
Abstract
The generally accepted concept that astrocytes are highly resistant to hypoxic/ischemic conditions has been challenged by an increasing amount of data. Considering the differences in functional implications of protoplasmic versus fibrous astrocytes, the authors have investigated the possibility that those discrepancies come from specific behaviors of the two cell types. The reactivity and fate of protoplasmic and fibrous astrocytes were observed after permanent occlusion of the medial cerebral artery in mice. A specific loss of glial fibrillary acidic protein (GFAP) immunolabeling in protoplasmic astrocytes occurred within minutes in the area with total depletion of regional CBF (rCBF) levels, whereas “classical” astrogliosis was observed in areas with remaining rCBF. Severe disturbance of cell function, as suggested by decreased GFAP content and increased permeability of the blood–brain barrier to macromolecules, was rapidly followed by necrotic cell death, as assessed by ultrastructure and by the lack of activation of the apoptotic protease caspase-3. In contrast to the response of protoplasmic astrocytes, fibrous astrocytes located at the brain surface and in deep cortical layers displayed a transient and limited hypertrophy, with no conspicuous cell death. These results point to a differential sensitivity of protoplasmic versus fibrous cortical astrocytes to blood deprivation, with a rapid demise of the former, adding to the suggestion that protoplasmic astrocytes play a crucial role in the pathogenesis of ischemic injury.
Most of our knowledge about ischemia-related astrogliosis comes from models of transient blood deprivation, either global or focal. Global ischemia leads to selective neuronal loss, without brain infarction (reviewed in Schmidt-Kastner and Freund, 1991). In such cases, astroglial reactivity is delayed, associated with neuronal degeneration, and possibly associated with secondary hyperplasia (DeLeo et al., 1987; Gotti et al., 1990; Petito et al., 1990; Schmidt-Kastner et al., 1990; Gehrmann et al., 1992; Ordy et al., 1993; Petito and Halaby, 1993; Stephenson et al., 1995; Conway et al., 1998). Astrogliosis, featured by increased volume and increased immunoreactivity for glial fibrillary acidic protein (GFAP), persists in damaged areas for weeks after ischemia (Petito et al., 1990), without conspicuous cell loss. Similar characteristics are found in focal ischemia, which, in contrast to global arrest of blood flow, leads to severe brain infarction. In this case, astrogliosis occurs in periinfarct areas (Yamamoto et al., 1986; Yamada et al., 1994; Li et al., 1995; Liu et al., 1999), or in remote areas of secondary neuronal degeneration (Wu and Ling, 1998). It results in the formation of a glial scar surrounding the final area of pan necrosis (Chen et al., 1993; Schroeter et al., 1995; Duggal et al., 1997). These observations have led to the postulate that, thanks to their unique energy capacities, brain astrocytes can resist hypoxic/ischemic conditions better than neurons or other glial populations (Petito et al., 1998), a hypothesis supported by in vitro data (Goldberg et al., 1987; Tombaugh and Sapolsky, 1990; Petito et al., 1991; Goldberg and Choi, 1993; Kelleher et al., 1993; Lyons and Kettenmann, 1998).
However, the dogma of a strong resistance of astrocytes to ischemic conditions is challenged by an increasing amount of data corroborating the suggestion that astrocytic death can precede neuronal loss and aggravate ischemia-induced brain injury (Plum, 1983; Siesjö, 1984; Garcia et al., 1995). In models of transient (Garcia et al., 1993) or permanent (Liu et al., 1999) middle cerebral artery occlusion (MCAO), astrocytic demise precedes neuronal degeneration in selected brain areas. In vitro studies demonstrate that astrocytes are more vulnerable than neurons to degradation promoted by impairment of ATP synthesis (Fukuda et al., 1998). Other data show that although astrocytes are relatively resistant to hypoxia, they are highly sensitive to coupling of acidosis and hypoxia, a paradigm that parallels the progressive deterioration observed in the ischemic penumbra (Swanson et al., 1997; Bondarenko and Chesler, 2001). Finally, dying astrocytes may play a prominent role in the propagation and amplification of ischemia-induced brain lesions via gap junctions (Lin et al., 1998).
Although differences among ischemic models used in various studies may contribute to the discrepancy of astroglial responses observed, further consideration must be given to the existence of subpopulations with regional specificity at both molecular and functional levels (Denis-Donini et al., 1984; Miller and Raff, 1984). Although this dichotomy is currently questioned, astrocytes have been divided into two classes on the basis of their morphology, development, and cell surface antigens (Raff and Miller, 1984). Fibrous astrocytes, or type 2 astrocytes, are predominantly found in the white matter, and have a typical stellate morphology, whereas protoplasmic (type 1) astrocytes account for most of the astrocytes in the gray matter. Whether subpopulations of astrocytes respond to neurologic disease processes with particular sensitivity have not yet been determined.
The present study examines the responses and fate of protoplasmic and fibrous astroglial in a severe model of focal cerebral ischemia. Our results indicate that the two populations of astrocytes respond very differently in those conditions, the former only exhibiting rapid and massive cell death. This involvement of a vast and specific astroglial population should likely be taken into consideration in the development of therapeutics counteracting the effects of stroke in cortical areas.
MATERIALS AND METHODS
Experiments were conducted in respect to the French and European Community guidelines for the care and use of experimental animals.
Ischemia model
C57Bl/6 male mice (25 g, Janvier, France) were anesthetized with chloral hydrate (500 mg/kg) and received an electrocoagulation of the left MCA as previously described (Guégan et al., 1996). Sham-operated animals were subjected to the same surgical procedure without MCAO. Surgery was performed with the animals spontaneously breathing. Body temperature was continuously monitored with a homeothermic blanket control unit (Harvard Apparatus, Les Ulis, France), and was maintained at 37.0° ± 1°C until 1 hour after surgery. Animals were killed at 15 and 30 minutes, and at 1, 3, 12, and 24 hours after surgery. Nonoperated (NOp) animals were used as naive controls.
Immunohistochemistry
Eighteen operated (three for each time point), and two NOp mice were perfused by intracardiac administration of 4% paraformaldehyde in 0.1 mol/L phosphate-buffered saline (pH 7.4) under 4% chloral hydrate anesthesia. Brains were removed, post-fixed for 24 hours in the same fixative, cryoprotected and frozen. Twenty-micron-thick horizontal or coronal sections from NOp, sham-operated, and operated mice were made on a cryostat. Endogenous peroxidases were quenched with 3% H2O2, and nonspecific binding of antibodies was prevented by blocking for 1 hour with 1% serum albumin in phosphate-buffered saline. Sections were then processed for GFAP (cow polyclonal anti-GFAP antibodies, 1/10,000; Dako, Denmark) immunohistochemistry with a 24-hour incubation in phosphate-buffered saline containing 0.3% Triton X-100, followed by detection with Vectors' SG kit (Vector Laboratories, Burlingame, CA, U.S.A.). For double-labeling studies, sections were processed for active caspase-3 (p20, 1/2,000; R&D Systems, Minneapolis, MN, U.S.A.) immunohistochemistry using anti-rabbit Cy3-labeled secondary antibodies (1/1,000; NEN, Boston, MA, U.S.A.), followed by incubation in 0.1 mol/L glycine buffer pH 3.34 to remove any residual immunoglobulin G (Nakane, 1968), and were processed for GFAP immunohistochemistry using monoclonal anti-GFAP antibodies (Boehringer, France) with secondary antibodies coupled to fluorescein isothiocyanate (1/400, NEN). Sections were examined with a Zeiss fluorescence microscope, and images were acquired with a CoolSnap color camera (RS Photometrics, Tucson, AZ, U.S.A.). Double-labeled sections were examined with an Axiovert 135S Zeiss confocal microscope (LSM Micro Systems, Jena, Germany).
Biochemical analysis
GFAP content was analyzed at 1 and 3 hours after MCAO by Western blotting. Brain hemispheres from 12 operated (6 for each time point) and 3 NOp mice were frozen in dry ice, crushed, and lysed in 1 mL of ice-cold 50 mmol/L Tris buffer containing 1 mmol/L EDTA, 150 mmol/L NaCl, 0.5% Triton X-100, 1 mmol/L phenylmethylsulfonyl fluoride, and 5 μL of protease inhibitors cocktail (Sigma, St. Quentin Fallavier, France). Samples were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis and were transferred to a PVDF membrane (Guégan et al., 1999). Blots were probed with cow polyclonal anti-GFAP antibodies (as above, 1/2,000), followed by enhanced chemiluminescent reaction (ECL+, Amersham, Orsay, France). Blots were routinely stripped in a denaturating buffer (Tris HCl 0.5 mol/L, pH 6.8, sodium dodecyl sulfate 10%, β-mercaptoethanol 0.8%), and re-probed with anti-α-tubulin antibodies (1/5,000, Sigma) for 1 hour at room temperature as a loading control. Quantitative results are normalized to α-tubulin content, as described (Harrison et al., 2000).
Regional cerebral blood flow
Regional cerebral blood flow (rCBF) was measured with the iodo-14C-antipyrine (14C-IAP) quantitative method (Sakurada et al., 1978), on three mice 1 hour after MCAO. A polyethylene tubing was inserted into a femoral artery for tracer injection, and a second catheter was inserted into the contralateral femoral artery for sampling of arterial blood. Fifty minutes after MCAO, 5 μL of iodo-14C-antipyrine (dose MBq/kg) were injected during 30 seconds, and blood samples were collected at intervals of 2 seconds. After euthanasia by a high dose of pentobarbital (60 mg/kg), brains were rapidly removed and frozen in isopentane chilled to −80°C with dry ice. Tissues were sectioned with a cryostat in 20-μm slices and heat-dried. One of four adjacent sections was stained with cresyl violet for structural analysis, and the other sections were exposed to a Kodak film along with standards of known radioactivity. Regional cerebral blood flow was calculated from film optical densities of brain autoradiograms and standards corrected for arterial blood radioactivity.
Electron microscopy
Four mice (three operated and one NOp) were perfused with a chilled mixture of 2% paraformaldehyde and 3% glutaraldehyde in 0.1 mol/L cacodylate buffer (pH 7.4). Brains were cut at 100 μm with a Vibratome (Vibratome Co., St. Louis, MO, U.S.A.), and sections were washed in phosphate buffer and either stained for GFAP immunohistochemistry (one section of two), or directly postfixed in 1% osmium tetroxide for 1 hour. Sections were dehydrated in an ascending series of ethanol to 100%, and embedded in Taab 812 resin (Taab Laboratories, Reading, U.K.). Ultrathin sections were cut with an LKB UM5 ultramicrotome (Leica, Queil-Malmaisen, France), counterstained with uranyl acetate and lead citrate, and examined on a Philips (Limeil-Brevannes, France) CM10 electron microscope. Sections treated for GFAP immunocytochemistry as already described were washed and processed from the osmium tetroxide step to embedding and ultrasectioning. Ten randomly selected sections including core or penumbra areas were examined for each animal.
Leakage of the blood–brain barrier
Permeability of the blood-brain barrier was investigated 1 hour after MCAO (n = 3 for each molecule) for two molecules of different molecular weights (MW), the horseradish peroxidase (HRP, MW 40 kd) and immunoglobulin M (IgM) (MW 450 kd). Injection of 0.15 mL of a 5% solution of HRP (Type IV, Sigma) was performed 15 minutes before perfusion into the retroorbital vessel. Leakage of HRP into the brain parenchyma was visualized by incubating cryostat sections in 3,3′-diaminobenzidine tetrahydrochloride (0.01%, Vector Laboratories, France) with 0.002% hydrogen peroxide in Tris buffer (pH 7.6) for 20 minutes. Extravasation of systemic IgM into brain parenchyma was revealed by peroxidase immunohistochemistry as already described, after incubation of cryostat sections with biotinylated antibodies against mouse IgM (1/300, Vector Laboratories, France) for 2 hours.
RESULTS
GFAP immunolabeling and protein levels
In contrast to other brain regions, such as the hippocampus or the striatum, which mainly contain fibrous astrocytes, the somatosensory cortex contained a majority of protoplasmic (type 1) astrocytes, of characteristic bushy aspect with numerous short and highly ramified processes (Fig. 1). Fibrous (type 2) astrocytes were present in the deepest part of layer VI, close to the corpus callosum, and at the external border of the cortex, right below the pia matter (Figs. 1A and 1B). GFAP-IR was lost as early as 15 min following MCAO in layers II to IV (Fig. 1B). In contrast, protoplasmic astrocytes of layers V and VI had higher intensities of GFAP labeling. Hypertrophy and increased GFAP labeling was also observed in fibrous astrocytes of the corpus callosum, whereas no change was yet detected in fibrous astrocytes of the brain surface. Similar images were obtained 30 minutes after occlusion (not shown). The intensity of GFAP labeling in protoplasmic astrocytes of layers V to VI further increased at 1 and 3 hours after MCAO, but no recovery was observed in the upper layers.
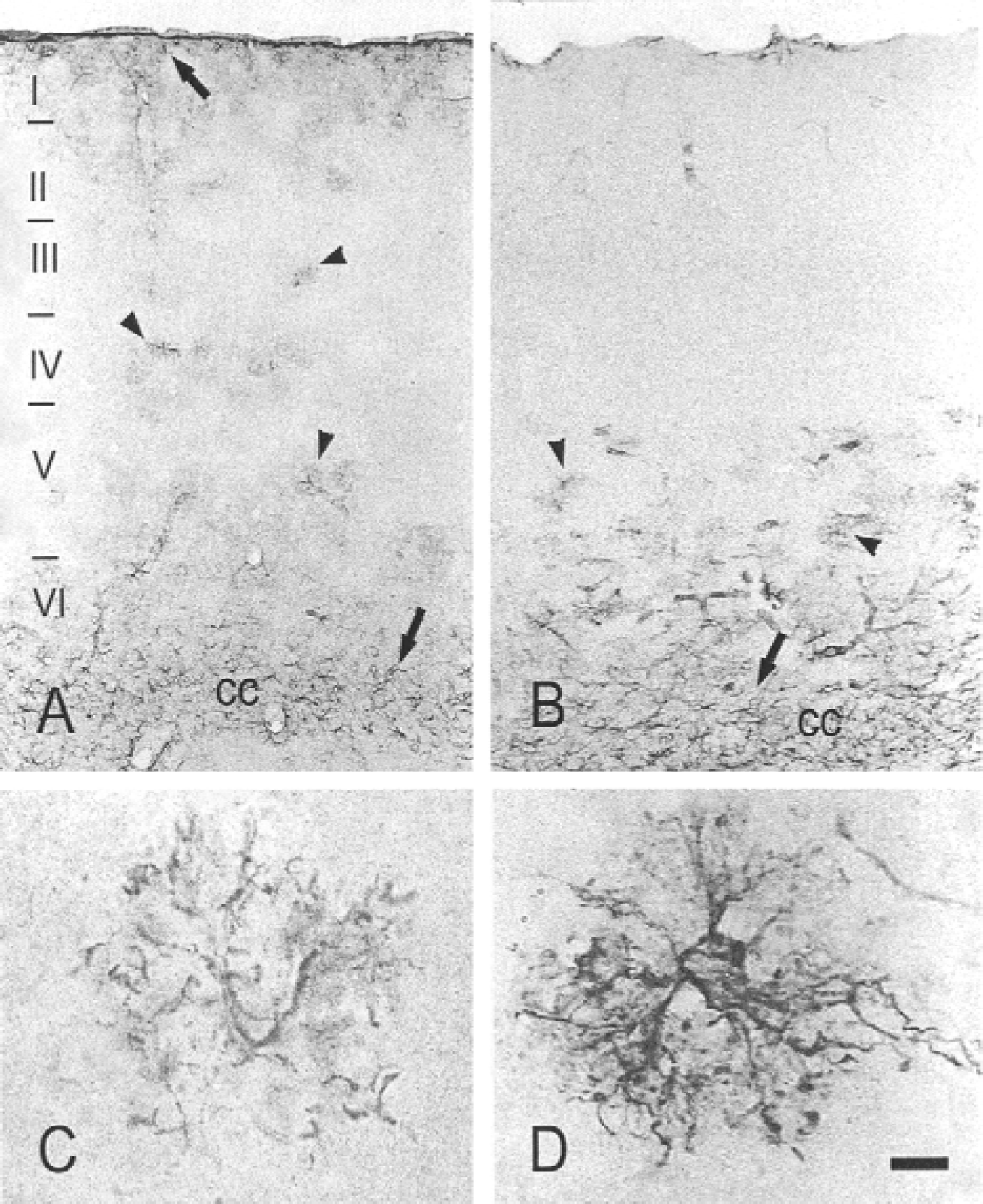
Glial fibrillary acidic protein (GFAP) labeling in the somatosensory cortex of normal mice
One hour after MCAO, dramatic structural alterations indicative of cell death, namely, disrupted processes, soma condensation, and nuclear labeling were observed in a number of reactive astrocytes of layers V to VI (Fig. 2A). Degeneration of protoplasmic astrocytes extended to the entire MCA territory between 6 and 24 hours after MCAO (Fig. 3).
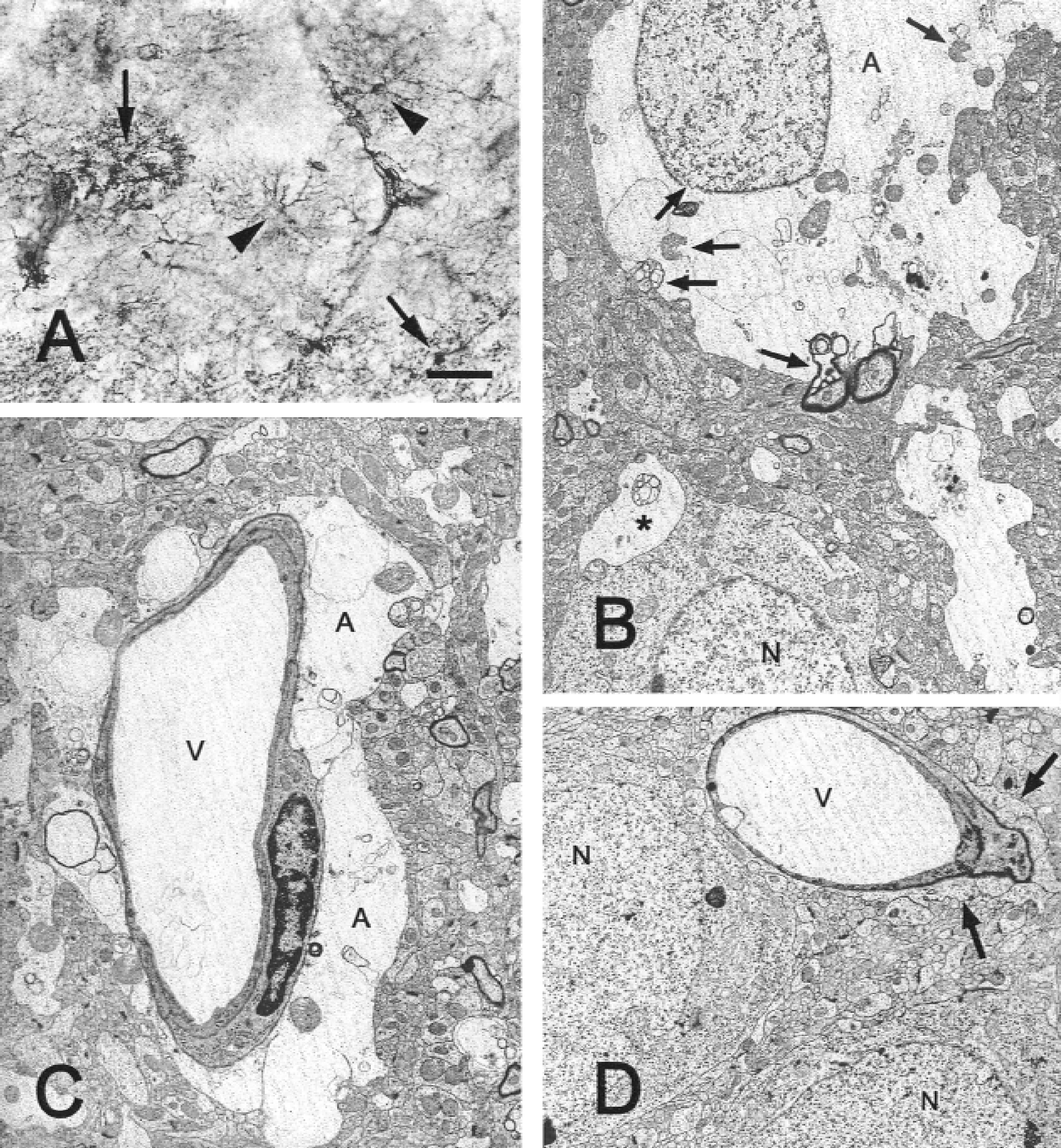
Astroglial structural alterations 1 hour after middle cerebral artery occlusion.
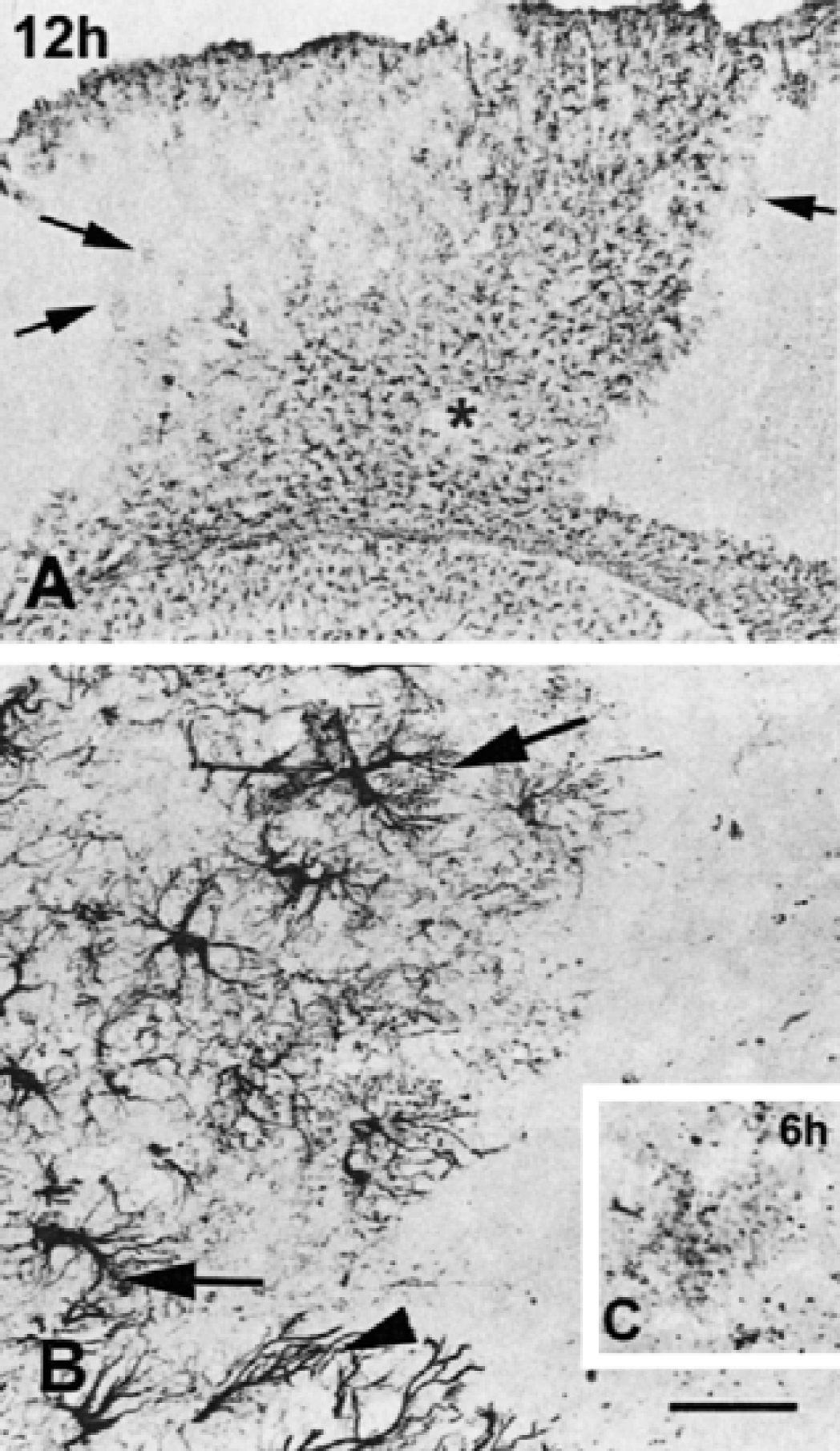
Reactivity of astrocytes 12 hours (
In contrast to protoplasmic astrocytes, fibrous astrocytes of layer I only responded to ischemia by a slight and transient hypertrophy at 3 hours, and had resumed their normal aspect at 24 hours (Fig. 4). Fibrous astrocytes of the corpus callosum maintained their reactivity, featured by hypertrophy and increased GFAP labeling up to 24 hours after MCAO (Fig. 3). At that time, hypertrophied fibrous astrocytes were seen in the surrounding, unlesioned tissue. Interestingly, astrocytes with morphologic features intermediate between fibrous and protoplasmic were also seen on both sides of the scar at 6 and 24 hours (Fig. 3B).
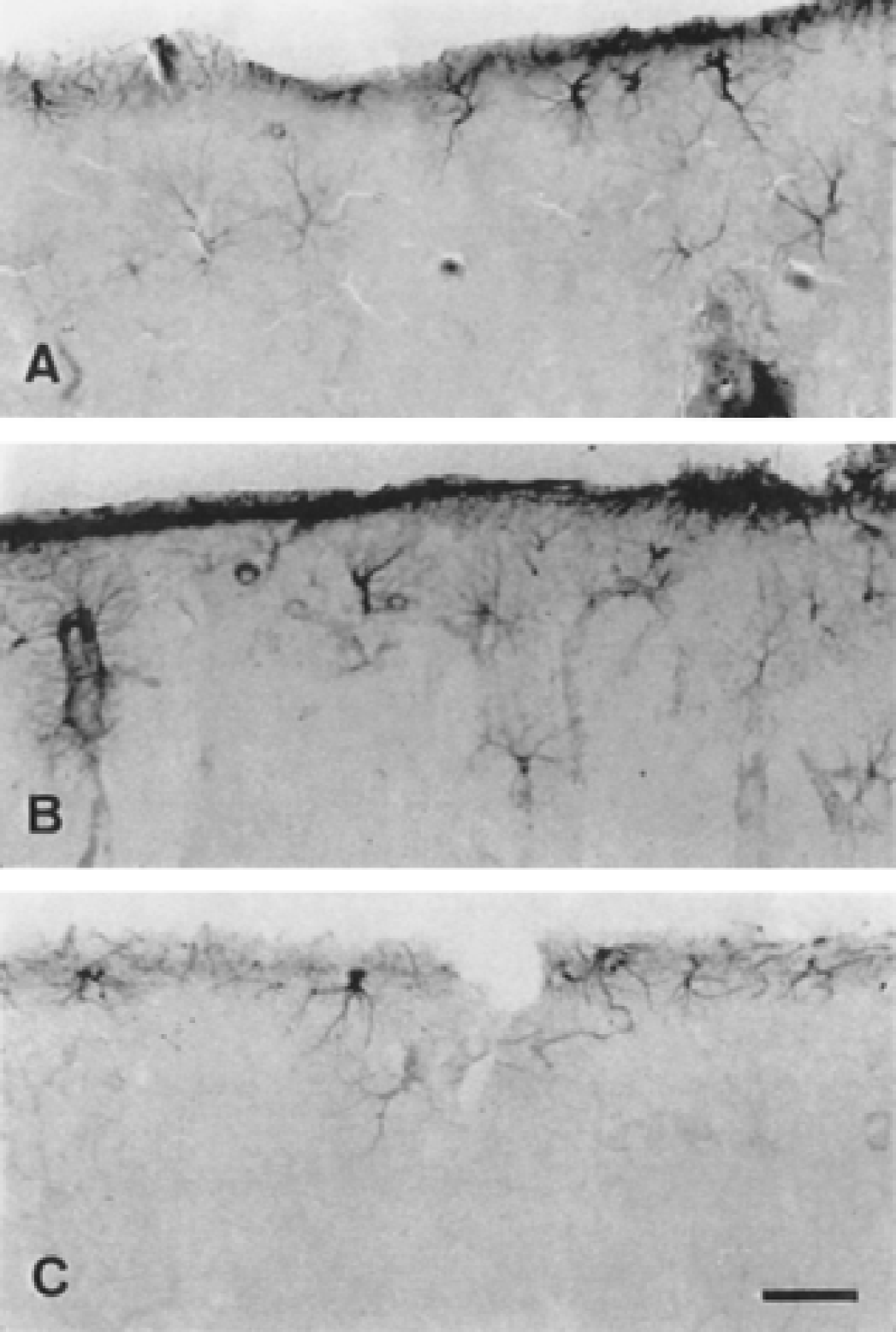
Reactivity of fibrous astrocytes forming the brain surface is observed only at 3 hours after middle cerebral artery occlusion (MCAO), with no conspicuous structural alterations or cell death.
A significant decrease (54.81%) of GFAP protein levels was observed in the lesioned hemisphere 1 hour after MCAO (Fig. 5). Levels were restored at 3 hours after MCAO, in agreement with the increased number of reactive astrocytes observed with GFAP immunohistochemistry in the periinfarct area.
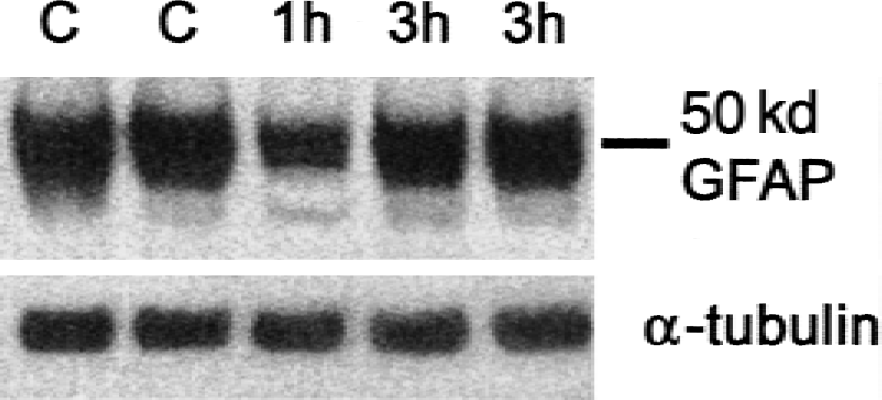
Western blotting of glial fibrillary acidic protein (GFAP) in sham-operated (C) and ischemized animals, 1 and 3 hours after middle cerebral artery occlusion. A significant decrease of GFAP is observed at 1 hour. Values are expressed in arbitrary units against α-tubulin, as mean ± SD (n = 6). One-factor analysis of variance with unpaired Student's t-test: *P < 0.01 versus sham.
Ultrastructural features of astrocytes in the lesioned area
In the core, electron microscopy confirmed the existence, at 1 hour, of a major swelling of the astrocytic soma (Fig. 2B) and perivascular endfeet (Fig. 2C). Swelling was consistently correlated with signs of irreversible cell damage, such as a rupture of mitochondrial, nuclear, and cytoplasmic membranes, and an electron lucent cytoplasm. No such features were observed in naive animals, in tissue retrieved from the same area (Fig. 2D).
Regional cerebral blood flow
Analysis of rCBF levels 1 hour after MCAO delineated several areas within the artery territory (Fig. 6; Table 1). Regional cerebral blood flow levels were depleted by 98% in area a, which can be regarded as the infarct core, and encompasses layers I to IV, as shown by correlated analysis of adjacent cresyl violet–stained sections. This area was surrounded by a thin rim of tissue with residual blood flow (30–20%, areas b and c), which involved deep layers V to VI. More peripheral areas (d) and areas outside the occluded MCA territory (e, f) displayed normal rCBF.
Regional cerebral blood flow measured by the 14 C-IAP technique
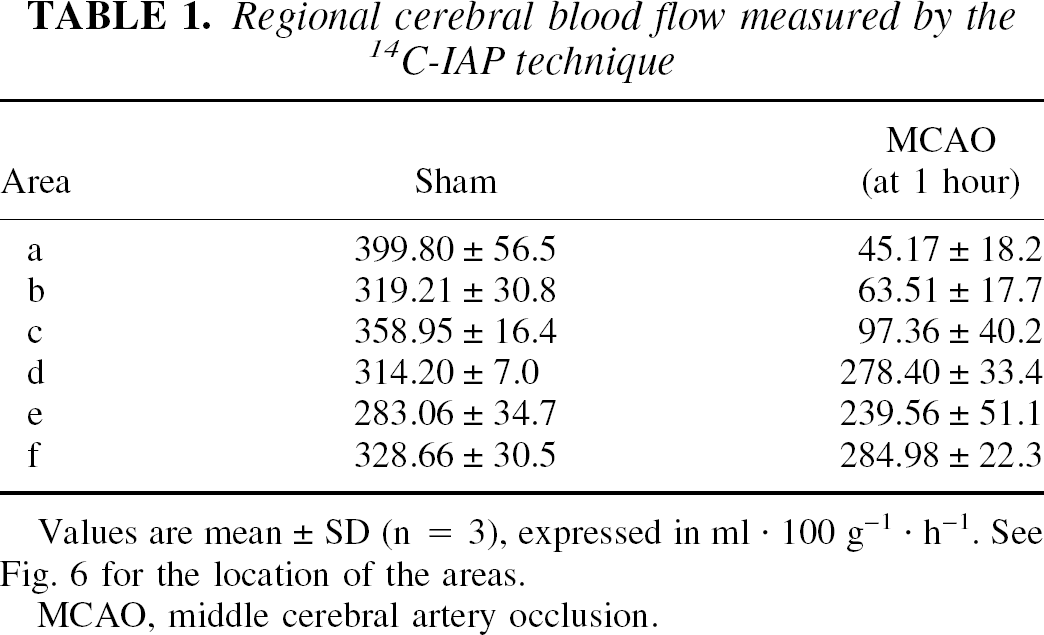
Values are mean ± SD (n = 3), expressed in ml · 100 g−1 · h−1. See Fig. 6 for the location of the areas.
MCAO, middle cerebral artery occlusion.
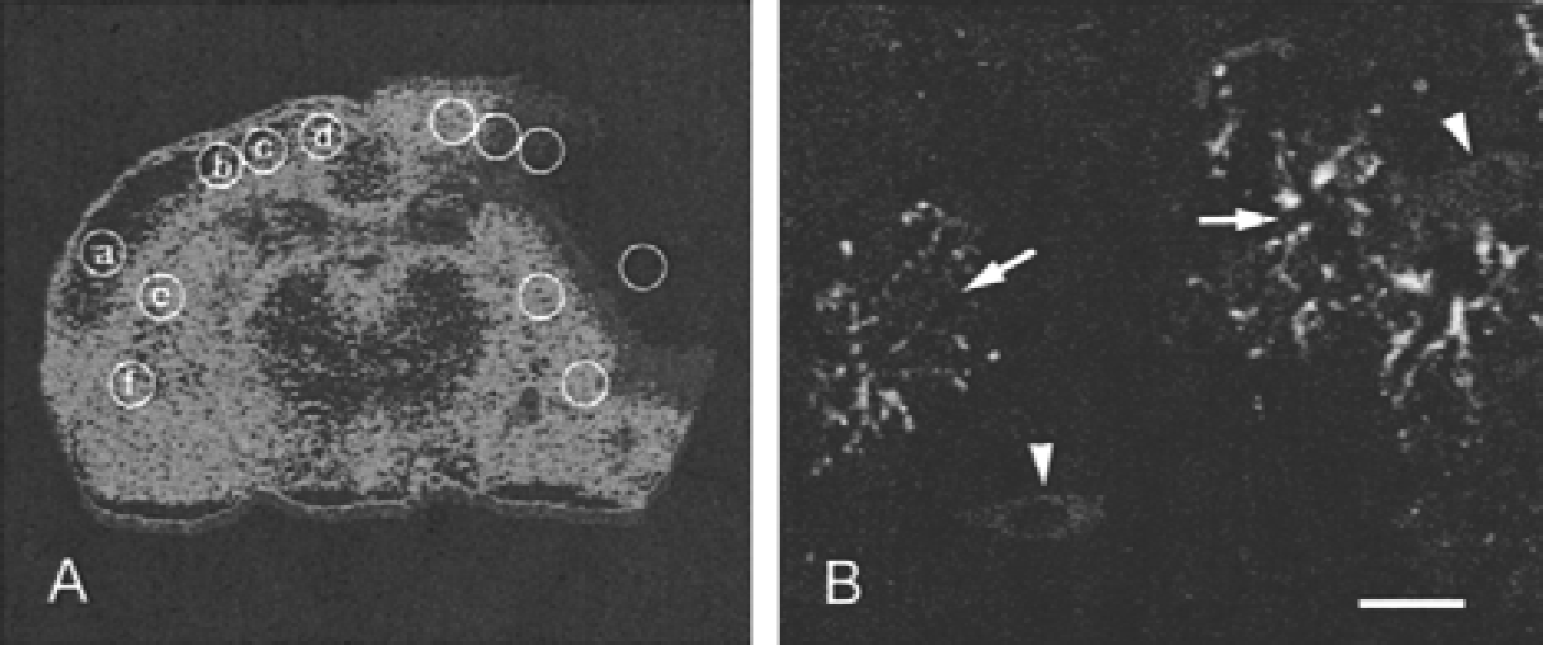
Illustration of regional cerebral blood flow (rCBF) with iodo-14C-antipyrine 1 hour after middle cerebral artery occlusion. Quantification of rCBF values for each region (a–f) is indicated in Table 1. The core area (a) is almost totally devoid of blood supply. Regions b and c feature the penumbra, with residual blood flow. Regions d, e, and f are outside the ischemized area.
Active caspase-3 immunoreactivity
According to previous descriptions in this model (Benchoua et al., 2001), active caspase-3 (p20) immunoreactivity was observed in neurons of the core and of the penumbra (Fig. 6B). Despite the presence of astrocytes with dramatic structural alterations in layer VI, no colocalization of the p20 protein with GFAP was detected with confocal microscopy.
Alterations of the blood–brain barrier
The permeability of the blood–brain barrier had similar alterations for the two molecules used as markers, because both HRP (40 kd) and IgM (450 kd) labeling was seen in the cortical parenchyma 1 hour after MCAO (Figs. 7A and 7B, respectively).
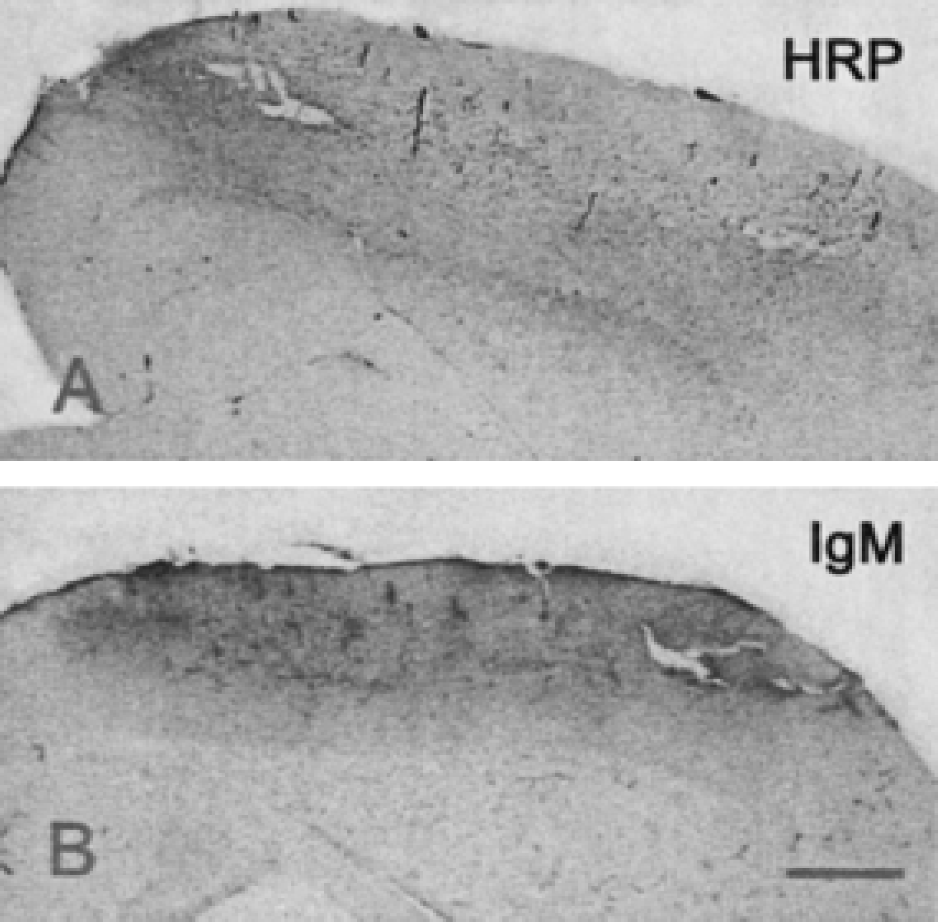
Leakage of the blood–brain barrier for horseradish peroxidase (HRP)
DISCUSSION
In this study, we investigated the pattern of reactivity and fate of protoplasmic and fibrous astrocytes in cortical areas supplied by the MCA in a model of severe focal ischemia. We show that protoplasmic cortical astrocytes undergo irreversible necrotic alterations within the first hour of MCA occlusion in the area totally deprived of blood supply. Protoplasmic astrocytes located in the adjacent areas with residual blood flow followed a more classic astrogliotic pattern. In contrast, fibrous astrocytes displayed a transient reactivity, and formed the glial scar that surrounds the final area of pannecrosis (summarized in Table 2). This reveals a peculiar sensitivity of cortical protoplasmic astrocytes to severe ischemic conditions. Associated with data obtained on transient models of blood flow arrest and in other brain regions, our results suggest that failure to rapidly restore blood flow in the MCA territory may accelerate degeneration of the main cortical astrocytic population, with evident repercussion on neuronal survival and infarction.
Schematic representation of the evolution of glial fibrillary acid protein (GFAP) contents and immunohistochemical labeling in the various parts of the ischemic lesion

MCAO, middle cerebral artery occlusion; ND, not determined; + to +++, graduations of estimated value.
Death of cortical protoplasmic astroglia rapidly follows MCAO
The very first glial reaction observed in our study was a loss of immunoreactivity for GFAP, the main component of gliofilaments, in protoplasmic astrocytes of the ischemized cortex. The presence of GFAP in protoplasmic astrocytes is still a matter a debate, based on the difficulty of labeling astrocytes in the gray matter using anti-GFAP antibodies. Recent immunohistochemical studies suggest the existence of two pools of astrocytes, one with a high GFAP content, and one with nondetectable GFAP (Walz, 2000).
Immunohistochemical characterization of protoplasmic astrocytes is subjected to concerns of technical nature, such as detection limits and aldehyde fixation. In addition, antibodies may not recognize a soluble, nonfibrillar form of GFAP (Stichel et al., 1991), which could account for GFAP contents in nonlabeled astrocytes of the gray matter. Therefore, data obtained with GFAP immunohistochemistry were confirmed in this study by both immunoblotting and electron microscopy. Furthermore, in almost all brain injury types, GFAP levels and number of GFAP-labeled cells consistently increase (Jabs et al., 1999), and astrocytes with low GFAP contents become detectable, a feature not observed in the infarct core in this study and in a transient model of MCAO (Chen et al., 1993).
The immediate loss of GFAP immunoreactivity may evoke rupture of gliofilament bundles due to dramatic cell swelling, a very early event in ischemia (Garcia et al., 1977; Garcia and Lossinsky, 1979; Jenkins et al., 1979; Kalimo et al., 1981). Cell swelling is considered a detrimental sign of astrocytic viability, but is also a dynamic process that may resolve within seconds after reperfusion (Hossman, 1976; Kimelberg and Ransom, 1986). In our case, no recovery of the GFAP labeling was observed in the central area of the lesion, and after 1 hour, marked swelling of all astrocytic compartments was associated with advanced disintegration of cytoplasmic elements rather than with an increased number of glycogen particles, as described in transient ischemia, in which swelling is reversible (Ito et al., 1997). In addition, even though hypertrophy and increased GFAP labeling were observed in both fibrous and protoplasmic astrocytes of the peripheral area, only protoplasmic astrocytes displayed ruptured cell processes and severe somatic shrinkage after 1 hour.
Structural alterations suggest astrocytic dysfunction. Loss of GFAP immunoreactivity has been correlated with loss of GFAP mRNA (Liu et al., 1999) and disruption of the intermediate filaments (Schmidt-Kastner et al., 1993). In turn, loss of GFAP mRNA associates with loss of ATP in the ischemic core (Yamashita et al., 1996), and with loss of key enzymes of astrocytic function (Liu et al., 1999). Absence of GFAP may increase the susceptibility of astrocytes to cytotoxic edema (Ding et al., 1998), leading to cell death, and mice with deletion of the GFAP gene have a higher susceptibility to MCAO (Nawashiro et al., 2000). A correlation between loss of GFAP and functional impairment is also suggested in this study by the decline of protein contents at 1 hour. The recovery of GFAP levels at later stages may reflect the progression of astrogliosis at the periphery of the lesioned area, and the hypertrophy of both protoplasmic and fibrous astrocytes in the periinfarct area and at the brain surface.
Astrocytes endfeet are in intimate contact with endothelial cells (Peters et al., 1991), from which they are separated by the basal lamina, and participate to the control of the blood–CNS interface (Stewart and Coomber, 1986). Entry of serum proteins into brain parenchyma has been described as a delayed phenomenon that occurs several days after global ischemia (Schmidt-Kastner et al., 1990), in relationships with astrogliosis. Breakdown of the blood–brain barrier for albumin, a low-molecular-weight compound, occurs several hours after focal ischemia in mice (Mandai et al., 1997). In our model, leakage for HRP and IgM was rapid, and initially encompassed the core area. The loss of one of these major functions of astrocytes in the brain therefore correlates with the time course and extent of structural alterations, suggesting that astrocyte demise 1 hour after MCAO was both structural and functional.
The ultrastructural features observed in the core 1 hour after MCAO signed irreversible commitment to death by necrosis (Wyllie et al., 1980). This hypothesis was further suggested by the lack of activation of caspase-3 in the penumbral area, where the persistency of GFAP allowed double labeling. Caspase-3 belongs to the family of cysteine proteases involved in the executive phase of apoptosis (Li and Yuan, 1999; Hengartner, 2000), and strong activation of caspase-3 correlates with apoptotic death of astrocytes (Takuma et al., 1999; Zhang et al., 2000). Although neurons containing active caspase-3 were abundant in layer VI, no labeling of GFAP-containing cells, either protoplasmic or fibrous, was observed.
Protoplasmic astrocytes are exquisitely sensitive to ischemia
Astrocytic demise was observed within the entire extent of the arterial territory, with the exception of a thin zone at the periphery and in deep cortical layers. Analysis of rCBF and comparison with GFAP immunohistochemistry revealed that the area devoid of GFAP labeling had severely depleted rCBF. This area can therefore be considered as the infarct core, whereas the surrounding area, with remaining GFAP-labeled cells and residual blood flow, can be described as penumbral. This points to the existence of a threshold in the viability of protoplasmic astrocytes with regard to CBF, which was not observed for fibrous astrocytes of the upper layers. Although penumbral microcirculation can maintain cell viability and induce a “classic” astroglial reaction, dramatic depletion of rCBF rapidly leads to impairment of astrocytic structure, and likely, function. In contrast with the suggestion that propagation of astrocytic damage by gap-junctions coupling (Giaume and McCarthy, 1996) may help the propagation of ischemic damage (Lin et al., 1998), early astrocytic demise seemed, in our case, instead related to the severity of blood deprivation and absence of reflow.
For ill-defined reasons, specific classes of astrocytes show heterogeneous levels of GFAP contents. For example, lower numbers of GFAP-positive cells occur in neocortical areas (Petito et al., 1990; Bernaudin et al., 1998), and GFAP is expressed at higher levels in fibrous astrocytes of the white matter than in protoplasmic astrocytes (Eddleston and Mucke, 1993). Whether varying GFAP levels reflect particular metabolic or functional demands and functions has still to be defined. Most of the studies about ischemia-related astrogliosis concern astrocytes of the fibrous type, mainly from hippocampus and striatum. Reports about the cortex describe observations performed days or weeks after ischemia in rat models with reperfusion (Schmidt-Kastner et al., 1990; Chen et al., 1993; Duggal et al., 1997). No specific data were available about protoplasmic astrocytes, which are characteristically present in the cortex. We show that, in contrast with hippocampal or striatal fibrous astrocytes, which may survive 2-hour periods of transient focal ischemia (Chen et al., 1993; Duggal et al., 1997), dramatic CBF depletion rapidly leads to the death of protoplasmic astrocytes in the cerebral cortex. This suggests a differential sensitivity of the two types of astrocytes to ischemic insults, according to their regional, and likely functional, specificity.
Interestingly, although fibrous astrocytes were not normally seen in cortical layers II to VI, they were specifically involved in the secondary formation of the glial scar surrounding the infarcted cortical tissue. Protoplasmic astrocytes bordering the lesioned area are progressively superseded by astrocytes with a stellar morphology. Morphologic changes of cortical astrocytes including protoplasmic and GFAP-containing (putatively fibrous) have been described after trauma (Singh and Mathew, 1989; Raivich et al., 1999). Those data have led to the concept of the existence of a continuum between the two extreme forms of astrocytes, indicating the great degree of flexibility of this neural cell type (Walz, 2000). This may correlate with our observation that astrocytes at the border of the scar included the combined structural characteristics of fibrous and protoplasmic types. It confirms that cortical astrocytes display substantial adaptive structural plasticity in response to brain injuries.
CONCLUSIONS
Astrocytes play an important role in maintaining neuronal function (Kimelberg and Ransom, 1986). In vitro, they protect neurons from a variety of insults, including those resulting from oxidative stress (Desagher et al., 1996), a primary mechanism of ischemia-induced cell loss. We show that in contrast with the commonly held assumption that astrocytes are the most resistant cells to ischemic conditions, protoplasmic astrocytes may indeed suffer altered cerebral blood flow rapidly and irreversibly. Our results stress the importance of rapid reperfusion in reversing glial damage and confirm that, in a number of clinical situations, glial dysfunctions may have major implications in the pathogenesis of infarction.
Footnotes
Acknowledgments:
The authors thank Drs. S. Juliano and M. Peschanski for critical review of the manuscript, and Dr. E. Cremer and J. Guichard (INSERM U474, Paris) for their help with electron microscopy.