
Abstract
The pathobiology of traumatic brain injury (TBI) includes activation of multiple caspases followed by cell death with a spectrum of apoptotic phenotypes. There are initiator (e.g. caspase-2, −8, and −9) and effector (e.g. caspase-3 and −7) caspases. Recently, caspase-2 and −8 have been shown to regulate cell death via provoking cytochrome c release from the mitochondria upstream of caspase-9. Here, we show that an intracerebral injection of the pan-caspase inhibitor boc-Aspartyl(OMe)-fluoromethylketone (BAF; 1 μmol) 1 min after TBI in rats reduces caspase-3-like activity, terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) and tissue damage, and cytochrome c release in ipsilateral cortex at 24 h versus vehicle. To investigate whether either caspase-2 and/or caspase-8 activation may contribute to cytochrome release, the effect of BAF treatment on caspase-2 and caspase-8 proteolysis was also examined. boc-aspartyl(OMe)-fluoromethylketone treatment inhibited proteolysis of caspase-2 but not caspase-8 24 h after TBI in rats versus vehicle. However, BAF with or without nerve growth factor (12.5 ng/hx14 days intracerebrally via osmotic pump) did not result in differences in motor function, Morris water maze performance, hippocampal neuron survival, nor contusion volume at 14 days. These data suggest that BAF treatment reduces acute cell death after TBI by inhibiting mitochondrial release of cytochrome c, possibly via a mechanism involving initiator caspases; however, BAF appears to delay cell death, rather than result in permanent protection.
Introduction
The pathobiology of traumatic brain injury (TBI) includes activation of multiple caspases followed by cell death with a spectrum of apoptotic phenotypes (Beer et al, 2001; Clark et al, 2000; Keane et al, 2001; Knoblach et al, 2002; Larner et al, 2004; Pike et al, 1998; Yakovlev et al, 1997). There are both initiator (e.g. caspase-2, −8, and −9) and effector (e.g. caspase-3 and −7) caspases (Eldadah and Faden, 2000). Recently, initiator caspases have been shown to be upstream regulators of cytochrome c release from the mitochondria. Under conditions of cell stress, caspase-2 permeabilizes the outer mitochondrial membrane facilitating cytochrome c release, an event requiring caspase-2 proteolysis but independent of its enzymatic activity (Lassus et al, 2002; Robertson et al, 2004). Activated caspase-8 can also result in cytochrome c release via truncation of the proapoptotic Bcl-2 family member Bid, with translocation of truncated Bid (tBid) into mitochondria (Henshall et al, 2001).
Several studies have now shown mitochondrial cytochrome c release in multiple rodent models of TBI, including controlled cortical impact (Sullivan et al, 2002), cryogenic injury (Morita-Fujimura et al, 1999), and impact-acceleration injury (Buki et al, 2000). Recently, we reported release of cytochrome c into cerebrospinal fluid in infants and children after severe TBI (Satchell et al, 2005), extending this phenomenon from experimental models to humans after TBI. Inhibition of cytochrome c release can be achieved using the immunophilin Cyclosporin A (Snyder et al, 1998). Cyclosporin A administration has been shown to result in many salutary effects after TBI, including preservation of mitochondrial function (Sullivan et al, 1999), and reductions in axonal (Buki et al, 1999) and cortical (Scheff and Sullivan, 1999) damage.
Relatively selective caspase-3 inhibitors such as N-benzyloxycarbonyl-Asp(Ome)-Glu(Ome)-Val-Asp(Ome)-fluoromethylketone (DEVD) have been reported to improve functional outcome and reduce neurological damage after experimental TBI (Clark et al, 2000; Knoblach et al, 2004; Yakovlev et al, 1997), as has the nonselective tri-peptide caspase inhibitor N-benzyloxycarbonyl-Val-Ala-Asp(OMe)-fluoromethylketone (VAD) (Knoblach et al, 2002). Another nonselective, pan-caspase inhibitor is the uni-peptide boc-aspartyl(OMe)-fluoro-methylketone (BAF). Systemic and local administration of BAF has been shown to be protective after cerebral ischemia (Cheng et al, 1998). boc-aspartyl(OMe)-fluoromethylketone prevents neuronal death in vitro (Allen et al, 1999), but requires nerve growth factor (NGF) rescue afterward for neurons to be functional (Werth et al, 2000). We are unaware of published reports where BAF was tested after experimental TBI in vivo.
The objective of this study was to determine the acute and chronic effects of local administration of the pan-caspase inhibitor BAF after controlled cortical impact (CCI) in adult male rats. Long-term studies included rats with or without NGF rescue, to parallel in vitro studies by other investigators (Werth et al, 2000).
Materials and Methods
Experimental Model of Traumatic Brain Injury
All studies were approved by the University of Pittsburgh Animal Care and Use Committee. Adult male Sprague–Dawley rats weighing 280 to 400 g had free access to food and water before and after surgery. A model of CCI with imposed secondary insult (moderate hypoxemia) was used as previously described (Clark et al, 1997). Briefly, rats were anesthetized with 4% isoflurane (Anaquest, Memphis, TN, USA) in O2. The trachea was intubated with a 14-gauge angiocatheter, and the lungs were mechanically ventilated with 2.0% isoflurane/66% N2O/balance O2. A femoral arterial catheter was inserted for continuous monitoring of blood pressure and intermittent arterial blood sampling. A rectal probe was inserted to monitor core temperature. A craniotomy was made over the left parietal cortex. A 0.003-inch outside diameter temperature probe (Physiotemp Corp., Clifton, NJ, USA) was inserted through a burr hole into the left parietal cortex to continuously monitor brain temperature. Rats were then warmed to a brain temperature of 37°C ± 0.5°C and allowed to equilibrate under anesthesia (1.1% isoflurane/66% N2O/balance O2) for 30 mins. After removal of the bone flap, injury was produced using the CCI device (Lighthall, 1988). For all studies depth of penetration of 2.5-mm, a velocity of 4.0 m/secs, and a duration of deformation of 50 ms were used. To produce moderate hypoxemia, air and oxygen were blended to achieve an FiO2 of 0.11 (1.1% isoflurane/74% N2O/19% air/6% O2) 1 min after CCI. This produces a PaO2 in the rats of 44 ± 1 mm Hg and a 40% reduction in mean arterial blood pressure by 30 mins (Clark et al, 1997). Hypoxemia was maintained for a total of 30 mins during which time the bone flap was replaced. Brain temperature was maintained at 37°C ± 0.5°C during this period. After 30 mins lines and monitors were removed, anesthesia was discontinued, and rats were allowed to awaken. Rats were extubated, placed in supplemental oxygen for 30 mins, and then returned to their cages. Sham rats received identical anesthesia, instrumentation and monitoring, and craniotomy, but did not undergo CCI or hypoxemia.
In Situ Detection of Caspase Activity
Caspase activation in situ was detected using the fluorescent probe fluorescein-5-isothiocyanate-VAD-fluoro-methylketone (FITC-VAD.fmk). Coronal sections were washed then incubated in a 1:50,000 dilution of FITC-VAD.fmk (Promega, Madison, WI, USA). Bis-benzimide was also applied to label nuclei. Slides were then coverslipped and examined using a fluorescent microscope. Images were collected using excitation/emission wavelengths of 494/520 (green) and 346/460 (blue) Λ for FITC-VAD emission and bis-benzimide, respectively.
Acute Effects of boc-aspartyl(OMe)-fluoromethylketone Injection After Controlled Cortical Impact
To determine the acute effect of BAF after CCI, rats (n = 4/group) were randomized to receive vehicle (20% dimethyl sulfoxide, DMSO, in artificial cerebrospinal fluid, aCSF = 127 mmol/L NaCl, 2.55 mmol/L KCl, 1.26 mmol/L CaCl2, 2.01 mmol/L MgCl2, pH 7.2 to 7.4), or 100, 500, or 1000 nmol BAF (Enzyme Systems Products, Livermore, CA, USA). Immediately after CCI, 5 μL of vehicle or BAF was injected intracerebrally via a Hamilton syringe, directly into the center of the contusion to a depth of 2.5-mm at a rate of 1 μL/min, with the remainder of the protocol as above.
Caspase-3-Like Enzyme Activity: Caspase-3-like enzyme activity was measured using the ApoAlert kit (Clontech, Palo Alto, CA, USA). Rats treated with 1000 nmol BAF chosen based on dose–response studies above or vehicle (n = 5/group) were anesthetized and perfused with ice-cold saline. The ipsilateral and contralateral cortex and hippocampus were dissected and stored at −70°C for batch analysis. Protein was extracted from each sample as described for Western blot analysis of whole-cell lysates (Clark et al, 2000). Fifty microliters of reaction buffer containing 10 mmol/L DTT were added to each sample (50 mg protein) and placed on ice. Each sample was incubated with 5 μL of the caspase-3 substrate DEVD-p-nitroanalide at 37°C for 1 h. Cleavage of the chromophore from the substrate was detected spectrophotometrically at a wavelength of 405 nm. The trauma-induced increase in caspase-3-like enzyme activity was determined by subtracting values obtained from the ipsilateral hemisphere from the contralateral hemisphere, and normalized per microgram protein calculated using a Bradford based assay.
Terminal Deoxynucleotidyl Transferase-Mediated dUTP-Nick End Labeling and Tissue Damage: At 24 h after CCI BAF or vehicle treated rats (n = 4/group) were anesthetized, perfused with 4% paraformaldehyde, and killed. The brains were removed, post-fixed in 4% paraformaldehyde, and paraffin-embedded. Brains were then cut into 5-μm coronal sections through the contusion, and mounted on glass slides. Sections were then processed for terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) and counter-stained with cresyl violet as described (Clark et al, 2000). Histologic assessment, performed by observers masked to treatment group, included qualitative scoring of TUNEL in cortex (5 = significant tissue loss with TUNEL, 4 = moderate tissue loss with TUNEL, 3 ≥ 20 TUNEL positive cells/40 x field, 2 = 11 to 20 TUNEL positive cells/40 x field, 1 = 1 to 10 TUNEL positive cells/40 x field, 0 = no TUNEL), and counting the number of surviving and TUNEL positive neurons in the CA1 and CA3 hippocampal regions.
Separation of Cellular Proteins: Separation of cytosolic, nuclear, and mitochondrial proteins was performed as previously described (Zhang et al, 2002). At 24 h after CCI BAF or vehicle treated rats (n = 4/group) were anesthetized with 4% isoflurane via nose-cone, then perfused with 200 mL ice-cold saline. Brains were removed and the ipsilateral hippocampus and cortex including and adjacent to the contusion were dissected on ice. Each sample was homogenized in lysis buffer then centrifuged at 1025g for 15 mins at 4°C, with the supernatants containing mitochondrial and cytosolic proteins and the pellets containing nuclei. Nuclear pellets were resuspended in buffer, incubated on ice for 15 mins, and centrifuged at 16,000g at 4°C for 20 mins. These supernatants were transferred and stored with 50 μL/mL glycerol at −80°C. The mitochondrial/cytosolic fractions were centrifuged at 735g at 4°C for 10 mins, these supernatants were further centrifuged at 10,000 g at 4°C for 15 mins to pellet mitochondria. The supernatants from mitochondrial pre-parations were centrifuged at 100,000g at 4°C for 1 h, and the final supernatants containing cytosolic proteins were stored at −80°C in 10% glycerol. The mitochondrial pellets were resuspended in 3% Ficoll solution containing 120 mmol/L mannitol, 30 mmol/L sucrose, and 25 μmol/L ethylenediaminetetraacetic acid (EDTA), and were layered on top of a 6% Ficoll solution containing 240 mmol/L mannitol, 60 mmol/L sucrose, 50 μmol/L EDTA. Ficoll gradients were centrifuged at 16,000g at 4°C for 25 mins and the pellets containing mitochondria were resuspended with cell lysis buffer. Protein concentration of lysates was determined with a Bradford-based protein assay (Bio-Rad). The suspensions were stored at −80°C until batch analysis.
Western Blot Analysis: Electrophoretically separated proteins were incubated in primary antibodies against cytochrome c (Clone 7H8.2C12; BD Pharmingen, San Diego, CA, USA) at a dilution of 1:5000, apoptosis-inducing factor (AIF; D20; Santa Cruz Biotechnology, Santa Cruz, CA, USA) at a dilution of 1:200, caspase-2 at a dilution of 1:200 (H-119, Santa Cruz Biotechnology), or caspase-8 at a dilution of 1:200 (SC 7890, Santa Cruz Biotechnology) for 1 h at room temperature. Washed membranes were incubated in the appropriate horseradish peroxidase-conjugated secondary antibody and immunoblotted proteins were detected using chemiluminescence (NEN, Boston, MA, USA). To assess efficiency of separation of cellular compartments and rule out nonspecific release of mitochondria-associated proteins, immunoblotting for cytochrome c oxidase (Clark et al, 2000) was also performed. Protein abundance was determined by measuring the relative optical density of the respective protein bands using a Kodak Image Station 440CF (Kodak, Rochester, NY, USA).
Functional and Histopathologic Effects of boc-Aspartyl(OMe)-fluoromethylketone Injection After Controlled Cortical Impact
To determine the chronic effect of BAF with or without NGF rescue after CCI, rats were block randomized to one of eight treatment groups: Group 1, sham + vehicleBAF + vehicleNGF; Group 2, sham + BAF+ vehicleNGF; Group 3, sham +vehicleBAF + NGF; Group 4, sham + BAF+ NGF; Group 5, CCI +vehicleBAF +vehicleNGF; Group 6, CCI + BAF + vehicleNGF; Group 7, CCI + vehicleBAF + NGF; Group 8, CCI + BAF + NGF. For each sham group n = 5 and for each CCI group n = 10. Immediately after CCI or sham injury 1000nmol BAF or vehicleBAF (20% DMSO) was injected intracerebrally as described above. All rats then underwent placement of an osmotic pump (ALZET Osmotic Pump Model 2002; 235.5 μL volume, 0.5 μL/h infusion rate, 14-day duration; Alza, Palo Alto, CA, USA) as per the manufacturer's instruction. The pump was placed subcutaneously into the rat's back and connected to a brain infusion catheter through a 1-mm burr hole drilled through the center of the bone flap. The bone flap with infusion catheter was then lowered into the ipsilateral hemisphere to a final depth of 2.5 mm, and was held in place using cyanoacrylate glue and 2 anchor screws in the left parietal bone outside the craniotomy. The osmotic pumps contained 2.5S NGF (25 μg/mL in aCSF; Harlan Bioproducts for Science, Indianapolis, IN, USA) or vehicleNGF (25 μg/mL mouse albumin in aCSF; Sigma, St Louis, MO, USA), to deliver 12.5 ng/h NGF. The NGF dose was based on previous studies showing protective effects of NGF after experimental TBI (Dixon et al, 1997; Sinson et al, 1997).
Functional outcome assessment included beam balance and beam walking tests of motor function on days 1 to 5 and spatial memory acquisition using the Morris water maze on days 8 to 14. Assessment of gross vestibulomotor function using a beam-balance task was performed by an observer masked to experimental condition. Rats were placed on a suspended, wooden beam (1.5-cm wide) and the duration that the rat remained on the beam (up to 60 secs) was measured. Training consisted of three trials that served as baseline measures. Assessment of finer components of vestibulomotor function and coordination using a beam-walking task was performed by an observer masked to experimental condition. Two days before injury, rats were trained to escape a bright light and loud noise by traversing a narrow wooden beam (2.5 × 100.0 cm2) with interspaced pegs and entering a darkened goal box at the opposite end of the beam. Rats were placed at one end of the beam close to the source of light and noise. The noise and light were terminated when the rat entered the goal box. Performance was assessed by measuring the time required to traverse the beam and enter the goal box.
A spatial memory acquisition paradigm of the Morris water maze was used for cognitive assessment. The maze employed a pool (180 cm diameter) filled with water to a depth of 28 cm. A clear Plexiglas platform below the surface of the water was used as a hidden goal platform. The pool was located in a room with numerous extra-maze cues that remained constant during the experimental studies. A 1-week acquisition block consisting of 4 daily trials over 7 consecutive days was used. On each acquisition trial, an observer masked to experimental condition placed each rat in the pool facing the wall. Rats began a trial from each of the four possible start locations in random order. The location of the platform was held constant for each rat. The first 5 days consisted of an aquisition task in which the platform was hidden beneath the surface of the water. The final 2 days consisted of a visible (cued) platform task in which the platform was raised 2 cm above the water surface. The visible platform task controls for potential nonspecific deficits in visual and motor function. Rats were given up to 120 secs to find the hidden platform. If the rat failed to find the platform after 120 secs, it was placed on the platform. Rats were allowed to remain on the platform for 30 secs before being placed in a heated incubator between trials (4-mins inter-trial interval).
After completion of functional outcome testing (day 14), rats were anesthetized, perfused with heparinized saline and 4% paraformaldehyde. Brains were removed, post-fixed at 4°C, and cryoprotected in 30% sucrose. Coronal sections (10-μm) were cut through the entire brain at 1-mm intervals from the occiput using a cryostat, mounted on glass slides, and stained with cresyl violet. In each serial section, the margins of the contusion and each hemisphere were outlined using an image analysis system (MCID, Imaging Research, St Catherine's, ON, USA) by an observer masked to experimental group. Contusion volume was calculated as the sum of each corresponding area in the 1-mm serial sections. Dorsal hippocampal neuron survival in brain regions underlying the contusion was determined in a coronal section underlying the center of impact, approximately 2.8-mm posterior to bregma. Neurons were counted in the entire anatomic CA1 and CA3 hippocampal regions by an observer masked to experimental group. Normal appearing neurons with a well-defined nucleus and cell body were counted in each hemisphere, and were discriminated from non-neuronal cells based on size and morphologic criteria as previously described (Clark et al, 1997, 2000). Neuronal cell counts are expressed as the percentage of surviving neurons in the ipsilateral versus contralateral hemispheres.
Statistical Analysis
Data are presented as mean ± s.e.m. or as individual data points for nonparametric data (TUNEL score). A t-test was used for single time points or single-dose versus vehicle comparisons after passing tests of normality and equal variance, otherwise a Mann-Whitney test was used. Analysis of variance (ANOVA) with Bonferroni's post hoc test was used for other comparisons involving parametric data. ANOVA on ranks with Dunn's post hoc test was used for nonparametric data. Functional outcome tests were analyzed using two-way repeated measures ANOVA with Tukey's post hoc test. A P < 0.05 was considered significant.
Results
Increased In Situ Caspase Activity and Cytosolic Cytochrome c after Controlled Cortical Impact
The regional pattern of caspase activation was determined using FITC-VAD.fmk, which fluoresces on cleavage by caspases, as well as other proteases including calpains (Bizat et al, 2005). Increased cleavage of FITC-VAD.fmk in situ was detected in injured brain but not in naïve controls (Figure 1). Increased cleavage of FITC-VAD.fmk was most prominent in the contusion core, with the majority of cells positively labeled (Figure 1C). Scattered cells with increased cleavage of FITC-VAD.fmk were also seen in the pericontusional cortex and ipsilateral hippocampus (Figures 1B, 1E and 1F). Increased cleavage of FITC-VAD.fmk by this method was not detected in the contralateral hemisphere in any group.
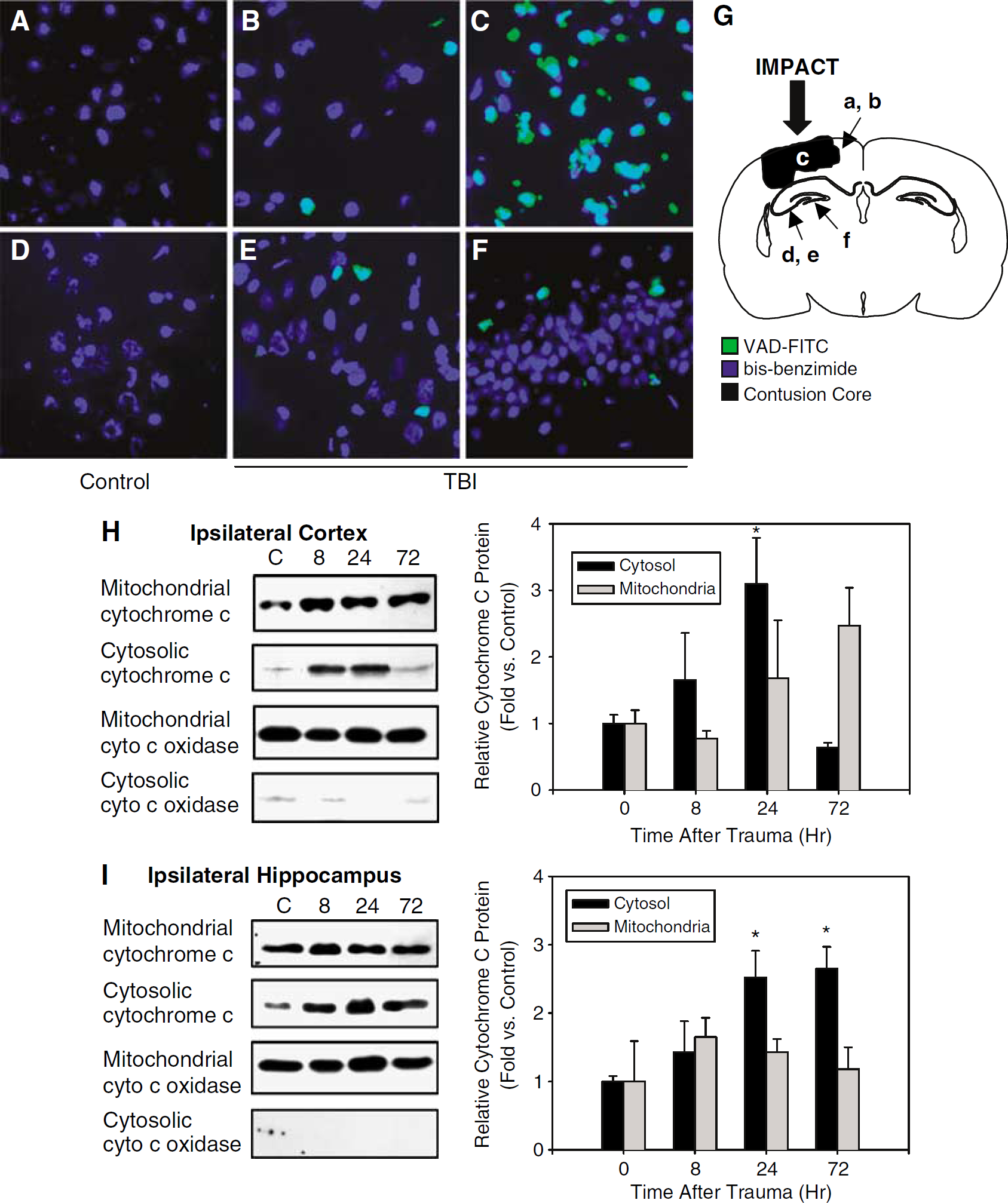
In situ detection of fluorescein-5-isothiocyanate-Val-Ala-Asp-fluoromethylketone (VAD-FITC; green) cleavage 24 h after controlled cortical impact (CCI) with secondary hypoxemic insult in rats. VAD-FITC cleavage was used as an indication of caspase and/or calpain activation. Sections were also stained with bis-benzamide (blue) to identify nuclei. In situ VAD-FITC cleavage was not detected in control tissue (
We also evaluated the time course of the release of cytochrome c into cytosol after CCI. Western blot analysis on subfractionated protein samples showed increased cytosolic cytochrome c in ipsilateral cortex at 24 h and ipsilateral hippocampus at 24 and 72 h after CCI compared with naïve controls (Figures 1H and 1I; P < 0.05). Baseline cytosolic cytochrome c was greater in the naïve hippocampal compared with the naïve cortical samples, although the magnitude of change after CCI was similar (3.1 ± 0.7-fold increase in cortex at 24 h versus control; 2.5 ± 0.4-fold increase in hippocampus at 24 h versus control). This was not associated with a concomitant reduction in relative abundance of mitochondrial cytochrome c; however, and therefore while consistent with mitochondrial release of cytochrome c after CCI these data are also consistent with increased cytochrome c production and increased cytosolic expression. Cytosolic levels of the integral mitochondrial membrane protein cytochrome c oxidase were minimally detectable in all groups, suggesting that increased cytosolic cytochrome c did not merely represent non-specific release of mitochondrial proteins.
boc-Aspartyl(OMe)-fluoromethylketone Reduces Caspase-3-Like Activity, Cytochrome c Release, and Terminal Deoxynucleotidyl Transferase-mediated dUTP Nick-end Labeling After Controlled Cortical Impact
A BAF dose-finding study was performed using caspase-3-like activity, TUNEL and tissue damage as the primary end point. Doses of 100, 500, and 1000 nmol were administered intracerebrally immediately after CCI. Only the 1000 nmol dose reduced caspase-3-like activity and TUNEL and tissue loss in injured cortex after CCI compared with vehicle treatment, as such the 1000 nmol dose was used for the remainder of the outcome studies. A dose of 1000 nmol BAF treatment reduced caspase-3-like enzyme activity by ~ 60% in ipsilateral cortex and hippocampus compared with vehicle treatment (Figure 1A; P = 0.03, t-test). BAF (1000 nmol) treatment reduced TUNEL and/or brain tissue damage in the ipsilateral hemisphere compared with vehicle treatment (Figures 2B–2D; P =0.02, Mann–Whitney rank-sum test). Cell counts in the CA1 and CA3 hippocampal regions were also performed in these sections. While significant hippocampal cell loss was observed in both CA1 and CA3 hippocampus at 24 h after CCI versus naïve controls, there was no effect of BAF treatment versus vehicle (data not shown).
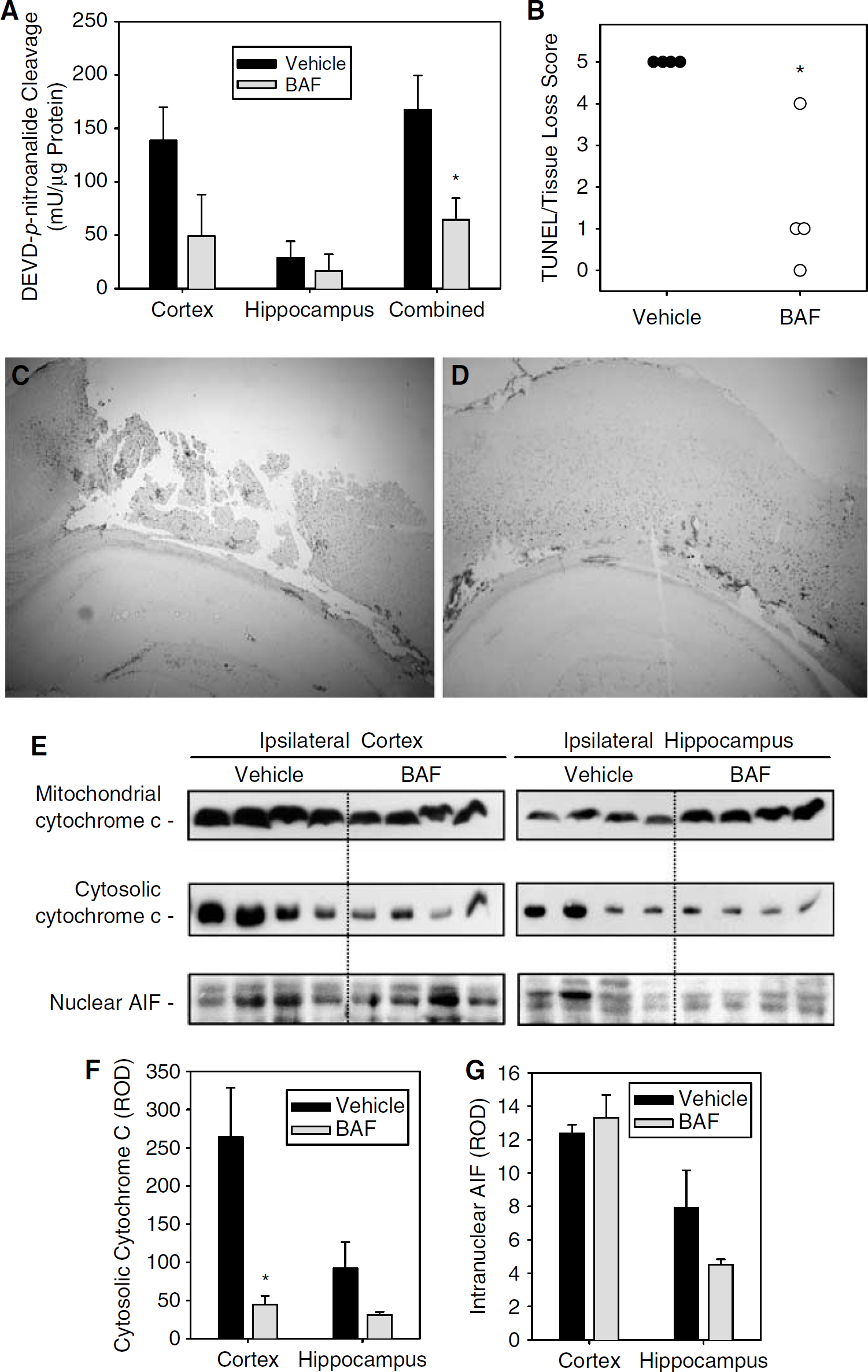
Biochemical and cellular effects of intracerebral BAF treatment after CCI with secondary hypoxemic insult in rats. (
Consistent with inhibition of caspase-dependent apoptotic cell death, BAF treatment reduced cytosolic cytochrome c abundance in ipsilateral cortex by >80% compared with vehicle treatment (Figures 2E and 2F; P < 0.05, t-test), but did not have a statistically significant effect in hippocampus. In contrast, BAF treatment did not have an effect on nuclear localization of AIF (Figures 2E and 2G), consistent with studies suggesting that AIF-mediated cell death is caspase-independent after TBI (Zhang et al, 2002). Caution is in order as sample sizes were limited and it is possible (or even likely) that larger sample sizes would reveal an effect of BAF treatment on cytosolic cytochrome c release and/or intranuclear AIF in hippocampus after CCI. However, it is evident that this dose of BAF does influence local biochemical changes after CCI.
boc-Aspartyl(OMe)-fluoromethylketone Reduces Caspase-2 But Not Caspase-8 Proteolysis After Controlled Cortical Impact
To determine whether the pan-caspase inhibitor BAF influenced caspases upstream of cytochrome c release, similar to in vitro studies (Lassus et al, 2002; Li et al, 1998; Robertson et al, 2004), cytosolic caspase-2 and caspase-8 proteolysis were evaluated 24 h after CCI in rats treated with 1000 nmol BAF or vehicle. Consistent with a regulatory role for caspase-2 in mitochondrial cytochrome c release, BAF treatment reduced caspase-2 proteolysis in ipsilateral cortex by > 50% compared with vehicle treatment, and also reduced caspase-2 proteolysis in ipsilateral hippocampus, but to a lesser degree than in cortex (Figures 3A and 3B; P < 0.05, t-test). In contrast, BAF treatment had no effect on caspase-8 proteolysis (Figures 3C and 3D). In this model, caspase-8 proteolysis was detected in injured cortex but not hippocampus at 24 h. However, in hippocampus, an apparent reduction in pro-caspase-8 in BAF versus vehicle treated rats after CCI was noted. Further study to investigate mechanisms of caspase-2 related cytochrome c release, and to verify a reduction in procaspase-8, with BAF (or other protease inhibitors) after TBI is warranted.
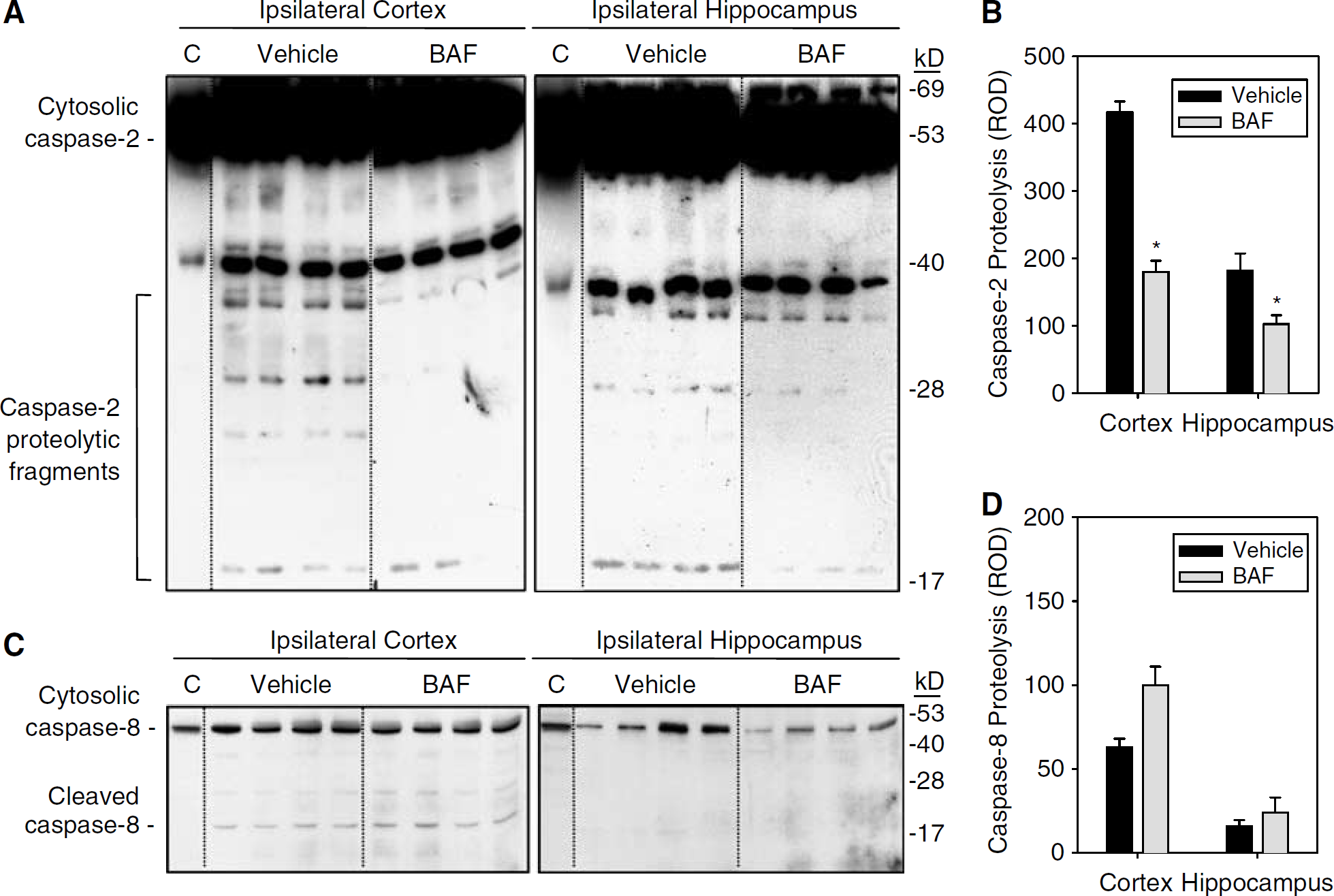
Effect of intracerebral BAF treatment on caspase-2 and −8 proteolysis after CCI with secondary hypoxemic insult in rats. Treatment with 1000 nmol BAF reduced proteolysis of caspase-2 (
boc-Aspartyl(OMe)-fluoromethylketone Does Not Influence Functional Outcome or Histologic Damage at 2 Weeks After Controlled Cortical Impact
To determine whether a single dose of BAF was sufficient to influence outcome after CCI, rats were randomized to receive 1000 nmol of BAF intracerebrally with or without NGF rescue. Nerve growth factor rescue was included based on in vitro studies showing that trophic factors are required to restore function in neural cells after serum deprivation (Werth et al, 2000), and in vivo studies demonstrating protective effects of NGF after TBI in rats (Dixon et al, 1997; Sinson et al, 1997). Sham rats were utilized in these functional outcome studies to examine for direct effects of intracerebral BAF (and NGF) on behavior.
All rats survived the injury, treatment, and 2-week outcome protocols. No differences were detected between treatment groups in sham injured rats; therefore, these data were combined for presentation in Figure 4 and statistical analysis. As expected, CCI produced typical motor dysfunction and impaired spatial memory acquisition assessed using the Morris water maze compared with sham injured rats (Figures 4A and 4B; P < 0.05 for all CCI groups versus sham in beam walking, and Morris water maze latency, two-way repeated measures ANOVA with Tukey's post hoc test); however, no differences were detected between treatment groups in CCI injured rats. Results using the beam balance test measuring the time up to 60 secs that the rat can balance on the suspended wooden beam (data not shown), were similar to the beam walking test measuring the time it takes for the rat to traverse the beam and enter the escape box. Similarly, no treatment effect was observed in contusion volume or CA1 and CA3 hippocampal neuron survival after CCI (Figures 4C and 4D).
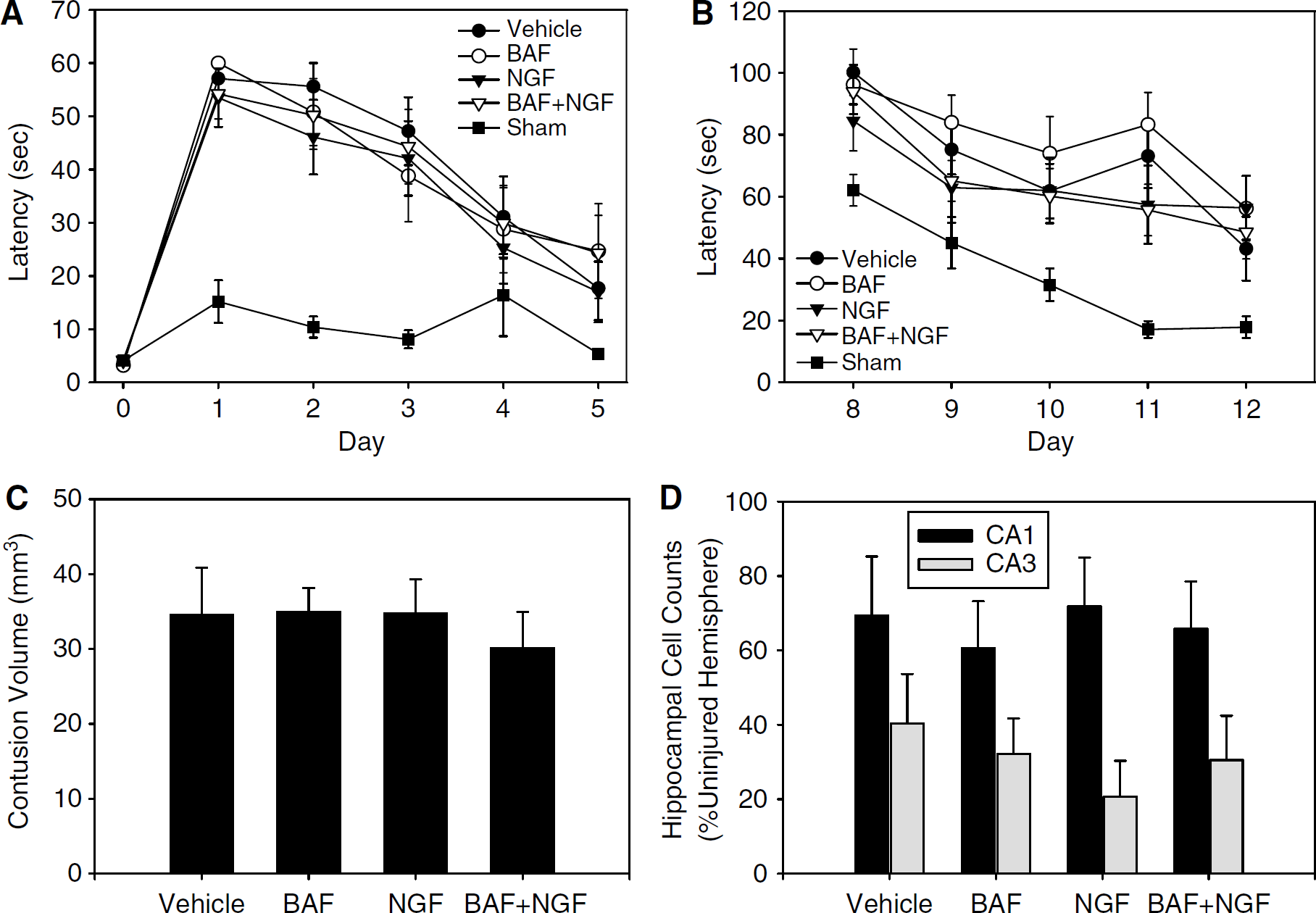
Functional and histologic outcome effects of intracerebral BAF treatment with or without NGF rescue after CCI with secondary hypoxemic insult in rats. Rats were treated with BAF (1000nmol x 1), NGF (12.5 ng/h x 14days), or appropriate vehicles in all combinations. Typical deficits in beam walking latency (
Discussion
The principal finding of this study is that cytochrome c release appears to be regulated in part by upstream caspase activation after TBI in vivo. Regulation of cytochrome c release by caspase-2 is supported by our findings showing that the pan-caspase inhibitor BAF reduces release of cytochrome c and caspase-2 proteolysis, as well as brain tissue loss and DNA fragmentation (TUNEL), acutely after CCI. The effect of BAF on cytochrome c release seemed to be more prominent in ipsilateral cortex versus hippocampus in this model of TBI. Despite a reduction in caspase-2 proteolysis, cytosolic cytochrome c, and caspase-3 activity in injured cortex with BAF treatment, these biochemical effects did not translate into improved functional or long-term histologic outcome—even when supplemented with NGF administration.
Recent in vitro studies have shown an important role for caspase-2 in regulation of cytochrome c release. Cytokine- and cell stress-induced apoptosis results in caspase-2-mediated permeabilization of the outer mitochondrial membrane (Lassus et al, 2002), disruption of cytochrome c binding to anionic phospholipids (Enoksson et al, 2004), and subsequent cytochrome c release. This phenomenon requires caspase-2 proteolysis but is independent of its enzymatic activity (Robertson et al, 2004). Caspase-2 deficient mice exhibit dysregulation of apoptosis during development, and interestingly can act as both positive and negative cell death effectors (Bergeron et al, 1998). A role for caspase-2 has been previously implicated in models of experimental TBI (O'Dell et al, 2000) and cerebral ischemia (Omori et al, 2002).
Caspase-8 may act upstream of cytochrome c release as well. Activated caspase-8 can proteolyze Bid to produce tBid, enabling mitochondrial translocation and cytochrome c release via the extrinsic Fas-mediated cell death pathway (Li et al, 1998). We did not observe an effect of BAF treatment on caspase-8 proteolysis in our model; although caspase-8 proteolysis was only detected in ipsilateral cortex, not hippocampus, at 24 h after injury. The Fas-pathway is operative after TBI in both experimental models and in humans (Beer et al, 2001; Franz et al, 2002; Keane et al, 2001; Qiu et al, 2002; Zhang et al, 2003). We also performed Western blots using several anti-Bid antibodies that were commercially available at the time of the study, but could not detect tBid in our model of TBI (data not shown). As such, we cannot rule out a role for Bid after TBI.
It is well established that cytochrome c release occurs after TBI in multiple experimental models (Buki et al, 2000; Lewen et al, 2001; Morita-Fujimura et al, 1999; Sullivan et al, 2002) and in humans (Satchell et al, 2005). Released cytochrome c engages with caspase-9 and apoptotic protease-activating factor-1 (Apaf-1) to form an ‘apoptosome’ that activates caspase-3 and results in cellular changes producing apoptotic cell death (Li et al, 1997; Zou et al, 1997). Several relatively selective caspase-3 inhibitors such as DEVD have been reported to be neuroprotective after experimental TBI, used primarily to target events downstream from cytochrome c release (Clark et al, 2000; Knoblach et al, 2004; Yakovlev et al, 1997). Fewer studies have tested the effects of less selective caspase inhibitors after TBI, which could be expected to have effects both upstream and downstream from cytochrome c release. Knoblach et al (2002) used the nonselective tri-peptide caspase inhibitor VAD and reported improvement in composite neurological scores and Morris water maze performance compared with vehicle treatment. boc-Aspartyl(OMe)-fluoromethylketone is a uni-peptide nonselective caspase inhibitor; however, despite impressive effects on cytochrome c release, caspase-2 proteolysis, caspase-3 activation, and early brain tissue preservation, a similar improvement in functional outcome was not observed in our study.
It is important to note that recent work has shown that the caspase inhibitors VAD and DEVD also bind to and inhibit calpains (Bizat et al, 2005; Knoblach et al, 2004). As such, a portion of the in situ cleavage of FITC-VAD (Figure 1) and ex vivo cleavage of DEVD-p-nitroanalide (Figure 2A) could have been secondary to activation of calpains after TBI. Furthermore, it is also possible that the effects of BAF after CCI are also in part due to calpain inhibition, or that the incomplete inhibition of DEVD-p-nitroanalide cleavage by BAF after TBI may be related to calpain activation. The latter would support testing a combination of relatively selective calpain and caspase inhibitors after TBI.
Despite a > 60% reduction in caspase-3 activity and > 80% reduction in cytosolic cytochrome c in injured cortex with BAF treatment, these biochemical effects did not translate into improved functional outcome or long-term histologic outcome assessed at 2 weeks. There are several possibilities for this lack of treatment effect. First, a single-dose of the pan-caspase inhibitor BAF given after injury may be insufficient to provide long-standing protection in cortex, where the treatment effects were most prominent. In our previous study, DEVD was administered intracerebrally over 24 h via an osmotic pump, resulting in decreased tissue damage but no effect on functional outcome (Clark et al, 2000). After fluid-percussion injury in rats, Yakovlev et al (1997) administered DEVD intracerebroventricularly before and after fluid-percussion injury, resulting in improved functional outcome, although an effect on histologic outcome was not reported. Second, it is possible that BAF may be less effective than other pan-caspase inhibitor such as VAD. In the study by Knoblach et al (2002), a single post-treatment dose of VAD (125 nmol/L) resulted in improved functional outcome. Third, it is possible that bioavailability of BAF was insufficient to effect cell death in hippocampus related to failure of drug delivery to subcortical regions, and that cell death in contused cortex was not amenable to treatment in this model. Fourth, pan-caspase inhibition may reduce cytochrome c release and caspase-3-mediated cell death, but not prevent cell death via caspase-independent, calpain-dependent, or necrotic pathways (Bizat et al, 2005; Knoblach et al, 2004; Lemaire et al, 1998; Los et al, 2002; Zhang et al, 2002). Fifth, caspase inhibitors may have important reparative and/or behavioral effects, related to effects on caspases or perhaps nonspecific effects on other enzymes, that would narrow the therapeutic window for this class of inhibitors. For example, caspase inhibitors have been shown to influence learning and memory (Dash et al, 2000), neurological functions very pertinent to preclinical trials in TBI, where the Morris water maze is a common outcome parameter (Hamm et al, 1992). Finally, more robust methods of cell counting, such as stereologic methods, may be necessary to more definitively determine the histopathologic effects of these inhibitors.
Based on in vitro studies testing combinations of BAF and NGF in neural cells after serum deprivation (Werth et al, 2000), we also included treatment arms using continuous infusion of NGF intracerebrally over the 14-day testing period. Despite in vivo studies demonstrating protective effects of NGF after fluid percussion injury (Sinson et al, 1997) and CCI (Dixon et al, 1997) in rats, NGF had no effect in our model, independent of whether BAF was also used. The dose and delivery paradigm used in our study was based on these previous reports (Dixon et al, 1997; Sinson et al, 1997), so the lack of effect was unexpected. It is possible that in this model of CCI with secondary hypoxemic insult that injury is either more severe, or different in scope than CCI alone, and not amenable to NGF treatment. It is also possible that NGF delivery in our study resulted in binding to low affinity p75 receptors that promote cell death, rather than trophic trk receptors (Kaplan and Stephens, 1994; Troy et al, 2002); although as mentioned this dosing regimen has been previously shown to be protective after both CCI and fluid-percussion injury (Dixon et al, 1997; Sinson et al, 1997).
In conclusion, this study suggests a role for caspase activation, possibly caspase-2, upstream of cytochrome c release in apoptosis induced by experimental TBI. Despite a lack of effect of singledose BAF treatment on functional or long-term histologic outcome, further study using different treatment paradigms or other caspase inhibitors still appears justified.
Footnotes
Acknowledgements
We appreciate generous support from the National Institutes of Health, National Institute of Neurologic Diseases and Stroke (RO1 NS38620 and P50 NS30318), the Department of Critical Care Medicine, University of Pittsburgh, and the Children's Hospital of Pittsburgh.