B is released from lysosomes after activation of specific apoptotic inducers, such as activation of death receptors of the tumor necrosis factor family (Foghsgaard et al., 2001;Guicciardi et al., 2000), during oxidative stress (Nilsson et al., 1997), growth factor starvation (Brunk and Svensson, 1999), or after p53 (Yuan et al., 2002) or sphingosine (Kagedal et al., 2001) activation. Inhibition of cathepsin B can fully rescue cells from tumor necrosis factor—induced apoptosis, demonstrating that the release of cathepsin B to the cytosol can act as a dominant executioner of apoptosis (Foghsgaard et al, 2001). Cathepsin B proteolytic effects include the activation of Bax and Bak, two proapoptotic members of the Bcl-2 family, and subsequent release of cytochrome c through permeabilization of the mitochondrial membrane (Boya et al, 2003;Guicciardi et al., 2000). Cytochrome c is a key activator of the apoptosome, which regulates the mitochondrial caspase activation pathway mediated by caspase-9 (Reed, 2001). As a whole, these results indicate that in addition to their role in autophagia and necrosis, lysosomal proteases also influence cell fate by recruitment of apoptotic components.
The involvement of cathepsin B in ischemic cell death has been demonstrated in mild models of transient focal (Seyfried et al., 1997) and global (Hill et al., 1997;Kohda et al., 1996;Yamashima et al., 1998) ischemia, where the protease participates to reperfusion-associated damage. Our results show that cathepsin B is also released from lysosomes in acute focal ischemia, in the absence of reperfusion, suggesting that lysosomal destabilization is part of the primary events that lead to cerebral infarction. This is confirmed by the neuroprotective effect of the cathepsin B inhibitor CA-074. The origin of cathepsin B release from lysosomes in an ischemic context remains to be clarified. In an attractive hypothesis, Yamashima and collaborators suggest that degradation of lysosomal membrane is induced by activation of calcium-dependent proteases of the calpain family after pathological increase of intracellular calcium contents (Yamashima et al., 1998). The cytoplasmic activity of cathepsin B observed in our study can result from the rapid intracellular calcium loading that follows arterial occlusion (Choi, 1995). Alternatively, cathepsin B release can be induced by caspase-8 (Guicciardi et al., 2000), which is activated in this model with a time course and distribution pattern similar to those observed for cathepsin B (Benchoua et al., 2001).
Activation of proinflammatory caspases, such as caspase-1 and caspase-11, or its human counterpart, caspase-4, in ischemia, has been mainly associated with blood recirculation (Ferrer and Planas, 2003;Kang et al., 2000;Rothwell, 2003). We show here that caspase-11 is also activated in the early stages of infarction, in the absence of reperfusion of the damaged territory, and confirm previous results on caspase-1 (Benchoua et al., 2001). The fact that caspase-11 can trigger the activation of caspase-1 in stroke has been proven in mice with genetic invalidation of the caspase-11 gene, which display defective caspase-1 activation after transient MCAO (Kang et al., 2000). That cathepsin B inhibition abolished pMCAO-induced activation of both caspase-11 and caspase-1 suggests the existence of a causal link between cathepsin B cytosolic release and proinflammatory caspase activation. Although the triggers of caspase-11 were previously ill-defined, several activators of caspase-1 have been described in ischemic insults, including formation of the inflammasome, a multiproteic complex that includes caspase-1 and caspase-5 (Tschopp et al., 2003), the Rip2/Cardiak/Rick pathway (Zhang et al, 2003), and caspase-8 (Benchoua et al, 2001). Resumption of caspase-1 activity with CA-074 treatment suggests that the cathepsin B—caspase-11 pathway is an important trigger of caspase-1 activation in the first steps of infarction.
Although not directly in the scope of this study, it is noteworthy that early activation of caspases by pMCAO was paralleled by increase in caspase precursor content. Transcriptional upregulation of caspase genes is well documented in apoptosis and has been reported in cerebral ischemia (Honkaniemi et al., 1996;Krupinski et al., 2000), including in a model of permanent MCAO (Harrison et al., 2000). However, to our knowledge, no data were available in the early steps of focal ischemic degeneration. We have previously described in this model a slight but significant decrease of caspase-3 mRNA 30 minutes after pMCAO in correlation with increases in enzymatic activity and p20 protein contents (Benchoua et al., 2001), which can explain upregulation of the proform. This suggests that arterial occlusion can rapidly trigger the processing of mRNA into caspase precursors.
In conclusion, the demonstration that cathepsin B directly activates caspase-11 and/or caspase-1 pathways after pMCAO adds to the proapoptotic functions of cathepsins in vivo, and confirms that apoptosis, necrosis, and autophagia, are interrelated mechanisms that can act in synergy or as a fail-safe system to precipitate cell death in acute pathologic conditions. This dual involvement also provides molecular clues to explain the composite morphologic features of cell death in focal ischemic lesions.
Footnotes
Acknowledgment
The authors thank Professor J. Yuan (Boston, MA, U.S.A.) for providing the anti—caspase-11 antibodies used in this study.