
Abstract
Mannose binding lectin (MBL) initiates complement activation and exacerbates tissue damage after systemic ischemia/reperfusion. We tested the hypothesis that MBL activates complement and worsens outcome using two levels of controlled cortical impact (CCI) in mice. After moderate CCI (0.6 mm depth), MBL immunostaining was detected on injured endothelial cells of wild-type (WT) mice and C3d was detected in MBL KO (deficient in MBL A/C) and WT mice, suggesting that MBL is dispensable for terminal complement activation after CCI. Brain neutrophils, edema, blood-brain barrier permeability, gross histopathology, and motor dysfunction were similar in injured MBL KO and WT mice. In mice subjected to mild CCI (0.2 mm), MBL KO mice had almost two-fold increased acute CA3 cell degeneration at 6 h (P<0.01 versus WT). Naive MBL KO mice had decreased brain volume but performed similar to WT mice in two distinct Morris water maze (MWM) paradigms. However, injured MBL KO mice had impaired performance in cued platform trials (P<0.05 versus WT), suggesting a transient nonspatial learning deficit in injured MBL KO mice. The data suggest that MBL deficiency increases susceptibility to CCI through C3-independent mechanisms and that MBL-deficient patients may be at increased risk of poor outcome after traumatic brain injury.
Introduction
Traumatic brain injury (TBI) induces an acute inflammatory response in injured brain that contributes to secondary tissue damage and neurologic dysfunction (Schmidt et al, 2005). Activation of complement plays a role in posttraumatic brain inflammation. Intracerebral complement activation occurs in traumatic human brain (Bellander et al, 2001; Schmidt et al, 2005; Stahel et al, 1998), and genetic or pharmacological inhibition of terminal complement components reduce brain neutrophil accumulation, blood—brain barrier damage, apoptosis, and neurologic dysfunction in experimental TBI models (Kaczorowski et al, 1995; Leinhase et al, 2006; Rancan et al, 2003). Complement activation may be mediated by three distinct pathways: The classical pathway (initiated by antigen-antibody aggregates and proceeding through C1q, C4, and C2), the alternative pathway (initiated by C3b and proceeding through factor B and factor D), and the lectin pathway (initiated by mannose binding lectin (MBL) and proceeding through C4 and C2).
Mannose binding lectin is a serum multitrimeric glycoprotein pattern recognition molecule in the collectin family that plays a role in innate immunity, including activation of complement and promotion of opsonophagocytosis (Takahashi et al, 2006). After systemic ischemia/reperfusion injury, the lectin pathway is initiated when a specific autoreactive IgM binds exposed tissue antigens and recruits serum MBL to form an IgM-MBL complex (Zhang et al, 2006). Tissue-bound MBL assembles a complex with and activates serum MASP (MBL-associated serine protease)-2. Activated MASP-2 cleaves C4 and C2, forming a C3-convertase to activate terminal complement. Whereas humans have a single functional MBL gene product, MBL-a and MBL-C can both initiate the lectin pathway in mice (Shi et al, 2004). In humans, MBL deficiency is the most common type of immunodeficiency, affecting as many as 10% to 30% of some ethnicities (Madsen et al, 1998; Sorensen et al, 2005). Single nucleotide polymorphisms in codon 52 (allele D), 54 (allele B), and 57 (allele C) result in dysfunctional MBL (Steffensen et al, 2000). Homozygotes for the B allele, the most common MBL single nucleotide polymorphisms, have increased susceptibility to and mortality from acute respiratory distress syndrome (Gong et al, 2007); however, a role for MBL in acute brain injury has not been reported.
Studies using mice deficient in MBL A/C (MBL KO) show that MBL plays a detrimental role in tissue damage and organ dysfunction in models of cardiac, skeletal muscle, renal, and intestinal ischemia-reperfusion by activating terminal complement and exacerbating reperfusion injury (Chan et al, 2006; Hart et al, 2005; Moller-Kristensen et al, 2005; Walsh et al, 2005). Although MBL has not been studied in a brain injury model, MBL mRNA expression has been detected in brain (Wagner et al, 2003), and MBL and other lectins have been hypothesized to play a role in brain development (Zanetta, 2003).
On the basis of the well-established role of MBL in systemic ischemia-reperfusion injury, we hypothesized that genetic deletion of MBL would reduce terminal complement activation, decrease histopathology, and improve functional outcome after controlled cortical impact (CCI) in mice. Contrary to this hypothesis, we found that MBL deficiency was associated with increased acute CA3 cell death and exacerbation of cognitive deficits in injured mice. The data suggest that MBL protects against traumatic cell death and may also be involved in the recovery of cognitive function after TBI.
Materials and methods
In all studies, investigators were masked to mouse genotype during surgery, data acquisition, and analysis. All animal experiments were performed under a protocol approved by the Subcommittee on Research Animal Care at the Massachusetts General Hospital.
Mice
Mice deficient in MBL A/C were generated as described previously (Shi et al, 2004) and backcrossed seven generations into C57Bl/6J. Microsatellite polymorphism analysis of representative mutant mice (Charles River Laboratories, Wilmington, MA, USA) showed greater than 96% C57Bl/6J DNA in the MBL KO mice (Moller-Kristensen et al, 2007). MBL KO mice do not have increased incidence of infectious disease at baseline and show no obvious developmental abnormalities (Shi et al, 2004). C57BL/6J mice from the same vendor as those used to construct the MBL KO (Jackson Laboratories, Bar Harbor, ME, USA) were used as controls. Male and female mice matched for age and weight (20 to 30 g) were used for experiments in equal numbers in MBL KO and WT groups.
Antibodies and Reagents
Rat anti-mouse MBL-A and rat anti-mouse MBL-C react specifically with mouse MBL-A and MBL-C and yield negative staining in peripheral tissues of MBL A/C knockout mice (Liu et al, 2001). Both MBL A/C antibodies were a kind gift of Dr Steffen Thiel (University of Aarhus, Denmark). Recombinant human MBL (rhMBL) (1 mg/mL) was a gift of NatImmune (Copenhagen, Denmark). Rhodamine- or FITC (fluorescein isothiocyanate)-conjugated Griffonia simplicifolia lectin I (1:200; Vector Laboratories, Burlingame, CA, USA) was used to identify cerebral vascular endothelium. Phycoerythrin-labeled rabbit anti-mouse CD11b (M1/70, 1:500) was obtained from BD Pharmingen (San Diego, CA, USA).
Controlled Cortical Impact
The CCI model was used as described previously with minor modifications (You et al, 2007). The trauma protocol was approved by the Massachusetts General Hospital Institutional Animal Care and Use Committee and complied with the NIH (National Institutes of Health) Guide for the Care and Use of Laboratory Animals. In all studies, wild-type and mutant mice were concomitantly subjected to CCI and other testing. Mice (8 to 16 weeks of age) were anesthetized with 4% isoflurane (Anaquest, Memphis, TN, USA) in 70% N2O and 30% O2 using a Fluotec 3 vaporizer (Colonial Medical, Amherst, NH, USA) and positioned in a stereotaxic frame. Anesthesia was maintained using 2% to 3% isoflurane. A craniotomy was made using a portable drill and 5 mm trephine over the left parietotemporal cortex, and the bone flap was removed. Mice were subjected to CCI using a pneumatic cylinder with a 3-mm flat-tip impounder, velocity 6 m/sec, set depth of 0.6 mm (moderate CCI) for immunohistochemical, behavioral, and volumetric studies. In our hands, a 0.6-mm set injury depth results in early destruction of CA3 tissue; therefore, an injury depth of 0.2 mm (mild CCI) was also used to investigate neuron degeneration in CA3 and to assess for injury level-dependent effects on overall lesion size and behavioral outcomes. In all studies, a 100-ms impact duration was used. After CCI, the scalp was sutured closed and the mice returned to their cages to recover from anesthesia.
Administration of Recombinant Human MBL
Mice were anesthetized and the jugular vein exposed by surgical cutdown in the midline neck region. For histochemical studies, 75 μg of FITC-labeled rhMBL in 75 μ1 volume was injected intravenously into the jugular vein over approximately 30 secs. The neck was sutured closed and the mice were subjected to CCI. In reconstitution studies for Morris water maze (MWM) testing, 200 μg rhMBL was administered intraperitoneally 1 day before and again on the day of CCI for a total of 400 μg per mouse. Control mice were administered equal volumes of phosphate-buffered saline (PBS).
Immunohistochemistry and Fluorescence Microscopy
Mice were killed at various times after CCI and the brains removed and frozen in nitrogen vapor. Cryostat brain sections (12 μm) were cut at 200 μm intervals from the anterior to posterior hippocampus. Cryostat sections were placed on poly-
Fluoro Jade B Staining
Fluoro Jade B (Chemicon, Temecula, CA, USA) solution was prepared from a 0.01% stock solution that was made by adding 10 mg of Fluoro Jade B powder to 100 mL of distilled water. To make up 10mL of staining solution, 0.4 mL of the stock solution was added to 9.6 mL of 0.1% acetic acid, resulting in a final dye concentration of 0.0004%. After fixing in 100% ethanol for 10mins at ambient temperature, the slides were air-dried and Fluoro Jade B solution was applied for 1 min. Slides were rinsed with distilled water and photographed using 488/522 nm filters.
Assessment of Blood—Brain Barrier Integrity
The integrity of the blood—brain barrier was evaluated using Evans blue dye. Evans blue (5mL/kg of a 2% solution in PBS) was administered through tail vein injection 2 h after CCI. Mice were killed at 24 h after CCI by transcardiac perfusion with 0.9% saline under deep general anesthesia. The brains were then removed and bisected at the midline. Injured and uninjured hemispheres were weighed and immersed in 3 mL N,N-dimethylformamide for 72 h, and the solution containing extravasated Evans blue was analyzed by spectroscopy (612 nm). The amount of Evans blue per gram brain tissue was calculated for each hemisphere from a standard curve. Blood—brain barrier permeability was expressed as nanogram Evans blue (left-right hemisphere) per gram brain tissue.
Assessment of Brain Edema
Brain edema was assessed by measuring brain water content using the wet-dry/wet brain weight method. Mice subjected to CCI were killed at 24 h and the brains removed and bisected at the midline to separate right and left hemispheres. The cerebellum and brain stem were removed. Uninjured brain tissue anterior and posterior to the contusion was removed by sectioning in the coronal plane. The wet weight of the remaining injured and uninjured hemispheres was obtained. Brain tissue was dried at 99°C for 48 h and dry hemispheric weights were obtained. Hemispheric brain water content was expressed as (wet-dry/wet) weight × 100%. Brain edema was expressed as the percentage difference in brain water content (L-R) hemispheres.
Assessment of Brain Neutrophil Counts
For each mouse, a brain section taken from the approximate center of the contusion was selected for analysis. Within this section, × 400 fields medial and lateral to the contusion were randomly selected and photographed using a CCD camera (SPOT Advanced, Diagnostic Instruments, Sterling Heights, MI, USA). Positively stained cells with the characteristic morphology of neutrophils were counted in each of the two fields. An average neutrophil count/ × 400 field was obtained for each mouse.
Assessment of Lesion Volume and Brain Tissue Loss
Morphometric image analysis (MCID, Imaging Research Inc., St Catherines, ON, Canada) was used to determine lesion volume (n = 6 to 7 per group) after CCI. Mice were deeply anesthetized with isoflurane, decapitated, and the brains removed and frozen in nitrogen vapor. Coronal brain sections (12 μm) were cut on a cryostat at 0.5 mm intervals from anterior to posterior and mounted on poly-
Semiquantitative Assessment of Acute Hippocampal Cellular Degeneration
Mice were killed at 6 h after CCI and the brains removed and frozen in nitrogen vapor. The 6 h time point was chosen because cell death is robust, but overt cell loss is not detectable at this early time after injury. Cryostat brain sections (12 μm) were cut at 200 μm intervals from anterior to posterior of the injured hippocampus and placed on poly-
Morris Water Maze Paradigm 1
An MWM task was used to evaluate spatial memory performance. The MWM apparatus consisted of a white pool (83 cm diameter, 60 cm deep) filled with water to a depth of 29 cm with several highly visible cues located on the walls of each of the four quadrants. Water temperature was maintained between 21°C and 25°C. A clear plexiglass goal platform 5 cm in diameter was positioned 0.5 cm below the water's surface approximately 15 cm from the southwest wall. Each mouse was subjected to 1 or 2 trials per day. For each trial, mice were randomized to one of the four starting locations (north, south, east, or west) and placed in the pool facing the wall. Mice were given a maximum of 60 (naive animals) or 90 sees (injured animals) to find the submerged platform. If the mouse failed to reach the platform by the allotted time, it was placed on the platform by the experimenter and allowed to remain there for 10 sees. Mice were placed in a warming chamber for at least 4 mins between trials. To control for possible differences in visual acuity or sensorimotor function between groups, two trials were performed using a visible platform raised 1 cm above the surface of the water. Performance in the MWM was quantitated by latency to the platform.
Morris Water Maze Paradigm 2
Mice were first subjected to a series of 12 visible platform trials, in which the platform location was randomly changed after each trial and no spatial references were used to facilitate navigation to the platform. In this paradigm, only one starting location was used per trial. The visible platform trials were performed over 2 days of testing (six trials per day). Maximum latency to the visible platform was 30 secs. Mice that failed to get on the platform after 30 secs were gently placed there by the investigator and allowed to remain for 10 secs. The 12 visible trials were followed by five sets of hidden platform trials as described above in MWM paradigm 1. After hidden platform testing, two sets of visible platform trials (in this case, using four starting locations per trial and a maximum latency of 30 secs) were again performed. Data from the first 12 visible platform trials, the hidden platform trials, and the last two visible platform trials were quantified by latency to the platform and analyzed separately.
Evaluation of Motor Performance
Vestibulomotor function was assessed using a wire grip test (You et al, 2007). Mice (n = 9 to 10 per group) were placed on a metal wire (45 cm long) suspended 45 cm above a foam pad and allowed to traverse the wire for 60 secs. The latency that a mouse remained on the wire within a 60-sec interval was measured, and wire grip scores were quantitated using a 5-point scale (You et al, 2007). The wire grip test was performed in triplicate and an average value calculated for each mouse on each day of testing.
Statistical Analyses
Data are expressed as mean ± s.e.m. Volumetric data, brain edema, blood—brain barrier data, and neutrophil counts were analyzed by rank sum test. Motor and Morris water maze test data were analyzed by two-factor repeated measures analysis of variance (group × time) followed by the Student-Neuman-Keuls test. For all comparisons, P <0.05 was regarded as significant.
Results
All mice survived mild (0.2 mm) and moderate (0.6 mm) CCI and appeared healthy during the experimental period. Baseline blood pressure and arterial blood gases did not differ between MBL KO and WT mice (Table 1).
Baseline physiological variables in wild-type and mutant mice
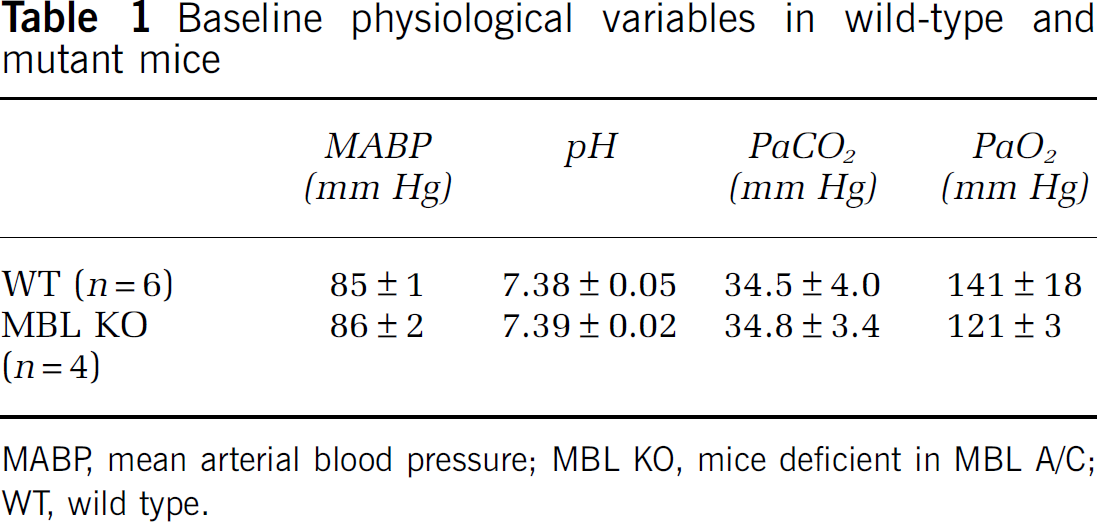
MABP, mean arterial blood pressure; MBL KO, mice deficient in MBL A/C; WT, wild type.
After moderate CCI, MBL-C colocalized with blood vessel endothelium in contused cortex and subcortical regions (hippocampus and thalamus) as early as 5 and 60 mins, and at 24, 48, and 72 h after injury (Figures 1A to 1C and data not shown). No MBL staining was observed in the contralateral (uninjured) hemisphere (Figure 1D). Mannose binding lectin antibodies did not react with injured MBL KO brain tissue (Figure 1E), and no labeling was observed for MBL in injured brain sections with omission of the primary antibody (data not shown). In contrast to endothelial cells, we did not observe MBL staining in neurons or glial cells in injured brain. Mannose binding lectin immunostaining was also not detected in naive mouse brain. In addition to MBL-C, we also observed MBL-A staining in injured vascular endothelium (Figure 1F). In agreement with the immunohistochemistry results, Alexa-Fluor 488-labeled rhMBL injected intravenously immediately before CCI labeled lectin-positive blood vessels in the injured cortex and subcortical brain regions of MBL KO mice at 60 mins (Figure 1G). Similar expression patterns of MBL labeling were also observed in the mild CCI paradigm (data not shown). Thus, MBL reacts with vascular endothelium early after CCI; however, MBL may also react with other cell types below the level of detection by our immunohistochemical protocols.
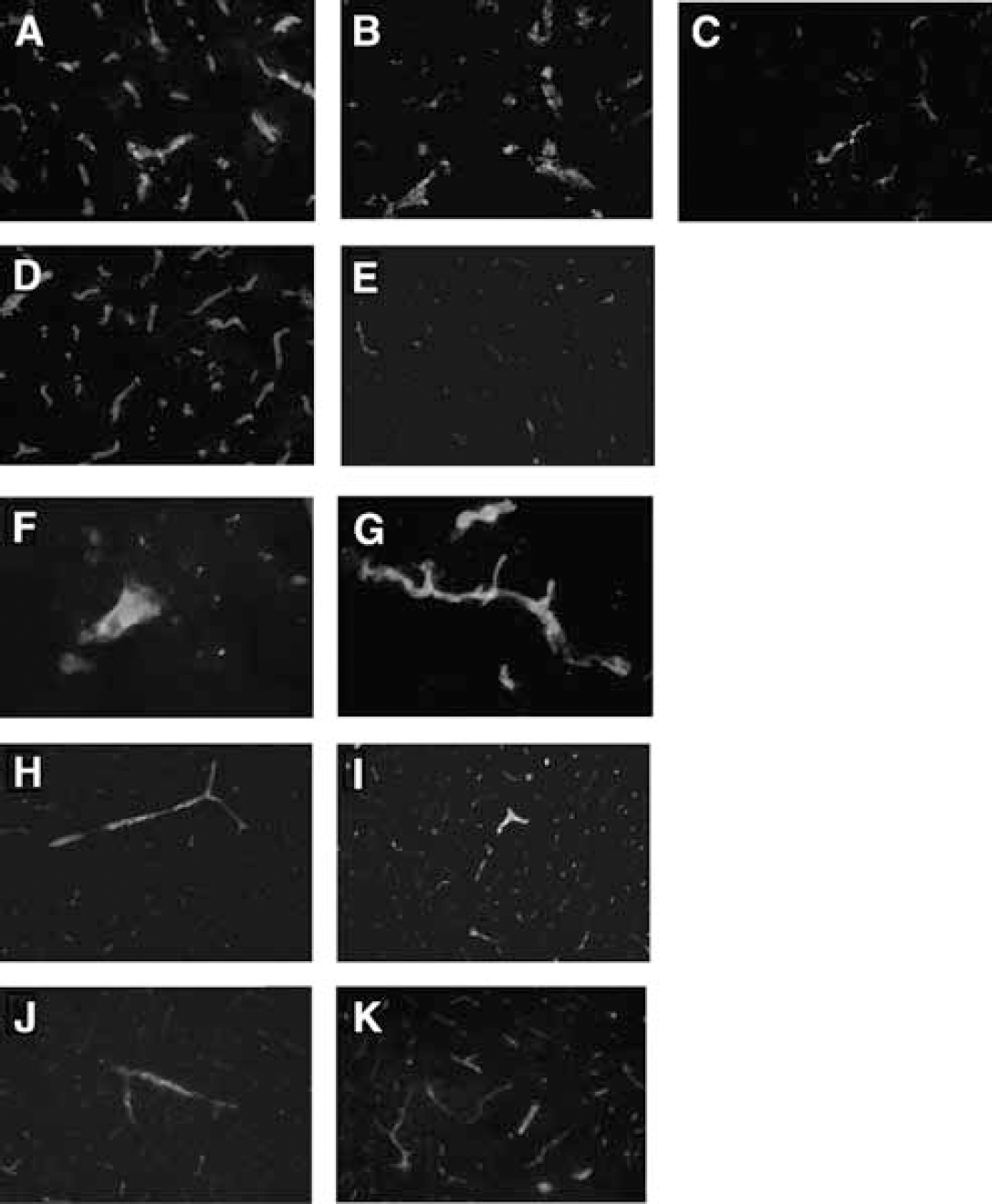
Immunohistochemical localization of MBL A/C, rhMBL, C3d, and C4c (depicted in green) in cortical endothelium (red) early after moderate controlled cortical impact (CCI). Mannose binding lectin-C colabeling with vascular endothelium was observed in injured cortical vessels as early as 5 mins (
To assess the activation of C3 in MBL KO mice, we used an antibody that specifically recognizes C3d (an activated C3 cleavage product) in injured mouse brain (You et al, 2007). After moderate CCI, robust immunostaining for C3d was observed on vascular endothelium of WT and MBL KO mice (Figures 1H and 1I), suggesting that C3 activation is independent of MBL in our CCI model. To examine whether C3 activation in MBL KO mice could occur through the classic pathway, we examined vascular endothelium for C4 deposition known to occur after CCI (You et al, 2007). Robust C4c immunoreactivity was observed after CCI in WT and MBL KO mice (Figures IJ and 1K), suggesting an intact classic pathway in MBL KO mouse brain (Shi et al, 2004). Thus, C3 activation after TBI may occur through the classic or alternative pathway in MBL KO mice.
On the basis of a possible functional interaction between MBL and brain vascular endothelium, we next examined the effect of MBL deficiency on posttraumatic blood—brain barrier permeability, leukocyte extravasation, and brain edema after moderate CCI. Blood—brain barrier permeability did not differ between MBL KO (30.6 ± 11.9 ng Evans blue per g brain, n = 6) and WT (31.4 ± 6.9 ng Evans blue per g brain, n = 6) mice. Brain edema at 24 h after injury (the time of peak edema in the CCI model), assessed as differences in the percentage brain water content between injured and non-injured hemispheres, did not differ between MBL KO (2.6 ± 0.2%, n = 5) and WT (2.7 ± 0.2%, n = 5) mice. Brain neutrophil counts in injured cortices also did not differ between MBL KO and WT mice (Figures 2A and 2B).
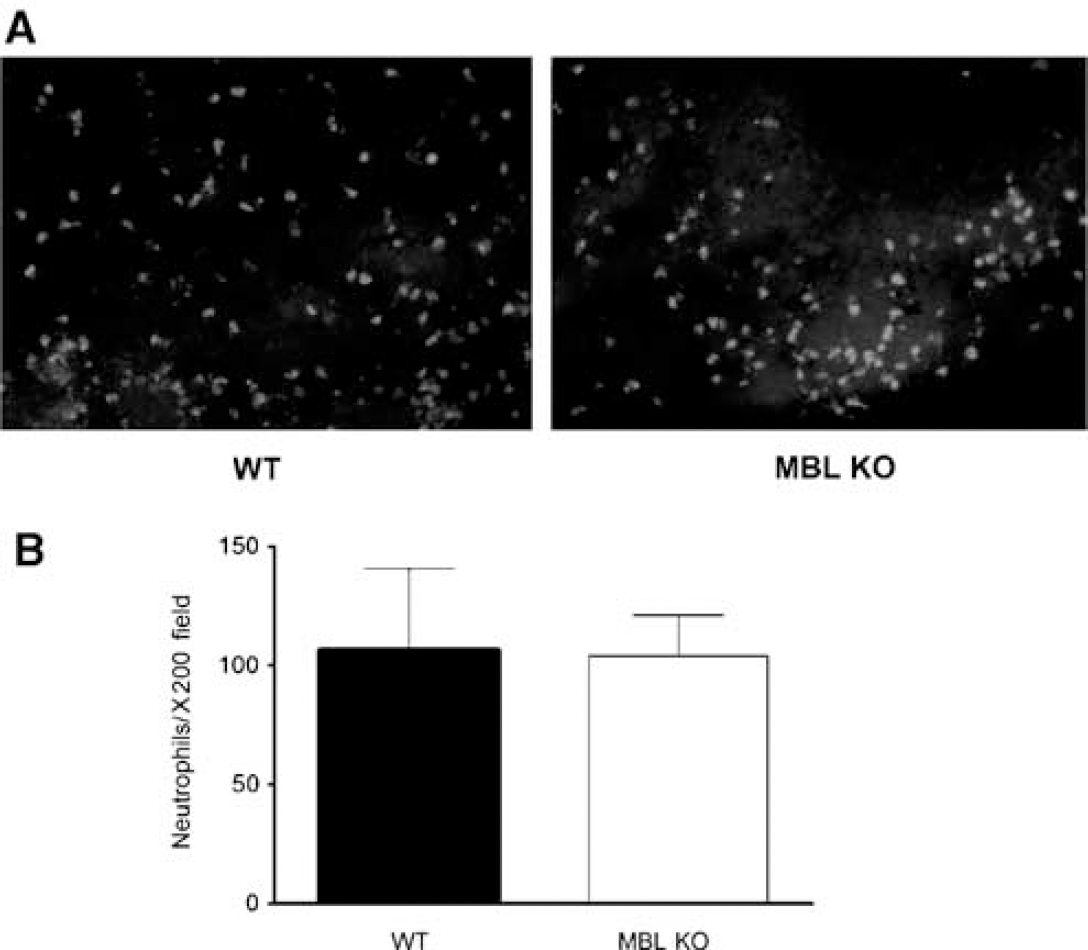
Mannose binding lectin deficiency does not affect brain neutrophil accumulation after moderate controlled cortical impact (CCI). (
We next evaluated the effect of MBL KO on posttraumatic brain tissue damage (Figure 3A). Figure 3B shows that in mice subjected to moderate CCI, posttraumatic brain lesion volume (2 weeks) was similar in MBL KO and WT mice. Similarly, lesion size did not differ in mice subjected to mild CCI (MBL KO 4.4 ± 0.2 mm3, WT 4.6 ± 0.4 mm3). However, MBL KO mice had approximately 15% decreased overall brain hemispheric volume compared to WT mice (P<0.05), suggesting that adult brain size is reduced by MBL A/C deficiency. At 8 weeks after CCI, brain tissue atrophy was similar between MBL KO and WT mice subjected to moderate CCI (Figure 3C).
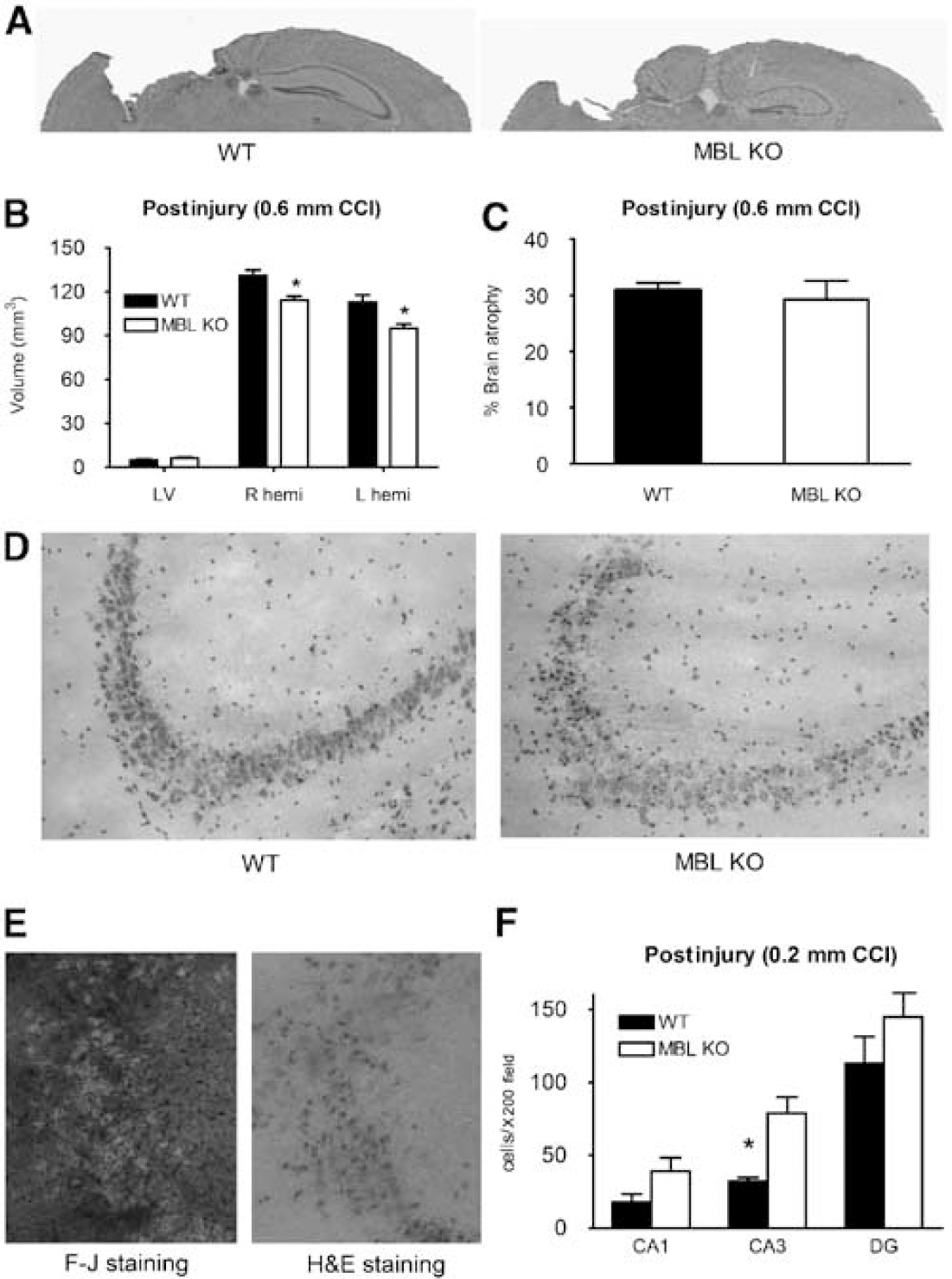
Histopathology of controlled cortical impact in MBL KO and WT mice. (
To evaluate a possible role for MBL in acute brain cell death, we assessed hippocampal neuron degeneration at 6 h after mild CCI, a time of robust neuronal injury that precedes overt cell loss in this region (Figures 3D to 3F). Acute neuronal degeneration in CA3 was almost two-fold greater in MBL KO versus WT mice (P<0.01), but acute cellular degeneration in CA1 and dentate gyrus did not differ between groups at this early time point. Of note, degenerating cells labeled by hematoxylin and eosin stain also labeled robustly with Fluoro Jade B (Figure 3E).
To determine the effect of MBL deficiency on functional outcome after CCI, we used two MWM paradigms and a wire grip test. In a standard MWM paradigm 1, naive MBL KO mice and C57Bl/6 control mice did not differ in their performance in hidden or visible platform trials (Figure 4A). After moderate CCI, latency to the hidden platform was similar among MBL KO, WT, and MBL KO mice reconstituted with rhMBL (Figure 4B). In subsequent visible platform trials, MBL KO mice had a trend toward impaired performance on trial 1 compared to WT and rhMBL-reconstituted MBL KO mice; however; the differences did not reach statistical significance in the repeated measures analysis of variance.
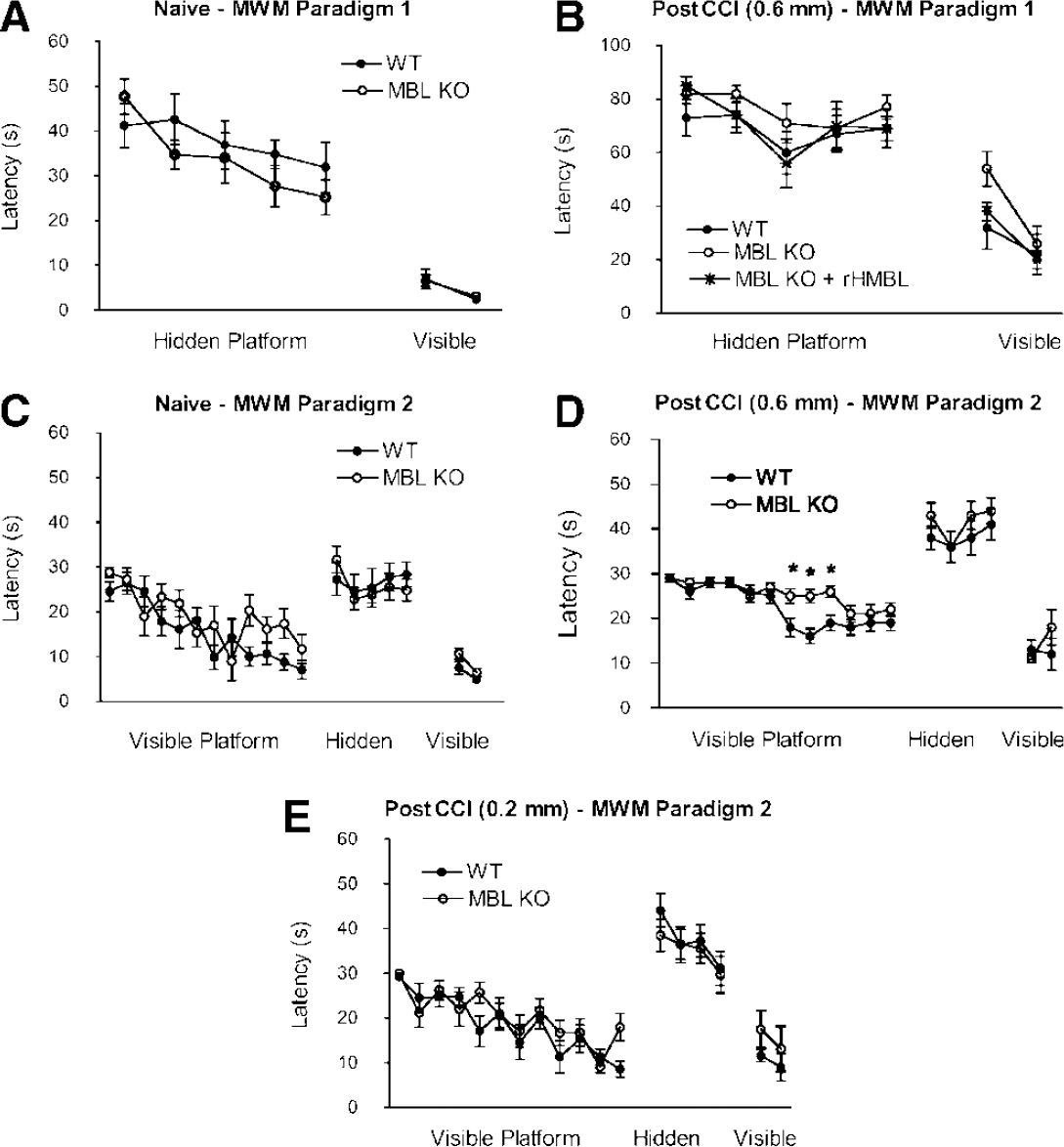
Increased cognitive deficits in MBL KO mice after controlled cortical impact (CCI). Naive (uninjured) and injured mice were assessed in two MWM paradigms at 8 to 14 days after two levels of CCI. (
To assess possible nonspatial learning deficits in injured MBL KO mice, we used MWM paradigm 2. Naive MBL KO and WT mice did not differ in their performance in visible or hidden platform trials (Figure 4C). However, after moderate CCI, performance in the visible platform trials (initiated before hidden platform trials) was significantly worse in MBL KO mice versus WT mice (P<0.05 for group, +P<0.01 on the days indicated (Figure 4D)). In subsequent hidden platform trials, both groups performed equally well. In contrast to moderate CCI, mild CCI did not produce a MWM deficit versus naive mice in WT or MBL KO animals (Figure 4E).
To assess whether impaired MWM performance in MBL KO mice could be attributed to a gross deficit in sensorimotor function after CCI, we used the wire grip test. Compared to naive mice, motor performance was impaired after moderate and mild CCI in both MBL KO and WT mice (P<0.05, Figures 5A and 5B). However, no differences between groups were observed in motor scores over the experimental period at either of the two injury levels used.
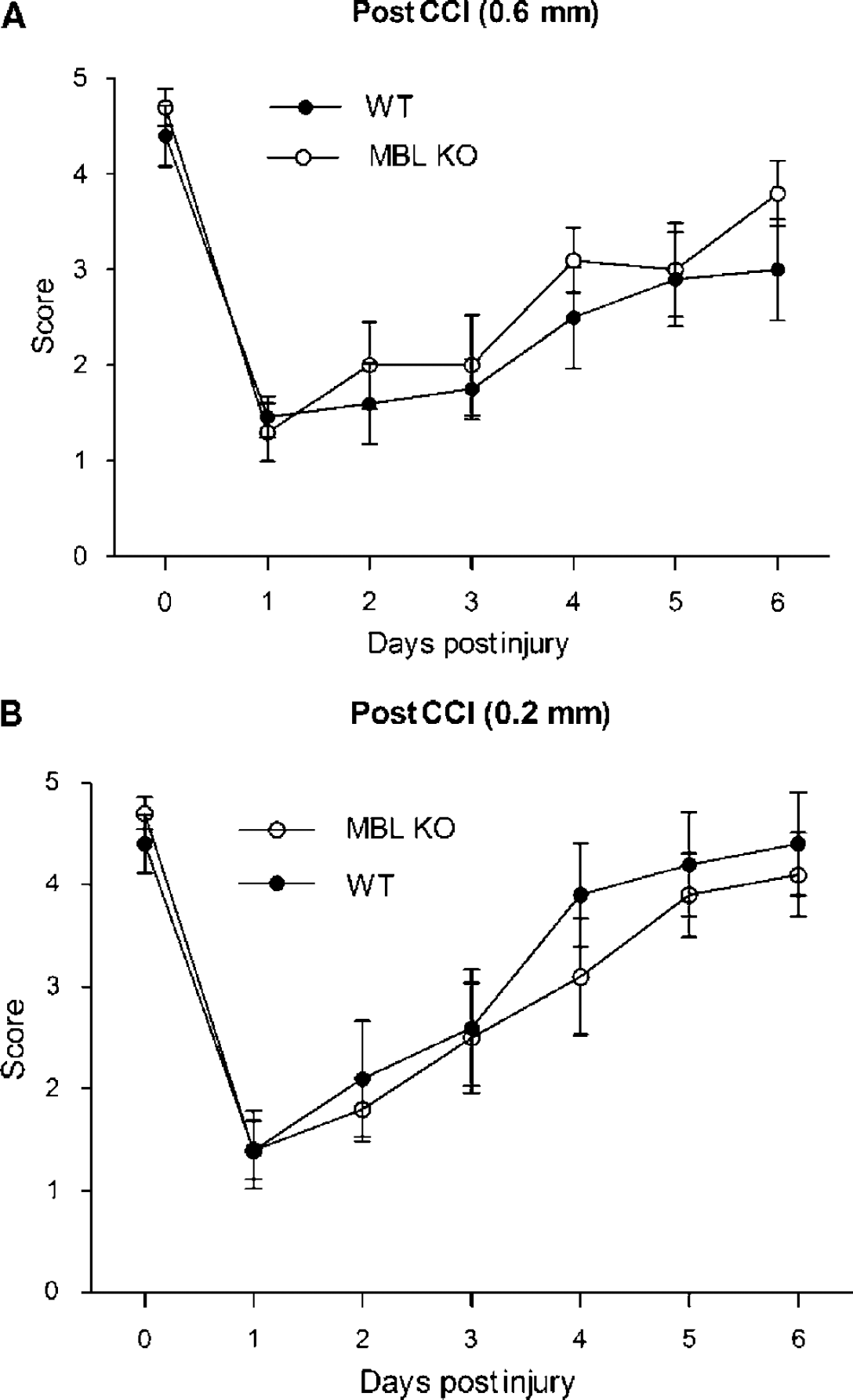
Effect of MBL KO on postinjury motor deficits after two levels of controlled cortical impact (CCI). (
Discussion
The major findings of this study are that MBL deficiency exacerbates acute CA3 cell death and cognitive dysfunction independent of complement activation after CCI and that MBL contributes to brain size in adult mice. To our knowledge, this is the first translational study of MBL in an acute brain injury model. The data suggest an endogenous neuroprotective role for MBL and a heretofore unknown functional linkage between innate immunity and outcome after TBI.
We found that mice deficient in MBL had increased CA3 neuronal degeneration as early as 6 h after CCI. This effect was specific to CA3, as the numbers of degenerating neurons in CA1 and dentate gyrus did not differ between groups. The CA3 region is particularly vulnerable to traumatic and excitotoxic cell death (Clark et al, 1997; Colicos et al, 1996), suggesting that endogenous MBL may function to inhibit excitotoxic neuronal death directly or indirectly after TBI. In closed head TBI models, C3 activation promotes neuronal cell death (Leinhase et al, 2006; Rancan et al, 2003). In contrast, MBL appears to be an endogenous neuroprotectant that limits cell death after CCI, perhaps by initiating antiapoptotic neuronal survival signaling pathways. Recent data have shown the protective effects of other members of the collectin family in this regard, such as antiapoptotic effects of surfactant protein A on pulmonary epithelial cells (White and Strayer, 2002). Alternatively, MBL may act indirectly by promoting phagocytosis of dying cells, in which case MBL deficiency could increase the numbers of remaining degenerative cells after CCI in CA3 (Takahashi et al, 2006). Although further work is needed to determine the mechanism(s) of MBL-mediated neuroprotection in CCI, the data showing robust activation of C3 in MBL KO mice suggest that inhibition of terminal complement is not the primary mechanism of protection.
Increased CA3 cell death after TBI has been observed with inhibition of inducible nitric oxide synthase in rats and mice (Sinz et al, 1999), in apolipoprotein E knockout mice (Han and Chung, 2000), and in GFAP knockout mice (Otani et al, 2006). This study suggests that MBL is also an endogenous neuroprotectant against TBI-induced CA3 cell death. Of note, female (but not male) transgenic mice overexpressing human copper/zinc superoxide dismutase had decreased CA3 cell death after experimental TBI (Igarashi et al, 2001). In this study, we used female and male mice to achieve sufficient power for robust statistical analysis of behavioral data. However, our study was not powered adequately to explore possible gender effects of MBL deficiency. This is an important issue, as interaction between gender and genotype may contribute to susceptibility to seizures after TBI (Kochanek et al, 2006) and gender may influence cell death after experimental TBI depending on the model used. Gender differences in CA3 cell death and hippocampal or cortical tissue damage were not observed in WT mice subjected to CCI (Bruce-Keller et al, 2007; Hall et al, 2005), but were reported in mice subjected to closed head diffuse TBI (Kupina et al, 2003). Further studies are needed to examine a potential interaction between MBL deficiency and gender in the CCI model as well as in TBI patients.
Our data suggest that the modest cognitive dysfunction observed in injured MBL KO mice quite likely involves deficits in nonspatial learning. Because spatial and non-spatial learning components may influence performance in hidden platform trials (Micheau et al, 2004), we used two separate MWM paradigms to identify more precisely the MBL KO injury phenotype (Gerlai et al, 2002). In MWM paradigm 2 (which began with cued platform trials), we found that acquisition of (nonspatial) procedural learning was impaired in MBL KO mice. This impairment was reversible over time and did not lead to increased deficits in subsequent hidden platform trials compared to those observed in injured WT mice. Taken together, the data are consistent with a modest nonspatial learning deficit in injured MBL KO mice. Although the exact mechanism of the nonspatial cognitive deficits in injured MBL mice remain to be identified, it is unlikely that increased CA3 cell death can account for impaired nonspatial learning, as CA3 lesions do not influence performance in visible platform trials in the MWM (Gerlai et al, 2002).
Because collectins, including MBL, are important for host defense against bacterial and viral pathogens, deficiency of MBL could conceivably exacerbate TBI by increasing susceptibility to infection (Moller-Kristensen et al, 2006; Shi et al, 2004; Wellmer et al, 2000). However, increased infection rate would not account for differences in CA3 cell death at 6h, and we did not observe sickness behavior or brain abscesses in injured MBL KO mice throughout the experimental period. There is some evidence that MBL might be important for recovery of synaptic function and hence overall cognitive performance after TBI (Lehmann et al, 1993). Our immunohistochemical data suggest that MBL may also influence endothelial signaling after TBI. How endothelial signaling may affect neuronal cell death or cognitive dysfunction after TBI is a relatively unexplored field. In Alzheimer's disease, vascular endothelial nitric oxide synthase (eNOS) downregulation and chronic brain hypoperfusion contribute to learning and memory deficits, as do alterations in levels of vasoactive mediators (e.g., VEGF, vascular endothelial growth factor) linked to learning and memory (de la Torre and Aliev, 2005; Tong et al, 2005; Wang et al, 2006). In addition, endothelial cells can directly affect neuronal transmission through the release of endothelial-derived nitric oxide (Garthwaite et al, 2006). Thus, MBL might influence postinjury cognitive function and cell death through a number of mechanisms involving endothelial-neuronal signaling.
In contrast to systemic ischemia/reperfusion models in which MBL KO abrogates C3 activation (Hart et al, 2005; Moller-Kristensen et al, 2005; Walsh et al, 2005), MBL KO did not prevent C3d deposition on vascular endothelium after CCI. In addition, posttraumatic brain neutrophil accumulation, known to be C3-dependent (Kaczorowski et al, 1995; You et al, 2007), was similar in MBL KO and WT mice. Thus, MBL A/C, and hence the lectin pathway, appears to be dispensable for terminal complement activation after CCI, in agreement with previous reports from our group and others that indicate a predominant role for the alternative pathway in TBI (Rancan et al, 2003; You et al, 2007). The lack of differences in postinjury cognitive function between C3 KO and WT mice also argues against a critical role for C3 in the MBL KO postinjury (cognitive) phenotype (You et al, 2007). Thus, MBL appears to influence outcome after TBI by mechanisms fundamentally different from those involved in systemic ischemia-reperfusion injury (Zhang et al, 2006). In line with this notion, we recently reported a C3-independent role for C4 in the pathogenesis of motor dysfunction and brain tissue damage after CCI (You et al, 2007).
We conclude that MBL limits CA3 cell death and may promote normal recovery of cognitive brain function after CCI. If so, patients with MBL gene polymorphisms associated with low or dysfunctional serum MBL might be at risk for worse histopathological and perhaps functional outcome after TBI. If so, administration of rhMBL may offer a novel treatment for this subpopulation of TBI patients. Similarly, the possibility that supraphysiological doses of MBL might improve outcome after TBI in patients regardless of serum MBL activity also warrants investigation.