
Abstract
Human C-reactive protein (CRP), the classic acute phase plasma protein, increases in concentration after myocardial infarction and stroke. Human CRP binds to ligands exposed in damaged tissue and can then activate complement and its proinflammatory functions. In contrast, rat CRP, which binds to similar ligands, does not activate complement. In the present study, systemic complement depletion with cobra venom factor in adult rats subjected to middle cerebral artery occlusion did not affect cerebral infarct size, indicating that circulating complement does not contribute to injury in this model. However, we have previously reported that administration of human CRP to rats undergoing coronary artery ligation caused a marked increase in size of the resulting myocardial infarction, associated with codeposition of human CRP and rat complement in the infarcts. In the present study, we show that adult rats subjected to middle cerebral artery occlusion and then treated with human CRP similarly developed significantly larger cerebral infarcts compared with control subjects receiving human serum albumin. Human CRP can thus contribute to ischemic tissue damage in the brain as well as in the heart, and inhibition of CRP binding may therefore be a promising target for tissue protective acute therapeutic intervention in stroke as well as in myocardial infarction.
Ischemic infarction is a potent stimulus for the acute phase response, in which a wide range of plasma proteins, mostly produced in the liver, increase dramatically in concentration to values that usually reflect closely the extent of tissue damage (Pepys and Hirschfield, 2003). Acute phase proteins include some coagulation factors, complement proteins, protease inhibitors, transport proteins, and various proteins of unknown function. Because this nonspecific acute phase response is highly conserved in evolution and invariably occurs after most forms of tissue injury, infection, and inflammation, it is generally considered to be beneficial by contributing to host defense, resolution of injury, and promotion of repair. However, as with specific adaptive immunity, the potential exists for enhancement of tissue damage by increased production of biologically active proteins. In particular, C-reactive protein (CRP), the classic acute phase protein, has avid specific binding reactivity for phosphocholine (Volanakis and Kaplan, 1971) and other ligands (Du Clos, 1989;Volanakis and Wirtz, 1979) that are differentially or more extensively exposed in or on damaged cells and tissues. Ligand bound human CRP can then potently activate the classic complement pathway, with cleavage and fixation of C3 (Kaplan and Volanakis, 1974). C3 is the most abundant complement protein and the major opsonin of the complement system, as well as the source of the chemotactic and proinflammatory anaphylatoxin, C3a. Although CRP and the plasma protein family to which it belongs, the pentraxins, are highly conserved in evolution with extensive amino acid sequence homology, the same flattened β-jelly roll fold (the so-called lectin fold), and the same cyclic pentameric arrangement of protomers, there are nonetheless major differences between the CRP molecules in different species. Thus, unlike human CRP, which activates human and rat complement, rat CRP does not activate complement in either species (de Beer et al., 1982a). Furthermore, rat CRP is glycosylated, contains two covalently linked subunits, and circulates at concentrations of approximately 300mg/L, increasing to approximately 900mg/L in the acute phase response (de Beer et al., 1982a). In contrast, human CRP is not glycosylated, has noncovalently associated protomers, and circulates at a median concentration of approximately 0.8mg/L in health, increasing to as much as 400 to 500mg/L at the peak of the most intense acute phase response (Pepys and Hirschfield, 2003). These differences make the rat an excellent model system for investigation of possible complement-dependent pathobiologic effects of human CRP.
Infarction of myocardial tissue has long been known to be a powerful acute phase stimulus (Kushner et al., 1978), and recent work shows that the peak CRP values in patients with acute myocardial infarction strongly predict outcome and survival both immediately and for months after the event (Anzai et al., 1997;Pietilä et al., 1996;Ueda et al., 1996). This may reflect, in part, the general correlation between infarct size and magnitude of the acute phase response (de Beer et al., 1982b). However, all human myocardial infarction lesions examined from approximately 12 hours after coronary occlusion contain CRP and activated complement codeposited in and around the lesion, and there is evidence from quantification of CRP-C4d complexes in these hearts that CRP is a major contributor to the overall level of complement activation (Lagrand et al., 1997;Nijmeijer et al., 2003). In experimental animal models, complement clearly makes a substantial contribution to the final infarct size after coronary artery ligation or ischemia-reperfusion injury (see Griselli et al., 1999 and earlier work cited therein). Possible mechanisms include the proinflammatory and opsonic activities of complement, as well as direct cytoxicity for ischemically damaged myocardial cells that would otherwise recover. CRP-mediated complement activation may thus contribute importantly to the eventual infarct size and outcome in human clinical acute myocardial infarction. This hypothesis is strongly supported by our finding that injection of human CRP into rats after ligation of the coronary artery leads to significant enhancement of the size of the resulting myocardial infarction, associated with codeposition of human CRP and rat complement in and around the infarct (Griselli et al., 1999).
There have been few studies of the acute phase response after human ischemic stroke, and interpretation of CRP values in this situation is complicated by the fact that most stroke patients are elderly and frequently have other intercurrent pathologies that can stimulate the acute phase response, especially bacterial infections, developing rapidly after the acute cerebral ischemic event. However, a recent small but meticulous and well-controlled analysis of CRP and other inflammatory markers in subjects with ischemic stroke has confirmed that there is a modest but highly significant acute phase response, even among patients without infection (Emsley et al., 2003). Also, CRP concentration has been reported to be an independent predictor of survival after ischemic stroke in humans (Muir et al., 1999). It is therefore possible that circulating CRP could enter the ischemic brain and contribute to cerebral tissue damage. After middle cerebral artery occlusion in the rat, cerebral infarct size is maximal at 24 hours, and we report in the present study that this was not affected by systemic complement depletion. However, in rats injected with human CRP immediately after performing middle cerebral artery occlusion and daily thereafter, cerebral infarct size at 72 hours was significantly increased compared with control animals receiving human serum albumin.
MATERIALS AND METHODS
C-reactive protein
Human CRP was isolated from malignant effusion fluids by calcium-dependent affinity chromatography and was greater than 99% pure by specific immunoassay for CRP, total protein estimation, and SDS-PAGE analysis of reduced denatured samples, as was previously reported (de Beer and Pepys, 1982). The CRP was composed entirely of intact pentamers, as shown by analytical gel filtration chromatography, and was 100% functionally intact, as shown by calcium-dependent binding to immobilized ligands, phosphocholine, and phosphoethanolamine. Electrospray mass spectrometry of the purified CRP yielded only protomers with the authentic Mr of 23,027, corresponding exactly to the known amino acid sequence of CRP including the N-terminal pyrrolidone carboxylic acid. The Gram-negative bacterial LPS content of the isolated purified CRP, measured by the kinetic chromogenic Limulus amebocyte lysate assay (BioWhittaker Europe S.P.R.L., Parc Industriel de Petit Rechain, B-4800, Verviers, Belgium) was 0.16 ng/mg CRP.
Experimental protocols
Male Fischer 344 rats weighing approximately 280 grams each were maintained on a 12-hour light/dark cycle with free access to food and water, apart from the 24 hours before the surgical procedures when food supply was restricted. The animals were anesthetized using 3% isoflurane in 30% oxygen and 70% air, and the left middle cerebral artery (MCA) was permanently occluded by microbipolar coagulation. Mean arterial blood pressure and heart rate were monitored during MCA occlusion using a Maclab 8-chart system, and physiologic variables of the animals were constantly monitored and maintained at normal levels; in particular, body temperature was maintained at 37 ± 1°C (Gill et al., 2002). After recovery from anesthesia, 20 mg/kg doses of human CRP in solution in 10 mM Tris buffered 0.14 NaCl containing 0.002 mM CaCl2, pH 8.0, were injected intraperitoneally at 5 minutes, 24 hours, and 48 hours after MCA occlusion in the experimental group of eight rats. A control group of seven rats received human serum albumin (Sigma Aldrich, Poole, Dorset, U.K.), 20 mg/kg intraperitoneally at 5 minutes, 24 hours, and 48 hours after MCA occlusion. All animals survived until 72 hours after the first injection of CRP or albumin, and their temperatures were monitored and maintained within the physiologic range throughout. At 72 hours, the animals were anesthetized, the brains were removed, and the volume of infarction was measured at the level of the cortex, the caudate nucleus, and the hemisphere as a whole, in 13 slices through the brain, using unbiased stereologic tools (Olympus, 8604 Volkerswil, Switzerland). No immunohistochemical studies were done.
In a separate experiment, conducted in precisely the same way, one group of rats received 250 U/kg of highly purified cobra venom factor (Pepys et al., 1979) in solution in phosphate-buffered saline (PBS) by intraperitoneal injection 24 hours before MCA occlusion. A control group received the same volume of PBS alone. All rats were bled before cobra factor treatment and 24 and 48 hours afterwards. Infarct size was measured as described previously in this article, 24 hours after MCA occlusion. Rat C3 was measured by electroimmunoassay using monospecific rabbit ant-rat C3 antiserum (de Beer et al., 1982a) and is expressed as percentage of the pretreatment value in each individual animal.
RESULTS
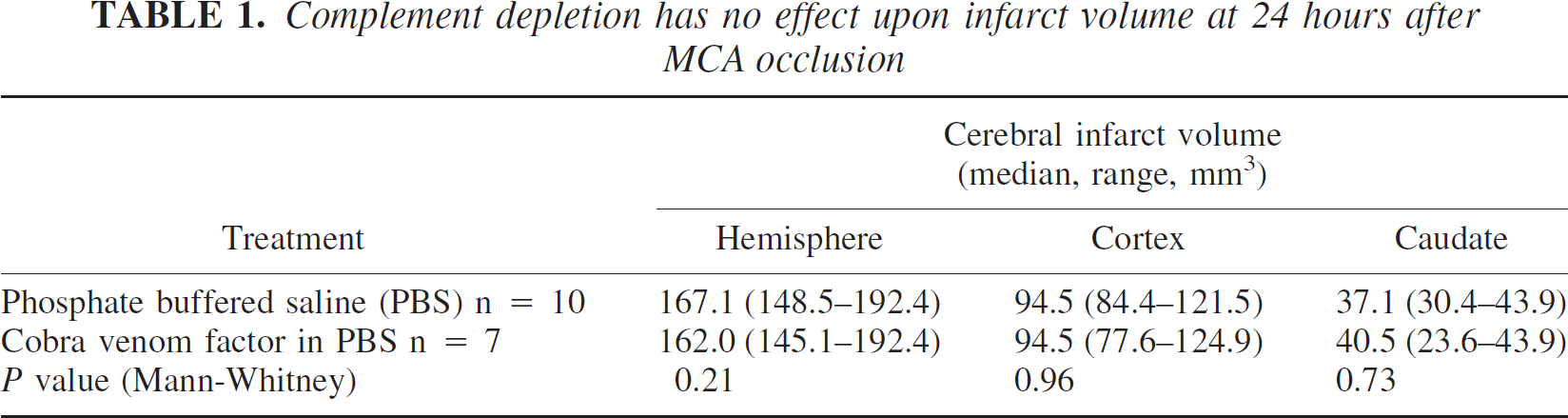
In the rats (n = 7) that received cobra venom factor 24 hours before MCA occlusion, median (range) circulating C3 was reduced to 9.4 (4.4–15.0)% of pretreatment values at the time of operation and was 3.5 (1.9–10.0)% at the time of infarct assessment 24 hours later. The low concentrations of C3 antigen in the samples at these times after cobra venom factor administration consisted entirely of inactive cleaved fragments, and no active native C3 was present. However, there was no difference between the infarct volumes in these animals and controls that were pretreated only with PBS (Table 1).
Complement depletion has no effect upon infarct volume at 24 hours after MCA occlusion
In contrast, rats receiving human CRP had significantly larger infarct volumes at 72 hours after MCA occlusion than the control animals that received human serum albumin (Mann-Whitney test) (Fig. 1). This was most marked in the cortex, and although the infarcts in the caudate nucleus were not different between the groups, the overall infarct volume in the whole hemisphere was significantly larger in the CRP-treated group.
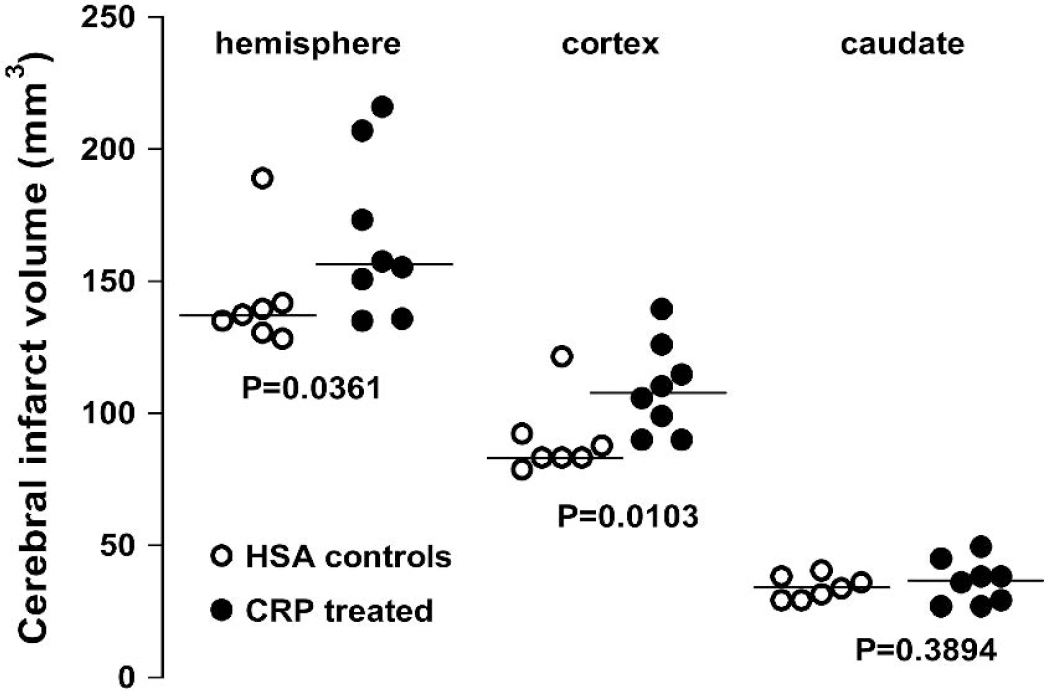
Human C-reactive protein increases cerebral infarct volume after middle cerebral artery occlusion. Each point represents the infarct volume in an individual rat measured within the hemisphere as a whole, the cortex, or caudate nucleus alone. Rats received a dose of 20 mg/kg of either human C-reactive protein or human serum albumin (in the control group) immediately after middle cerebral artery occlusion and then again at 24 and 48 hours. Infarct size was assessed at 72 hours after occlusion. Horizontal lines indicate the median values for each group. P values are from one-tailed Mann-Whitney tests.
DISCUSSION
Cerebral infarct volume in the present rat model is maximal at 24 hours after MCA occlusion (Gill et al., 1995, 1996). This is presumably too early for circulating complement to penetrate the lesion and contribute significantly to final infarct size. Indeed, our present findings confirm that cobra venom factor-treated rats, with profoundly depleted circulating complement both at the time of MCA occlusion and throughout the experiment, had lesions that were not significantly different from those in control animals. This result is consistent with previous work in a rabbit model of acute thromboembolic stroke (Lew et al., 1999). Another study in 21-day-old rats subjected to hypoxic-ischemic injury followed by reperfusion and examined at 48 hours and after 21 days also failed to show any effect of complement depletion either by cobra venom factor or soluble complement receptor type 1. In contrast, there are several reports of beneficial effects of complement depletion in different models of cerebral ischemia, including ischemia with short-term (2–4 hours) reperfusion in adult rats (Vasthare et al., 1998), ischemia with 23 hours reperfusion in adult mice (Huang et al., 1999), hypoxic ischemia with 24 hours reperfusion in 7-day-old rats (Imm et al., 2002), and hypoxic ischemia with up to 5 days reperfusion in 7-day-old rats (Cowell et al., 2003). However, it is important to note the very major differences between these various models and also from the model of MCA occlusion that we have used in adult rats. In particular, reperfusion after ischemic injury enables circulating complement to reach injured tissue much more freely than after terminal occlusion. Also, the mode of neuronal injury and cell death is known to be different in neonatal compared with adult animals (Ginsberg and Busto, 1989;Johnston and Silverstein, 1985). A role for autologous complement in the absence of external factors therefore remains controversial, especially as complement proteins may also be produced locally within the brain and may have beneficial as well as harmful effects upon neurons (van Beek et al., 2003).
In contrast, we have previously reported that intraperitoneal administration of human CRP to rats, producing sustained high circulating values of human CRP, was associated with increased size of myocardial infarcts after ligation of the coronary artery (Griselli et al., 1999). Human CRP was deposited, together with rat complement in and around the infarct, and the damaging effect of human CRP was completely abrogated by concurrent complement depletion of the rats (Griselli et al., 1999). Human CRP can thus enhance ischemic tissue damage by a complement-dependent mechanism. The present study shows that this mechanism may also operate in the brain, as rats receiving human CRP had significantly larger infarct volumes at 72 hours after MCA occlusion. Presumably ischemic damage to the blood brain barrier enables circulating plasma proteins, including the administered human CRP and autologous rat complement, to gain access to cerebral tissue.
CRP is known to bind to the plasma membranes and to small nuclear ribonucleoprotein particles in the exposed nuclei of damaged cells (Du Clos, 1989;Kushner and Kaplan, 1961). After such binding, human CRP but not rat CRP can then activate the classical complement pathway (de Beer et al., 1982a) and engage its opsonic, chemotactic, and proinflammatory functions. Complement may thereby directly injure ischemic cells that would otherwise have survived and promote additional tissue injury by increasing the intensity of the neutrophil and other cellular inflammatory responses to anoxic cell damage and death. The possible alternatives of direct tissue damage by human CRP or complement-independent, proinflammatory effects of human CRP by interaction with phagocytic cells are controversial (Pepys and Hirschfield, 2003), and, as noted previously in this article, complement depletion completely abrogated the effect of human CRP in the myocardial infarction model (Griselli et al., 1999). The precise mechanisms underlying human CRP-dependent pathogenicity clearly merit further investigation, for example using different doses and timing of CRP administration and testing the effects of complement depletion. However, the fact that human CRP can exacerbate ischemic tissue damage in the brain as well as in the heart identifies this protein as a potentially important novel therapeutic target.
Footnotes
Acknowledgments
The authors thank Mr. Remy Wybrecht and Ms. Marie-Therese for their technical expertise in conducting the animal studies and Ms. Beth Jones for expert preparation of the manuscript.