
Abstract
Although vascular dysregulation has been documented in patients with extracranial vascular disease, transient ischemic attacks, and stroke, the pathomechanisms are poorly understood. To model thromboembolic stroke in rats, photochemically induced nonocclusive common carotid artery thrombosis (CCAT) was used to generate a platelet thrombus in the carotid artery of anesthetized rats. After CCAT, platelet aggregates break off the thrombus, travel to the distal cerebral vasculature, damage blood vessels, and cause small infarctions. The authors hypothesized that deficits in the endothelial nitric oxide synthase (eNOS) pathway may be responsible for vascular dysfunction after embolic stroke. To examine the functional status of the eNOS system, they measured eNOS-dependent dilation after CCAT by applying acetylcholine through a cranial window over the middle cerebral artery. The authors also measured eNOS mRNA and protein in the middle cerebral artery to determine whether functional changes were caused by alterations in expression. eNOS-dependent dilation was reduced at 6 hours, elevated at 24 hours, and returned to baseline 72 hours after CCAT. Endothelial nitric oxide synthase mRNA increased at 2 hours and was followed by a rise in protein 24 hours after CCAT. Changes in the eNOS system may account for some of the observed vascular deficits in patients with cerebrovascular disease.
Keywords
Vascular damage after ischemia or embolic stroke is important as both a primary and secondary injury mechanism (Dietrich et al., 1993a; del Zoppo et al., 1998, 2000). Alterations in the structural, molecular, or physiologic characteristics of blood vessels may lead to blood–brain barrier permeability, edema, or hemodynamic perturbations, with devastating consequences to downstream tissue (Dietrich et al., 1993a; Faraci and Heistad, 1998). Hemodynamic perturbations are evaluated in patients diagnosed with either vascular stenosis (White and Markus, 1997) or a history of transient ischemic attacks to assess vascular health and stroke risk (Ringelstein et al., 1992; Gur et al., 1996; Markus and Cullinane, 2001). These perturbations are tested by measuring changes in cerebral blood flow elicited by vasodilating stimuli such as hypercapnia (Silvestrini et al., 2000), acetazolamide (Chollet et al., 1989), or controlled drops in mean arterial pressure (Nishimura et al., 1999). Although many clinical studies agree that deficits in hemodynamic response are important indicators for future stroke, the mechanisms underlying these deficits are poorly understood.
Dysfunctional endothelial-mediated vasodilation may be responsible for experimental and clinical observations of hemodynamic deficits. Nitric oxide (NO) is produced by Ca2+/calmodulin stimulation of the constitutively expressed enzyme endothelial nitric oxide synthase (eNOS) and regulates endothelial-dependent dilation by activating guanylate cyclase in vascular smooth muscle. Deficits in eNOS expression or function could account for some of the vascular lesions observed in cerebrovascular disease. For example, the nonselective NOS inhibitor NG-nitro-
A rat model of common carotid artery thrombosis (CCAT) was developed to study thromboembolic stroke. CCAT produces a photochemically induced platelet thrombus in the common carotid artery that releases emboli into the distal cerebral vasculature. Emboli interact with vessels, causing ultrastructural changes in endothelial cells, blood-brain barrier permeability, and infarction. Recent experiments that combined CCAT with either global ischemia (Dietrich et al., 1999a) or a second CCAT (Danton et al., 2002) found that a second insult resulted in severe lesions between 1 and 5 days after the initial insult. Inhibition of eNOS during CCAT resulted in larger infarcts, more severe behavioral deficits, and a greater number of embolic occlusions (Stagliano et al., 1997a,b). Thus, deficits in eNOS-dependent dilation after the initial insult may make the brain more vulnerable to a second insult by altering endothelial control of blood flow. Cerebral infarctions after single episodes of CCAT are usually less than 500-μm in diameter and would not be expected to cause significant hemodynamic changes in the early branches of the middle cerebral artery (MCA). Damage within the large vessel is most likely caused by a primary vascular insult.
This study tested the hypothesis that CCAT results in alterations of eNOS expression and function in the MCA. Carotid Doppler flow monitoring verified that the carotid thrombus resulted in embolic and hemodynamic deficits. A cranial window preparation was used to measure eNOS-dependent acetylcholine-induced vascular dilation. Reverse transcription polymerase chain reaction (RT-PCR) and Western blotting evaluated vascular changes in eNOS mRNA and protein content.
MATERIALS AND METHODS
Experiments were performed on male Wistar rats (n = 81) weighing 300 to 380 g, obtained from Charles River breeders (Wilmington, MA, U.S.A.). The Animal Care and Use Committee at the University of Miami School of Medicine approved animal protocols. All reagents were purchased from Sigma-Aldrich Corp. (St. Louis, MO, U.S.A.) unless otherwise specified.
Surgical procedures
Rats were anesthetized with 4% halothane, 70% nitrous oxide, and 30% oxygen. After induction, halothane was lowered to 1.5% and administered through a rodent mask (51610, Stoelting Co., Wood Dale, IL, U.S.A.). The right femoral artery was catheterized with PE-50 polyethylene catheters for monitoring and maintaining arterial blood gases and mean arterial pressure to within normal limits. The femoral vein was catheterized for administration of drugs. Rats were intubated, paralyzed with pancuronium intravenously for survival surgeries or D-tubocurarine for nonsurvival surgeries and ventilated mechanically (50095-1, Stoelting Co., Wood Dale, IL, U.S.A.) with 0.5% halothane. Body temperature was monitored and held between 36°C and 37°C throughout surgery via a rectal temperature probe and heating pad (CMA150, CMA Microdialysis AB, North Chelmsford, MA, U.S.A.).
Common carotid artery thrombosis
The common carotid artery was exposed by blunt dissection through a midline, ventral skin incision. Nerves and muscle surrounding the artery were retracted, and the cavity was filled with saline. Thirty minutes after blood gases were adjusted to within normal limits and the carotid artery was exposed, laser irradiation was begun. Common carotid artery thrombosis was produced via the photochemical method (Watson et al., 1987; Futrell et al., 1988). Erythrosin B (30 mg/mL in 0.9% saline) was injected via the femoral venous catheter (40 milligrams per kilogram) during a 2-minute period with an infusion pump (PHD2000, Harvard, Holliston, MA, U.S.A.). Simultaneously, a tunable argon laser (Innova 70-4, Coherent, Santa Clara, CA, U.S.A.) (wavelength, 514 nm; peak power 375 mW) was focused on the common carotid artery for 10 minutes. Carotid blood flow and the generation of emboli were monitored with a transit-time ultrasound probe (T206, Transonic Systems, Ithaca, NY, U.S.A.) operating at 3.6 MHz and placed distal to the site of thrombosis. Sham surgeries were performed exactly as described without irradiation. After irradiation, the catheters were removed and incisions were sutured. After surgery, rats were returned to their cages until killed.
Cranial window
Rats were divided into 4 groups; baseline rats received sham CCAT (n = 6) followed by a cranial window 24 hours later, and experimental rats received CCAT followed by a cranial window 6 hours (n = 6), 24 hours (n = 8), and 72 hours (n = 6) after CCAT. Rats were prepared as already described and were placed in a stereotaxic frame in the prone position. The cranial window was fashioned following modified, previously published procedures (Morii et al., 1986; Koenig et al., 1993). Briefly, a 4-mm diameter x 1-mm high base with 4 ports was cemented onto the bone overlying the distribution of the MCA. Using a Zeiss operating microscope (Carl Zeiss Microimaging, Thornwood, NY, U.S.A.), the bone inside the base was drilled out while saline was continually flushed over the drill bit to reduce heat and remove bone shavings. The MCA was then exposed by carefully removing the dura. To avoid damaging the surface of the brain on the bone edge and reduce bleeding while incising the dura, the rat was briefly hyperventilated to reduce intracranial pressure. A clear window of quartz glass was placed over the base, and the entire unit was sealed into place with dental acrylic. Artificial cerebrospinal fluid (155 mmol/L Na+, 2.9 mmol/L K+, 1.1 mmol/L Mg2+, and 132.8 mmol/L Cl−) was prepared, continually bubbled with 5% CO2, and infused at 0.7 mL/min into the window using a peristaltic pump (Ismatec 78017-10, Cole-Parmer, Vernon Hills, IL, U.S.A.) throughout the remainder of the experiment. Temperature of the infused artificial cerebrospinal fluid was maintained by passing the rubber tubing through a Liebig condenser (24/40, Fisher Scientific, Pittsburgh, PA, U.S.A.) heated by circulated hot water from a water bath (Lauda MT, Fisher Scientific). A heat lamp was used to maintain the temperature inside the window precisely between 36.5°C and 37°C. After preparation of the window, halothane was reduced to 0.5% and the animal was permitted to equilibrate during 30 minutes. Images were visualized through an operating microscope (82x, Zeiss) and images of the vascular field were obtained with a ½-inch CCD camera (Cohu, San Diego, CA, U.S.A.), observed on a monitor, captured, stored, and analyzed for diameter measurements (160 images per time point) using Image-Pro Plus (Silver Spring, MD, U.S.A.) interfaced to a dimension analyzer and a personal computer (C Squared, Ft. Lauderdale, FL, U.S.A.). Concentrations of acetylcholine (5 × 10−6, 5 × 10−5, and 5 × 10−4 mol/L) were dissolved in artificial cerebrospinal fluid, infused for 10 minutes, and images were taken at 2, 5, and 10 minutes. Vascular dilation to sodium nitroprusside (10−6 mol/L) was evaluated after the experiment to ensure viability of the preparation. Data were analyzed with a repeated-measures analysis of variance followed by Tukey's post hoc comparison where appropriate.
Middle cerebral artery dissections for Western blots and RT-PCR of eNOS expression
At the appropriate interval after CCAT, animals were anesthetized with 5% halothane and decapitated. The brain was quickly removed from the skull, and the MCA was cut away from adjacent pia. The region of the irradiated common carotid artery was also removed. Samples were placed in sterile tubes, frozen in liquid nitrogen, and stored at −80°C. All dissections were performed in cold saline.
Quantitative Western blots
Determination of relative levels of protein is performed by quantitative Western blot analysis. Vessels from the site of the thrombus and MCA were isolated 6, 24, and 72 hours (n = 5 in each group) after CCAT and rapidly frozen in liquid nitrogen. Total protein was isolated by homogenization in 50 mmol/L Tris, pH 8.0, 100 mmol/L NaCl, 0.1% sodium dodecyl sulfate, 0.01 mg/mL leupeptin, and 100 μmol/L PMSF, followed by boiling for 10 minutes. Proteins were concentrated approximately 10-fold by centrifugation with Microcon YM-10 filters. Protein concentration was determined using the Bio-Rad DC Protein Assay system.
Twenty-five micrograms of protein per sample was added to gel loading buffer containing 2% sodium dodecyl sulfate and boiled before loading onto an 8% polyacrylamide stacking gel and electrophoresed at 70 V in 25 mmol/L Tris, 250 mmol/L glycine pH 8.3, and 0.1% sodium dodecyl sulfate. After transfer to a polyvinyl difluoride transfer membrane (Polyscreen, NEN Research Products, Boston, MA, U.S.A.) in a 25 mmol/L Tris, 192 mmol/L glycine, pH 8.3, 10% methanol buffer for 2 hours at 200 mA, the blot was blocked overnight at 4°C in 5% nonfat dry milk in phosphate-buffered saline plus 0.1% Tween 20.
Incubation with a primary polyclonal antibody against eNOS (SC-654; Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.) was performed in the same buffer at 1:500 dilution at room temperature for 2 to 4 hours. After three 5-minute washes in 1x phosphate-buffered saline plus Tween, incubation with horseradish peroxidase–linked anti-rabbit secondary IgG (1:3,000) was done for 45 minutes at room temperature followed by washes as before. Detection of signal is performed with New England BioLabs (Beverly, MA, U.S.A.) Phototope-HRP Western Blot Detection Kit using LumiGLO reagent as per the manufacturer's protocol and visualized on X-ray film. Film was then scanned and digitized and densitometric measurements were made using the Scion program to quantify signals from each sample. After stripping of the antibody, the blot was re-incubated as already described with a monoclonal antibody against β-actin (1:5,000) (AC-15, Sigma-Aldrich Corp., St. Louis, MO, U.S.A.). The actin signal was used as an internal control for loading uniformity. Measurements of eNOS were divided by actin measurements for each sample. The mean and SD of each group were calculated and analyzed using analysis of variance followed by the LSD post hoc test.
Reverse transcription–polymerase chain reaction
The expression of eNOS in the vasculature of sham and experimental rats was measured by semiquantitative RT-PCR. Total RNA was extracted from isolated vessels 2, 24, and 72 hours (n = 5 in each group) by homogenization in a guanidinium thiocyanate lysis buffer, extracted with phenol, and precipitated in isopropanol. The RNA was pelleted by centrifugation and resuspended in buffer containing MgCl2 and RNAse-free DNAse. Fifteen minutes at 37°C degraded any contaminating DNA. The RNA was precipitated and resuspended again. RNA (0.2 μg) from each sample was reverse transcribed into cDNA using BRL's Superscript II RT according to the manufacturer's protocol with oligo-dT to prime the first strand synthesis. One fourth of the cDNA product was used in PCR with Perkin Elmer (Boston, MA, U.S.A.) Ampli-Taq Gold polymerase and gene-specific primers to produce a PCR product. The amplification conditions were determined for each set of primers such that the number of cycles fell in the linear range. eNOS was amplified as follows: 94°C for 9 minutes followed by 32 cycles at 94°C for 30 seconds, 57°C for 30 seconds, and 72°C for 30 seconds with a final 72°C elongation step for 7 minutes. Primers were designed based on published sequences and checked for specificity by BLAST search. The sequences for the eNOS primers amplified a 310-bp product and were forward: ACTGGCATTGCACCCTTCCGG and reverse, GTTGCCAGAATTCTCTGCACG. β-Actin primers (GS740, Promega, Madison, WI, U.S.A.) were used to amplify a 285-bp product under similar amplification conditions for 25 cycles. The PCR products were electrophoresed on a 1% agarose gel and photographed on an ultraviolet light box with a DNA mass ladder (Gibco-BRL) to determine relative amounts of amplimer synthesized. Measurements of eNOS PCR product were divided by actin values and analyzed by an analysis of variance followed by a Bonferroni corrected post hoc comparison.
RESULTS
Carotid blood flow
To ensure formation of a nonocclusive carotid stenosis, diameter-independent Doppler ultrasound (Drost, 1978) was used to measure blood flow three to four diameters downstream of the irradiated site. The blood column fluoresced and the carotid segment visibly constricted on irradiation. For the first 2 minutes, peak systolic blood flow increased and then began a slow decline (a representative recording is shown in Fig. 1A). Before CCAT, the carotid wave remains stable (Fig. 1B). During and after irradiation, occasional abrupt increases in flow corresponded to embolus release (Fig. C). Histologic analysis of the thrombus 5 minutes after irradiation found a 50 ± 5% (n = 5) stenosis of the arterial diameter (Fig. 1D). Carotid flow decreased during CCAT and partially recovered by 2 hours after irradiation (Fig. 1E).
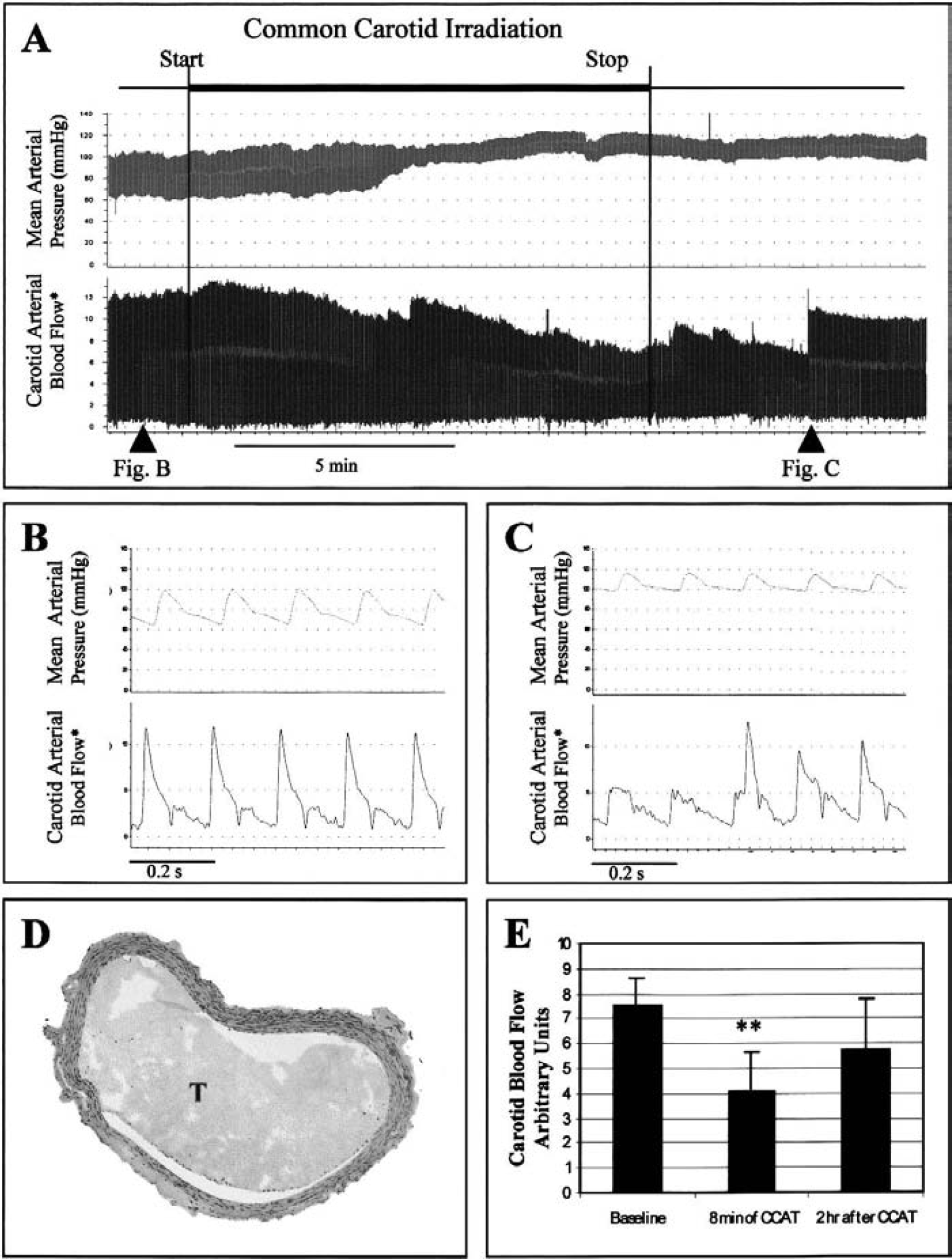
Common carotid artery thrombosis produces hemodynamic disturbances in the common carotid artery during and after irradiation. By monitoring carotid blood flow and embolus release by diameter-independent Doppler ultrasound, uniform thrombosis between rats may be confirmed.
Endothelial-dependent middle cerebral artery dilation
Endothelial-dependent dilation of the MCA was evaluated by applying acetylcholine to the MCA under observation through a cranial window. Dilation in response to acetylcholine was reported to be endothelium-dependent and prevented by NOS inhibition (Furchgott and Zawadzki, 1980; Kitazono et al., 1996). Baseline MCA diameters for sham, 6-hour, 24-hour, and 3-day groups were 171.2 ± 24.7, 148.4 ± 33, 160.9 ± 21, and 148 ± 34 μm, respectively. Therefore, differences in acetylcholine response cannot be accounted for by differences in baseline diameters. Dose-dependent dilation to acetylcholine was observed in sham, 24-hour, and 3-day animals (Fig. 2). Dilation in sham animals was dose dependent at all concentrations (P < 0.05). Dilation in 24-hour animals differed significantly (P < 0.001) between the low and middle as well as the low and high (P < 0.03) doses. Dilation in 3-day animals differed significantly between the high and low doses (P < 0.02). Dilation at 6 hours was minimal and not dose dependent. At each concentration, significant differences between groups of animals were observed (Figs. 2 and 3). Twenty-four-hour animals dilated significantly more than all other groups at all acetylcholine concentrations, whereas 6-hour animals failed to dilate at all acetylcholine concentrations. By 72 hours, there was a nonsignificant reduction in the dilatory response to acetylcholine. To determine whether molecules downstream of eNOS (such as guanylate cyclase) could account for our physiologic observations, we exposed animals to sodium nitroprusside as an indicator of endothelium-independent dilation. No significant differences were noted between sham (18.5 ± 10.1%), 6-hour (28.8 ± 12.6%), and 24-hour (27.3 ± 11.9%) groups. However, because sham animals tended to dilate less than the 6-hour or 24-hour animals, alterations in NO targets such as guanylate cyclase cannot be excluded.
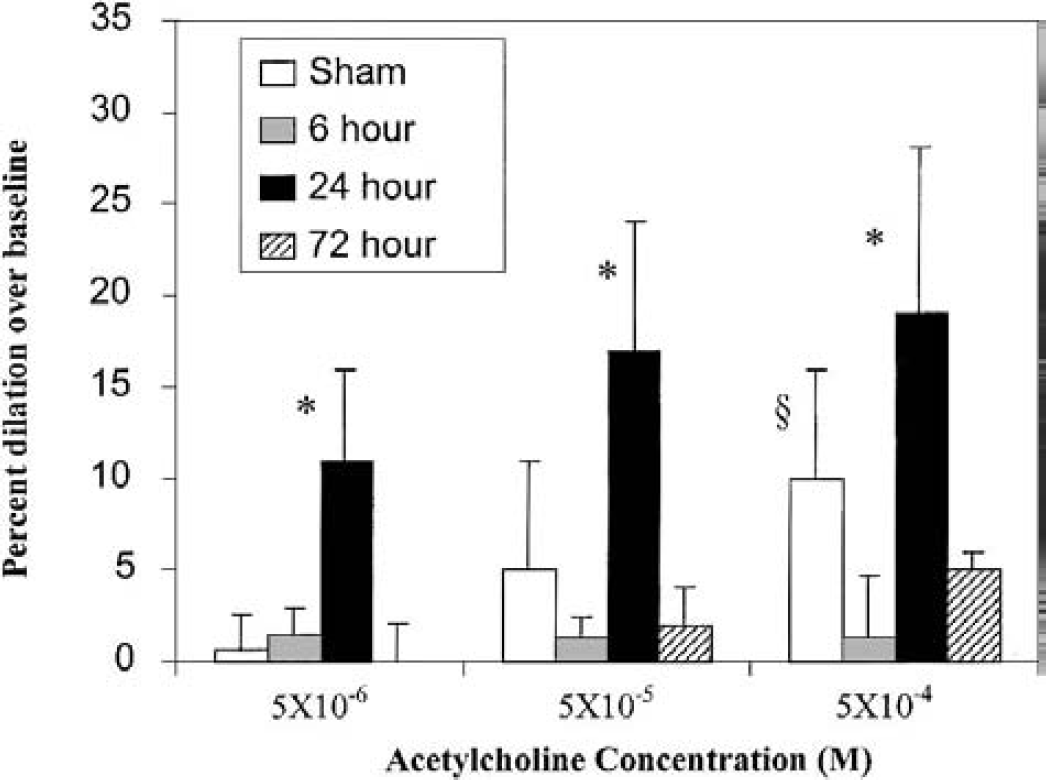
Acetylcholine-induced endothelial nitric oxide synthase–dependent dilation. Three increasing concentrations of acetylcholine were infused into a cranial window exposing the middle cerebral artery 24 hours after sham surgery, or 6, 24, and 72 hours after common carotid artery thrombosis (CCAT). Dose-dependent dilation was seen in sham, 24-, and 72-hour animals (see text). The two major findings are that vessels failed to dilate 6 hours after CCAT and dilated significantly more than all other groups 24 hours after CCAT. Data are plotted mean ± SD. *P < 0.04 compared with sham, 6 hours, and 3 days. §P < 0.02 compared with 6 hours.
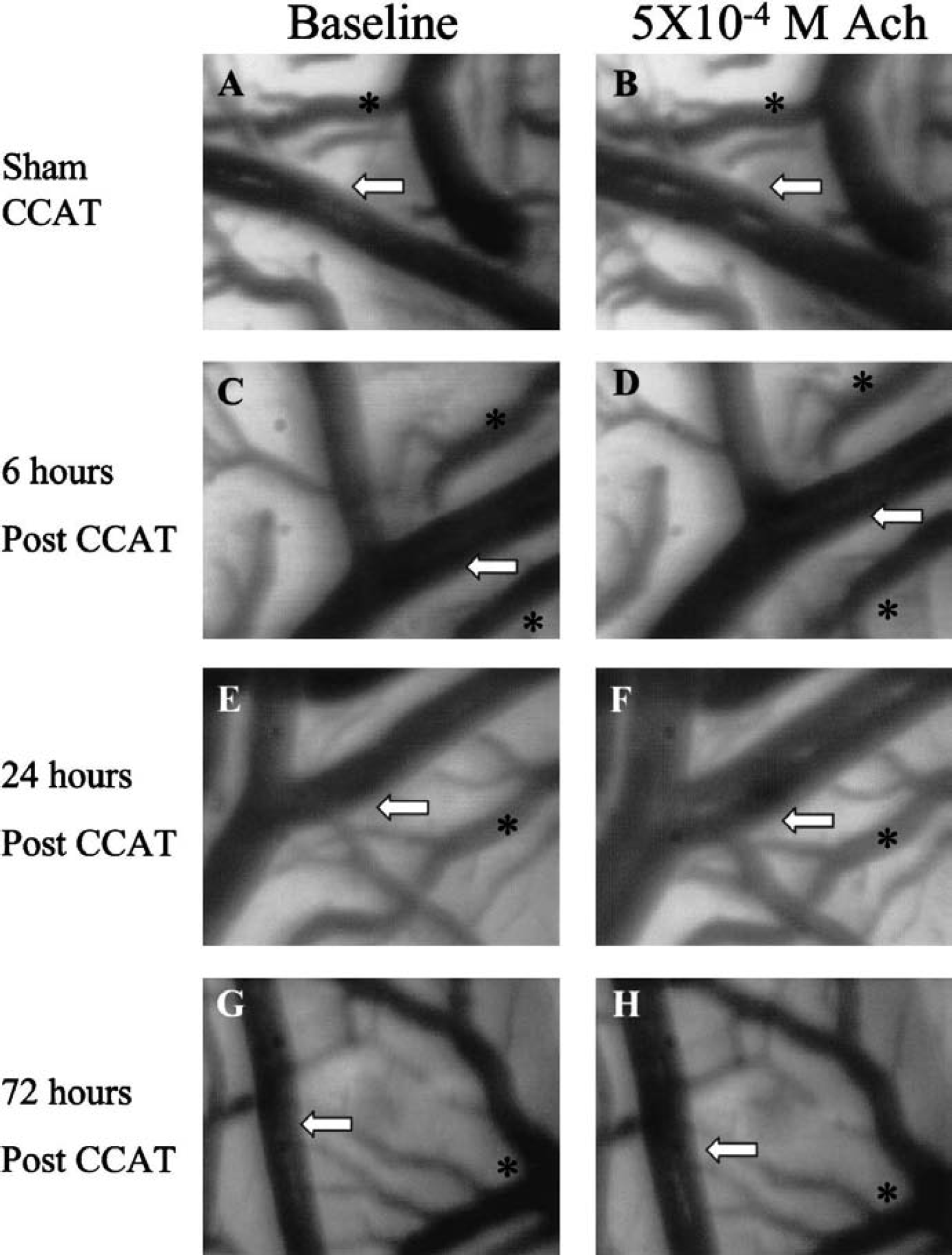
Representative examples of acetylcholine-induced endothelial nitric oxide synthase–dependent dilation at different time points after CCAT. Baseline images appear in the left column and vessels exposed to 5 × 10−4 mol/L acetylcholine appear in the right column. White arrows point to a reference position on the middle cerebral artery (MCA), and an asterisk marks a vein. Physiological MCA dilation in sham animals is observed
eNOS expression
Because eNOS expression is regulated in response to changes in blood flow and other stimuli, we evaluated eNOS expression after CCAT by RT-PCR and Western blotting to determine whether changes in eNOS may account for the alterations in eNOS-dependent dilation (Fig. 4). Optical density measurements of the eNOS RT-PCR product significantly increased (8 ± 2, P < 0.001) over baseline at 2 hours and returned to near baseline at 1 and 72 hours. eNOS protein remained stable at 6 hours, but was significantly elevated at 24 hours (2 ± 0.6, P < 0.05) with respect to baseline after CCAT and returned to baseline by 72 hours.
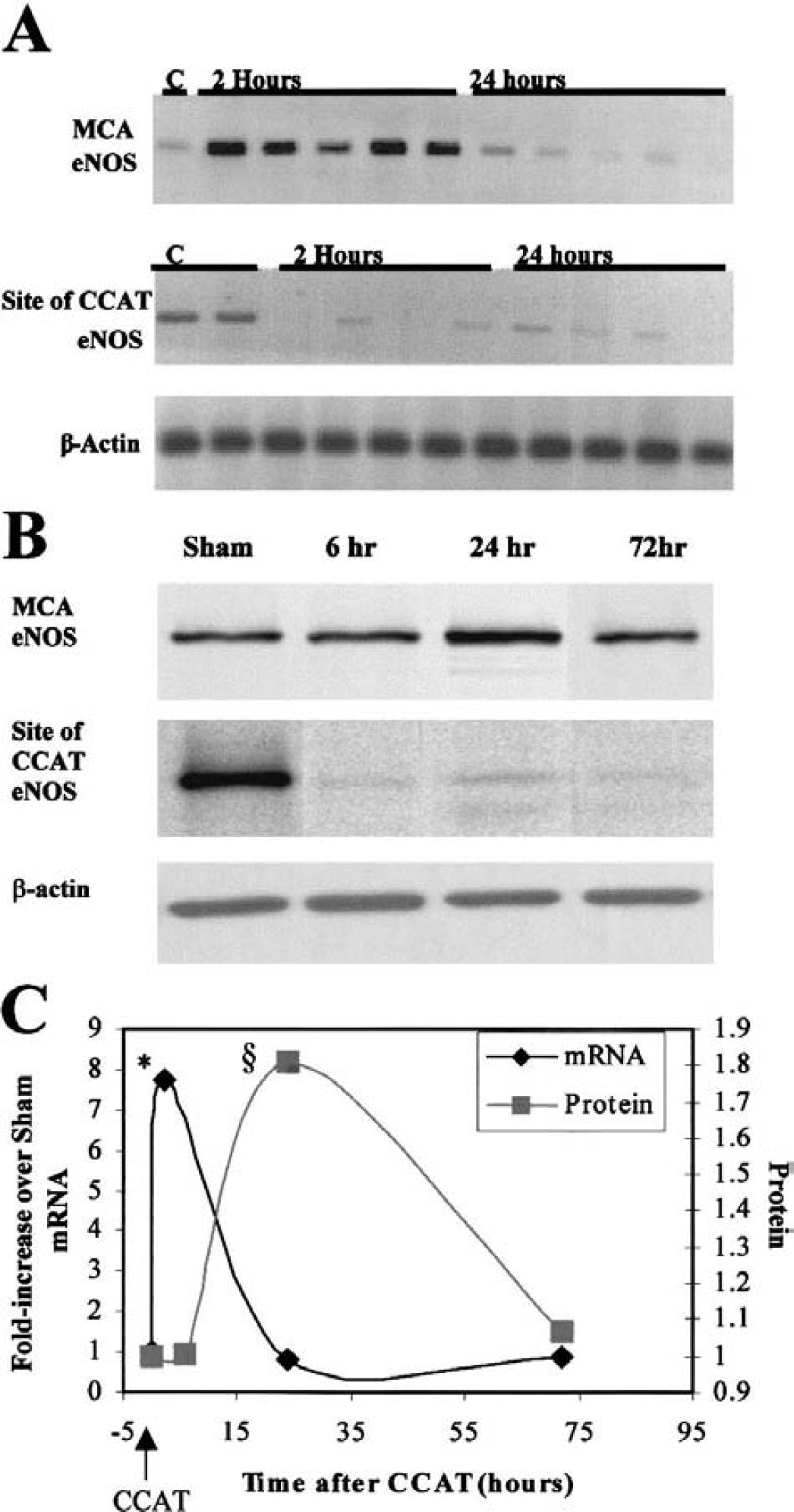
Endothelial nitric oxide synthase (eNOS) expression after common carotid artery thrombosis (CCAT) or control animals (C).
DISCUSSION
Activated platelets release protein-signaling molecules that influence endothelial cell physiology and gene expression. Molecular changes in endothelial cells have been observed in models of ischemia and inflammation in vivo (del Zoppo and Hallenbeck, 2000). Similar changes were also found in in vitro models that exposed cultured endothelial cells to activated platelets, suggesting that activated platelets alter the expression of endothelial genes (Lindemann et al., 2001; Bustos et al., 2001). CCAT produces activated-platelet emboli that are detectable in cerebral vessels for up to 3 hours (Dietrich et al., 1993b). Rats that either underwent CCAT or transfusion with thrombogenically activated blood collected downstream from platelet thrombi experienced ultrastructural, blood–brain barrier, and hemodynamic deficits (Dietrich et al., 1988; Dietrich et al., 1991). Deficits in cerebral blood flow were detected by the C14iodoantipyrine method and were resolved at 1 and 24 hours after thromboembolism (Dietrich et al., 1991; Stagliano et al., 1997b).
Alterations in the vascular response to platelet byproducts and surface proteins change depending on the condition of the endothelium and may be a therapeutically important complication of eNOS pathophysiology. For example, platelet-released serotonin dilates normal vessels but constricts vessels damaged by atherosclerosis or hypertension. Observations that physiologic vasodilation to serotonin is reduced in eNOS (−/+) mice and constriction occurs in eNOS (−/−) mice supports a role of eNOS involvement (Lamping and Faraci, 2001).
Reduced reactivity 6 hours after CCAT is not caused by reduced eNOS expression, because protein levels remain unchanged. Abnormal activity among protein regulators such as caveolin-1 as well as serine/threonine and perhaps tyrosine kinases may prevent eNOS from functioning normally early after injury (Fleming and Busse, 1999). Pathologic consequences of reduced eNOS activity include intravascular thrombosis from a reduction in NO-mediated platelet inhibition, embolic occlusions, or deficits in the autoregulatory response to hemodynamic stress. These data may explain the results of a previous study that found increased infarction volumes in rats with CCAT followed by transient hypotension (Dietrich et al., 1999b). Impaired eNOS-dependent dilation may disrupt cerebral autoregulation in response to hypotension, thereby reducing collateral flow and promoting severe infarction. Other investigators, using a model of subarachnoid hemorrhage, also found an acute upregulation of eNOS mRNA associated with slightly reduced protein expression and reduced reactivity, suggesting that this phenomenon is not unique to our model (Park et al., 2001).
The early upregulation of eNOS mRNA and subsequent increase in protein may result from either hemodynamic perturbations from the carotid stenosis or platelet–endothelial interactions. The delay between increases in eNOS mRNA and protein are consistent with published observations from other investigators (Iwase et al., 2000). Although persistent elevations in eNOS protein increase cerebrovascular reserve and protect against future episodes of ischemia, activation of neuronal nitric oxide synthase worsens ischemic disease by producing free radicals such as peroxynitrite that damage membranes of adjacent cells (Eliasson et al., 1999). However, eNOS upregulation may not be entirely beneficial. Improved collateral flow may obscure eNOS-generated free-radical damage that ultimately limits recovery. Cultured endothelial cells produced peroxynitrite on angiotensin II stimulation (Pueyo et al., 1998). The nonspecific NOS inhibitor Nω-nitro-
eNOS upregulation may be a molecular mechanism for cerebral hyperperfusion syndrome. After resection of arteriovenous malformations (Sakaki et al., 1992) or endarterectomy in cases of severe carotid stenosis (Bernstein et al., 1984; Penn et al., 1995; Meyers et al., 2000), patients are sometimes susceptible to dangerous increases in cerebral perfusion. Elevated eNOS may promote excessive dilation, making the vasculature permeable or vulnerable to hemorrhage.
In summary, despite a CCAT-induced upregulation of eNOS mRNA at 2 hours after CCAT, eNOS protein remains unchanged up to 6 hours after injury. eNOS-dependent vascular reactivity is significantly reduced 6 hours after CCAT. Potential consequences of eNOS dysregulation immediately after embolic stroke include more frequent trapping of emboli and the inability to autoregulate in response to hypotension. By 24 hours, protein expression and vascular reactivity have increased relative to baseline and are normalized by 72 hours. Potential pathologic consequences of upregulated eNOS include the production of peroxynitrite, leading to damage of adjacent cells or cerebral hyperperfusion syndrome. The changing temporal profile of eNOS expression may present a challenge to physicians attempting to treat vascular pathophysiology. Diagnostic tests of vascular reactivity may provide clues as to the status of the cerebral vasculature in a particular patient at a specific time. Clarifying the pathologic consequences of alterations in eNOS may facilitate identification of novel treatments for stroke.
Footnotes
Acknowledgments:
The authors thank Dale Pelligrino, Ph.D., for his helpful suggestions in preparing the cranial windows.