
Abstract
The authors investigated the effect of the C1 inhibitor (C1-INH), the only known inhibitor of complement C1, in a murine model of transient focal ischemia. Ischemia was induced by intraluminal occlusion of the middle cerebral artery. After 2 hours, reperfusion was produced by removing the nylon monofilament occluding the artery. The effect of 15 U C1-INH (intravenously) was evaluated in terms of general and focal neurologic deficits, ischemic volume, neutral red staining (to identify the brain areas subject to ischemic damage), and glial fibrillary acidic protein immunoreactivity (to show astrocytic response). Forty-eight hours after ischemia, C1-INH significantly improved general and focal deficits by 36% and 54%, respectively, and significantly reduced infarct volume (CI-INH, 6.69% ± 2.93%; saline, 24.24% ± 8.24%) of total brain. Neutral red staining further showed the strong protective effect of C1-INH in cortex, hippocampus, and striatum. Astrocyte activation induced by ischemia was not affected by C1-INH. These findings show that C1-INH displayed a potent neuroprotective action by effectively reducing ischemia—reperfusion injury.
The complement system is a proteolytic cascade of about 30 plasma proteins with an associated group of cell membrane proteins involved in a variety of immune-inflammatory responses (Gasque et al., 2000; Stahel et al., 1998). Complement activation by the classical, alternative, or lectin pathways results in the generation of anaphylatoxin peptides (C3a, C5a), opsonins (C4b, C3b), and the membrane attack complex, all of which play an important role in clearing microorganisms, immune complexes, and damaged cell products. C3a and C5a, potent chemoattractants and activators of polymorphonucleated cells, induce vasodilatation and vascular permeability, including blood—brain barrier permeability. In the CNS, local complement activation has been associated with the propagation and exacerbation of tissue injury in numerous degenerative and inflammatory disorders (Gasque et al., 2000; Stahel et al., 1998). Complement activation, however, not only leads to neuronal death, but may also contribute to brain tissue remodeling and repair by inducing the generation of antiinflammatory molecules, the activation of microglia phagocytosis, and the removal of toxic entities. Thus, complement activation may have a broad spectrum of consequences, making the overall effect in CNS disorders unclear.
Complement is activated in human stroke (Di Napoli, 2001) as well as in experimental models of cerebral ischemia (Huang et al., 1999; Schafer et al., 2000; Van Beek et al., 2000). Studies aimed at evaluating the effects of complement inhibition in cerebral ischemia and ischemia—reperfusion injury, however, have not reached univocal conclusions (Heimann et al., 1999; Huang et al., 1999; Liu et al., 1999; Vasthare et al., 1998). A detrimental role of complement in ischemic damage has been proposed by Heimann et al. (1999) who showed that complement inhibition significantly reduced brain tissue damage in a model of ischemic penumbra induced by photochemical cortical vein occlusion. In contrast, complement depletion induced by cobra venom failed to have any effect on brain injury in a rabbit model of thromboembolic stroke (Liu et al., 1999). Huang et al. (1999) reported that complement C3 inhibition by soluble complement receptor-1 (sCR1) induced only a moderate reduction of the ischemic lesion, a marked effect being obtained only when complement and selectinmediated adhesion were simultaneously inhibited.
C1 inhibitor (C1-INH) is the only known inhibitor of the C1s and C1r subcomponents of complement C1, and is of primary importance in the control of the complement classical pathway. C1-INH is currently used to manage angioedema attacks in patients with hereditary or acquired C1-INH deficiency, and in other critically ill patients (Buerke et al., 1995, 1998; Caliezi et al., 2000; Horstick et al., 1997; Kirschfink and Nurnberger, 1999; Lehmann et al., 2000). We have recently shown that C1-INH binds to damaged endothelial cells, and this action can improve the protective effect by increasing local bioavailability (Bergamaschini et al., 2001).
The inflammatory response that develops during an ischemic event involves major alterations of glia—neuron interactions, with important consequences on the evolution of the injury. Microglia and astrocytes are rapidly activated and their phenotype changed, and they begin to synthesize a wide range of factors. Microglial cells, the resident macrophages of the brain, rapidly transform into phagocytes and remove debris. Astrocytes have been reported to exert both harmful and beneficial actions in the injured tissue (Stoll et al., 1998).
Reperfusion, which occurs in human ischemia either spontaneously or as a result of pharmacological thrombolysis, has several important consequences on the extent of tissue injury and recovery. In particular, reperfusion allows the migration of inflammatory cells to the site of injury and increases availability of oxygen to damaged cells, possibly favoring oxygen radical induction (Dirnagl et al., 1999; Feuerstein and Barone, 1999). The aim of the present study was to evaluate the effect of C1-INH on neurologic deficits, ischemic volume, neurodegeneration, and the astrocytic response induced by ischemia—reperfusion in a model of transient focal ischemia induced by middle cerebral artery occlusion (MCAO) in the mouse (Connolly et al., 1996).
MATERIALS AND METHODS
Animals
Male CD1 mice (26 to 28 g; Charles River, Calco, Italy) were housed in cages (5 mice per cage) and maintained in a controlled environment (constant temperature, 21°C ± 1°C; relative humidity, 60%; regular light/dark cycle, 7 A.M.–7 P.M.) with free access to food (Altromin pellets for mice) and water. Procedures involving animals and their care conformed with institutional guidelines that are in compliance with national (D.L. n.116, G.U. suppl. 40; February 18, 1992) and international laws and policies (EEC Council Directive 86/609, OJ L 358,1; December 12,1987; NIH Guide for the Care and Use of Laboratory Animals, US National Research Council, 1996).
Electroencephalography
Electrocortical activity in the supply territory of the middle cerebral artery was monitored to verify of the occurrence of ischemia (Phillips et al., 2000; Wellons et al., 2000). Mice were anesthetized with ketamine (100 mg/kg) and xylazine (5 mg/kg). Two recording screw electrodes were implanted on the cortical surface at the following coordinates: L −3.5 mm and AP −0.5 mm; L −0.5 mm and AP −2.8 mm; nose bar −2.5 mm. A ground lead was positioned over the nasal sinus ensuring no contact with cerebral tissue. All the electrodes were cemented to the skull and insulated leads were routed to the recording apparatus (Instituto Battaglia Rangoni, Bologna, Italy) through a multipin socket.
Transient focal cerebral ischemia was achieved by MCAO, according to the method described by Connolly et al. (1996). Immediately after surgical implantation of EEG electrodes, mice were positioned supine on a heating pad equipped with a rectal probe to maintain a constant core temperature of 37°C ± 0.5°C (LSI-Letica, Barcelona, Spain). A midline neck incision was made and the carotid sheath was exposed. The following surgical steps were performed under an operating microscope (Nachet, Dijon, France) and using an optic fiber illuminator (Highlights 2100; Olympus, Milan, Italy). The left common carotid artery was exposed and the external carotid artery and its branches, including the occipital superior thyroid arteries, were isolated and cauterized. The pterygopalatine artery was ligated, and the external carotid artery was cauterized just proximal to its bifurcation. A 5–0 monofilament nylon suture, blunted at the tip by heat and coated in poly-L-lysine, was introduced into the internal carotid artery through the external carotid artery stump and advanced to the anterior cerebral artery to block its bifurcation into the anterior cerebral and middle cerebral arteries. The distance from the tip of the suture to the bifurcation of the common cerebral artery was 11 ± 0.5 mm. After a 2-hour ischemic period, blood flow was restored by carefully removing the nylon filament. This surgical procedure has been previously shown not to affect physiologic parameters (Connolly et al., 1996; Mattson et al., 2000).
Exclusion criteria
Successful induction of ischemia was indicated by an isoelectric EEG signal (Fig. 1). Preocclusion EEG amplitude (Fig. 1A) was compared with those measured at the onset of ischemia or before reperfusion (Figs. 1B and 1C), and mice showing ≤90% reduction were removed from the study (Phillips et al., 2000). Changes in EEG amplitude (microvolts) were quantified using an electronic calibrator and the mean value was calculated during 1-minute recording epochs.
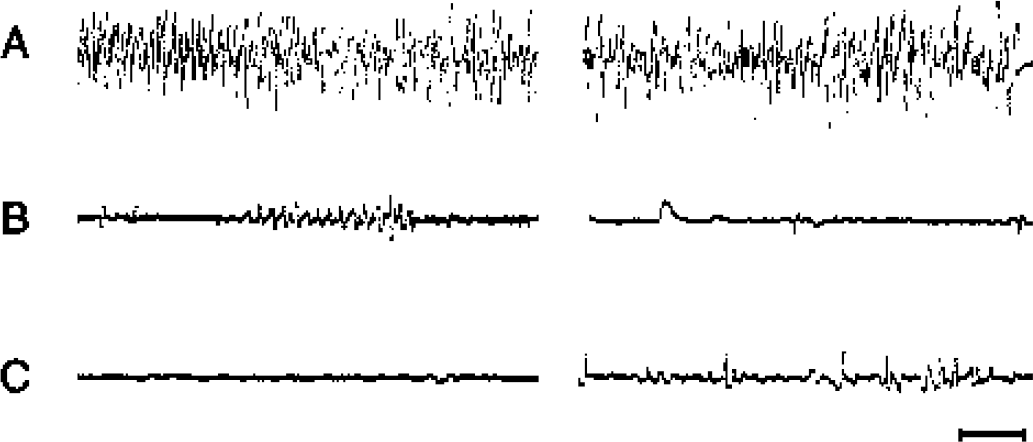
Representative EEG traces obtained in two mice before
Sham-operated mice
After surgical implantation of EEG electrodes, sham-operated mice received a midline neck incision that exposed the carotid sheath. The external carotid artery and its branches were isolated without being ligated or cauterized. Similar to ischemic mice, sham-operated mice were maintained at 37°C during surgery and recovery from anesthesia.
Drug treatment
Mice received a single intravenous injection of 15 U C1-INH (C1 Esterase Inhibitor [1 U corresponding to the activity of 1 mL normal plasma]; Baxter-Immuno, Pisa, Italy) or the same volume (150 μL) of saline 5 minutes after the onset of ischemia. This dose was chosen on the basis of an in vitro dose-response curve. Since the total blood volume in a mouse is about 2.5 mL, 1, 5, 10, 15, and 20 U C1-INH were added to 1 mL mouse plasma, and the lowest amount of C1-INH required to inhibit the activity of exogenous C1s (Immunochrom CI-INH reagent kit; Baxter-Immuno, Wien, Austria) was found to be 15 U/mL. Therefore, in subsequent experiments CD1 mice (n = 10) were treated with 15 U C1-INH and the effects on complement and coagulation were evaluated by measuring hemolytic activity (Total Haemolytic Complement Kit; The Binding Sites, Birmingham, U.K.) and activated partial thromboplastin time (Organon-Technica, Durham, NC, U.S.A.), respectively. A 90% to 95% inhibition of complement hemolytic activity with no effect on activated partial thromboplastin time was observed 30 minutes after C1-INH infusion, and complement hemolytic activity normalized 48 hours after C1-INH treatment. Microscopy evaluation of brain slices showed no evidence of hemorrhagic or thrombotic events in mice treated with C1-INH.
Evaluation of neurologic deficits
After 46 hours of reperfusion (48 hours after the induction of ischemia), each mouse was rated on two neurologic function scales unique to the mouse (Clark et al., 1997). Scores for both scales range from 0 (healthy) to 28 and represent the sum of the results of all categories for each scale. The general deficit scale evaluates hair (0–2), ears (0–2), eyes (0–4), posture (0–4), spontaneous activity (0–4), and epileptic behavior (0–12), whereas the focal deficit scale evaluates body symmetry (0–4), gait (0–4), climbing on a surface held at 45° (0–4), circling behavior (0–4), front limb symmetry (0–4), compulsory circling (0–4), and whisker response to a light touch (0–4). A trained investigator who was masked to the experimental conditions performed experiments.
Quantification of infarct size and volume
Immediately after neurologic deficit evaluation, brains were removed and 1-mm coronal slices were cut using a tissue chopper. The brain slices were stained with 2% (weight/volume) 2,3,5-triphenyltetrazolium chloride (TTC) in Dulbecco phosphate buffer at 37°C. The ischemic area, which was evidenced by the absence of TTC staining, was subsequently quantified by Analytical Imaging System (Imaging Research, St. Catherines, Ontario, Canada). The ischemic volume (%I) was then calculated according to the following equation (Swanson et al., 1990):
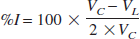
where VC is the volume of (normal) brain tissue in the control hemisphere, and VL is the volume of (normal) brain tissue the lesioned hemisphere, and
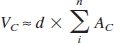
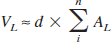
where d is the distance between sections, AC is the area of (normal) brain tissue in the control hemisphere on section i, and AL is the area of (normal) brain tissue in the lesioned hemisphere on section i.
Assessment of neurodegeneration
Mice were deeply anesthetized with equitensin (120 μL, intraperitoneally) and perfused via the ascending aorta with 20 mL 0.1 mol/L phosphate-buffered saline (PBS; pH 7.4) followed by 50 mL chilled paraformaldehyde (4%) in PBS. Brains were carefully removed from the skull, transferred to 20% sucrose in PBS, and stored at 4°C overnight for cryoprotection. The brains were then rapidly frozen via 3-minute immersion in isopentane at −50°C before being sealed into vials and stored at −70°C until use. The presence of neurodegeneration was evaluated by neutral red (Neutral Red Gurr Certistain; BDH, Poole, England) staining performed on 40-x m coronal sections (von Dorsche and Opitz, 1970).
Immunocytochemistry
Coronal sections (40 μm), prepared from perfused brains in the manner described previously, were used for assessment of astrocytic activation by glial fibrillary acidic protein (GFAP) immunostaining (De Simoni et al., 2000; Kalehua et al., 2000). Briefly, the sections were rinsed for 30 minutes in 0.4% Triton X-100 in 0.1 mol/L PBS followed by 15 minutes in 0.1% Triton X-100 and 3% fetal calf serum in PBS. The sections were incubated overnight with GFAP (mouse monoclonal, 1:250; Roche Molecular Biochemicals, Mannheim, Germany). The next day, sections were washed in PBS and incubated with biotinylated secondary antibody (goat anti—mouse IgG; Vector Laboratories, Burlingame, CA, U.S.A.) for 1 hour, washed, and incubated in avidin-biotin-peroxidase (Vectastain kit; Vector Laboratories). After reacting with 3'-3-diaminobenzidine tetrahydrochloride the sections were washed, mounted on gelatin-coated slides, dried, dehydrated through graded alcohols, fixed in xylene, and coverslipped using DPX mountant (BDH) before light microscopy analysis. For C1-INH immunostaining, slides were incubated with rabbit polyclonal anti—human C1-INH (1:1,000; Dako A/S, Glostrup, Denmark) followed by biotinylated secondary antibody (antirabbit IgG, Vector Laboratories) and avidin-biotin peroxidase.
RNA extraction and reverse transcriptase—polymerase chain reaction analysis
Total RNA was extracted from cortex and hippocampus according to the acid guanidium—phenol—chloroform method described previously (De Simoni et al., 2000). For polymerase chain reaction (PCR) analysis, total RNA (1 μg) was used as a substrate for single-stranded complementary DNA (cDNA) synthesis using MuLV reverse transcriptase (50 U/μL, Gene Amp RNA PCR Kit; Perkin Elmer, Emeryville, CA, U.S.A.), random hexamers (final concentration, 2.5 μmol/L) and dNTP mix (final concentration, 1 mmol/L each component). The transcription was carried out at 25°C for 10 minutes, 42°C for 20 minutes, and 99°C for 5 minutes. An aliquot (9 μL) of cDNA synthesis mix was used for the PCR reaction to amplify cDNA encoding for GFAP and β-actin as a reporter gene. Primers (0.2 μmol/L each) for the sequence of interest were used in a final volume of 50 μL. GFAP primers were as follows: forward, 5'-GCTAGCTACGAGAAGGTCC-3'; reverse, 5'-CCAG-TCTATACGCAGCCAGG-3', which result in a 283-bp PCR product (accession number AF332061). Primers for β-actin were as follows: forward, 5'-TGTGATGGTGGGAATGGGT-CAG-3'; reverse, 5'-TTTGATGTCACGCACGATTTCC-3', which result in a 514-bp product (Alonso et al., 1986). The PCR conditions were optimized for each set of primers. To ensure amplification of each cytokine within the linear range, each sample was amplified for 20, 22, 24, and 26 cycles in a DNA programmable thermal cycler (Perkin Elmer Cetus). To determine the relative amount of cytokines, PCR was run in 24 cycles for both genes. Denaturation was reached at 95°C for 30 seconds, annealing at 58°C for 30 seconds, and extension at 72°C for 30 seconds. In each PCR (RNA extraction, cDNA synthesis, and PCR), negative and positive controls were included. A portion of the PCR mixture (7 μL) was electrophoresed in a 1.5% (weight/volume) agarose gel, stained with ethidium bromide. The relative intensity of the bands was measured by an Image Quant analyzer (Molecular Dynamics, Sunnyvale, CA, U.S.A.). Data are expressed as the ratio of densitometric measurement (optical density) of the samples to the corresponding β-actin.
Experimental plan and statistical analysis
The experimental sets used for the study included (1) ischemic mice receiving C1-INH or saline (n = 8 or 10 per group, respectively), used for neurologic deficits and TTC staining, killed 48 hours after ischemia; (2) sham-operated mice and ischemic mice receiving C1-INH or saline (n = 3, 4, or 6 per group, respectively) for neutral red staining and GFAP immunocytochemistry, killed 48 hours after ischemia or sham operation; (3) sham-operated and ischemic mice receiving C1-INH or saline (n = 4, 5, or 6 per group, respectively) for reverse transcription (RT)-PCR analysis, killed 48 hours after ischemia or sham operation; and (4) ischemic mice receiving C1-INH or saline (n = 3 per group) for C1-INH immunostaining, killed 30 minutes after reperfusion.
Since intervals between neurologic deficit scores were not equal, data were expressed as median and 25th to 75th percentiles. In neurologic deficit and ischemic volume analysis, differences between ischemic mice receiving C1-INH or saline were analyzed by the Mann-Whitney test. The optical density ratio differences in the RT-PCR experiment were analyzed by the Kruskal-Wallis test.
RESULTS
Neurologic deficits were evaluated 48 hours after ischemia in mice receiving saline or 15 U C1-INH (Figs. 2A and 2B). CI-INH reduced general deficits by 36% (medians of saline- and C1-INH—treated mice, 11 and 7, respectively; Fig. 2A) and focal deficits by 54% (medians of saline- and C1-INH-treated mice, 24 and 11, respectively; Fig. 2B).
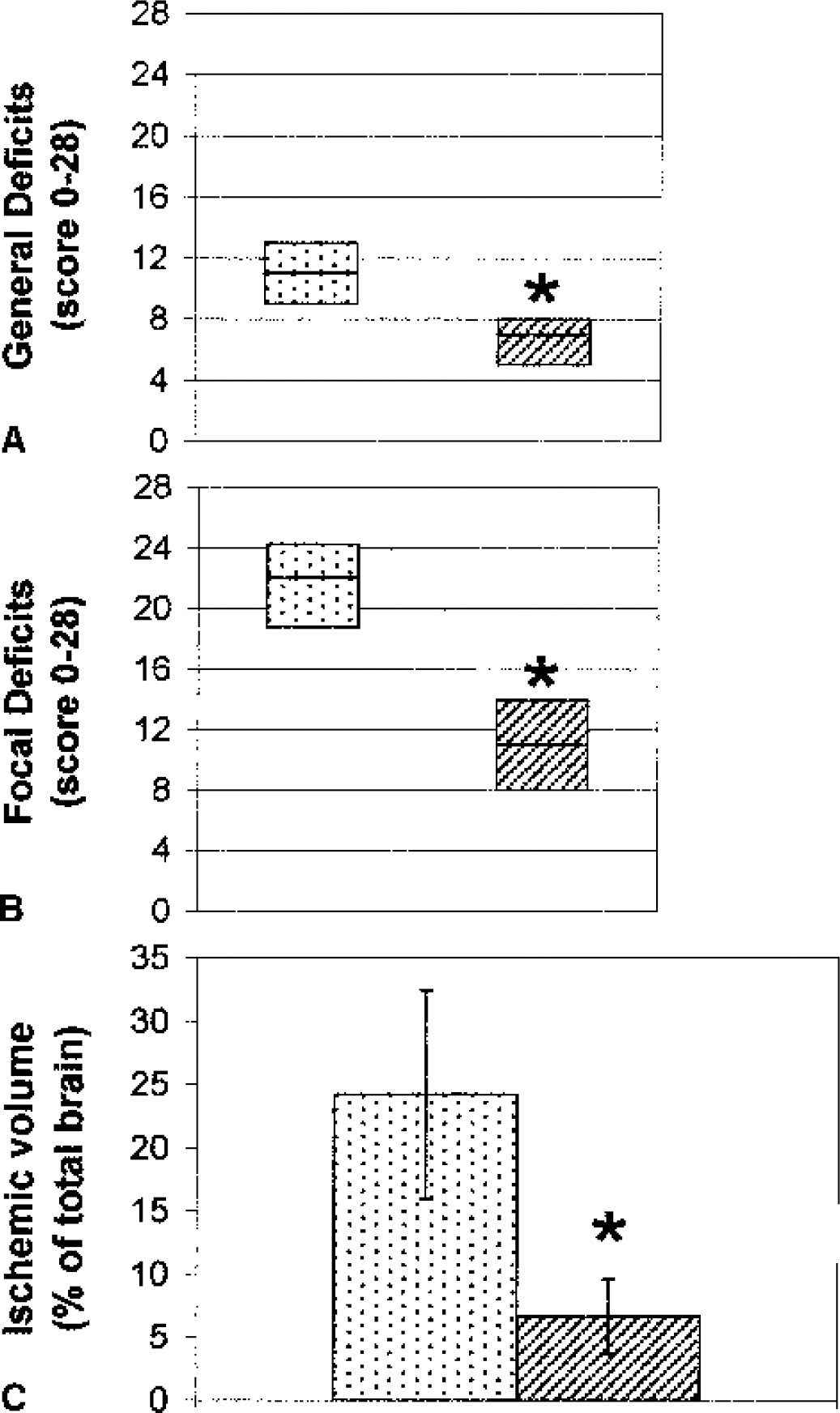
General and focal neurologic deficit score evaluation
Mice treated with C1-INH had a markedly reduced ischemic volume, as shown by TTC staining (24.24% ± 8.24% and 6.69% ± 2.93% in saline- and C1-INH—treated mice, respectively) corresponding to a 73% reduction (Fig. 2C). We analyzed the effect of ischemia and C1-INH treatment on neurodegeneration in detail in separate experiments. The ischemic insult induced marked neuronal cell loss, as observed in sections stained with neutral red, in different brain areas including the temporal cortex, striatum, hippocampus, and thalamus. In four of six mice, however, more extensive ischemic lesions were observed in selected areas, resulting in complete loss of brain tissue. In the areas surrounding the necrotic lesion or in animals not showing complete loss of tissue, a marked reduction of neuronal cell bodies was apparent (data not shown). Treatment with C1-INH prevented this decrease in neuronal cells. The general structure of the brain tissue, as shown by neutral red staining, was well preserved. Two of four mice showed small, scattered necrotic areas in the temporal cortex and striatum (data not shown).
We analyzed the extent of astrocytic activation after C1-INH administration by GFAP immunocytochemistry.
After 30 minutes of reperfusion, brain sections from mice treated with C1-INH demonstrated a definite immunoreactivity for the inhibitor, localized on endothelial cells and in the tissue surrounding brain capillaries, both in the ischemic and contralateral sides (Figs. 6A and 6B). This finding was present in every mouse examined (n = 3). No immunoreactivity could be observed in any ischemic mice treated with saline (Fig. 6C) or in nonischemic, nontreated mice (data not shown).
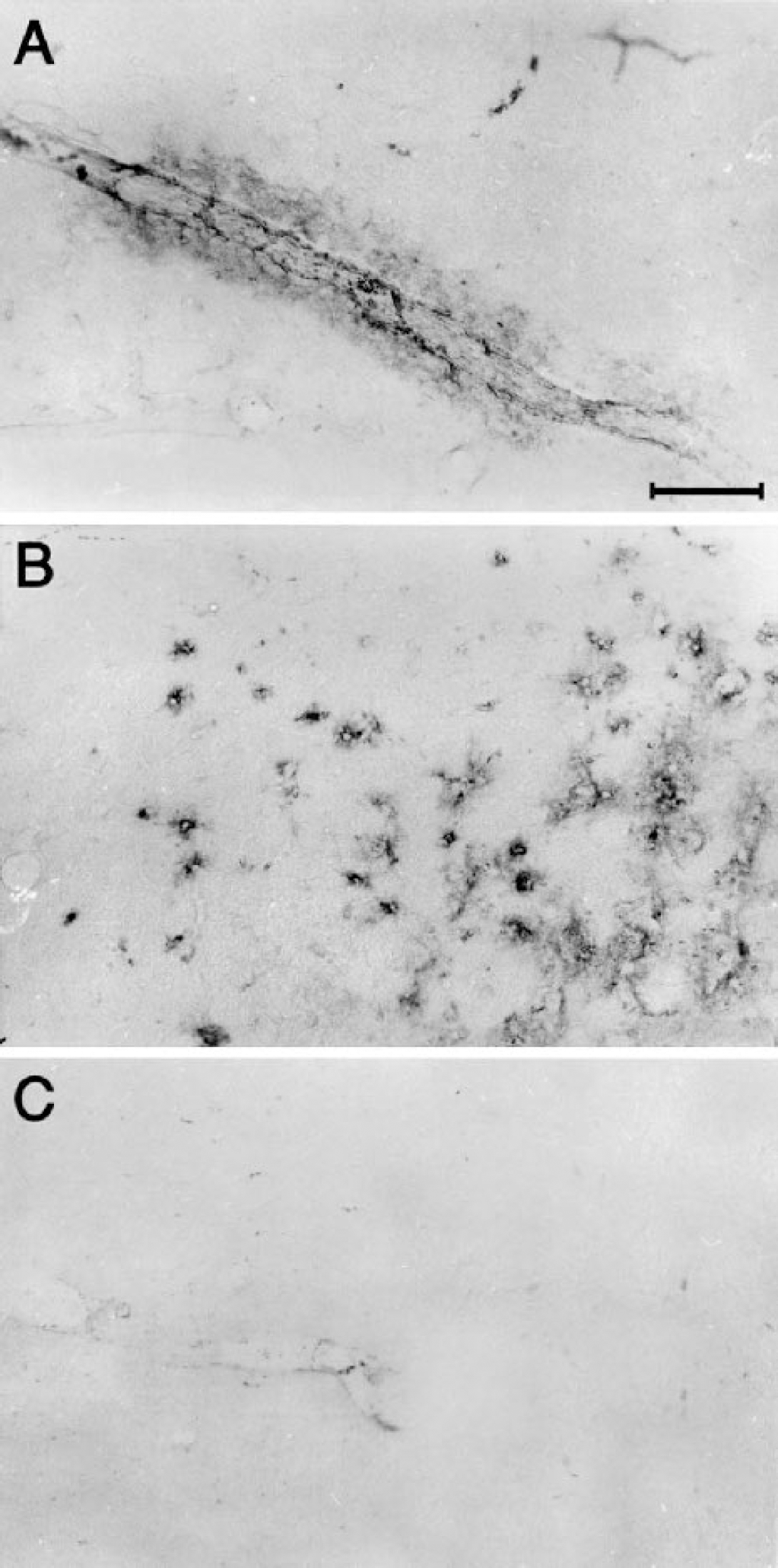
Photomicrographs of coronal brain sections (40 μm) showing a portion of piriform cortex
Forty-eight hours after MCAO, an increase in the number of GFAP-positive cells was apparent in several brain areas including hippocampus (Figs. 3 and 4), striatum, cortex, and corpus callosum. Ischemia induced a widespread activation of these glial cells, whose phenotypic changes from the quiescent to the activated state are marked by the enlargement of cell bodies and thickening of processes (Figs. 3A, 3B, 4A, and 4B). These features could be observed in both the lesioned and contralateral sides and were particularly evident in the hippocampus and corpus callosum (not shown). Forty-eight hours after ischemia, C1-INH administration did not inhibit astrocyte reactivity significantly in the same mice that showed a marked reduction of neuronal damage by neutral red staining. In fact, whereas a general decrease in the number and phenotype of stained astrocytes could be observed in selected regions (compare Figs. 3B and 3C), densely packed and thick processes (Fig. 3C) or dark cells with enlarged cell bodies (indicated by arrows in Figs. 3C and 4C) were still present throughout different brain sections, and RT-PCR analysis substantiated this observation. The GFAP messenger RNA (mRNA) evaluated in hippocampus (Fig. 5) and cortex (data not shown) was more activated in the lesioned side of ischemic compared to sham-operated mice. Treatment with C1-INH did not affect this increase, showing that astrocyte activation was still present in mice receiving the inhibitor (Fig. 5); similar results were obtained in the hippocampus and cortex.
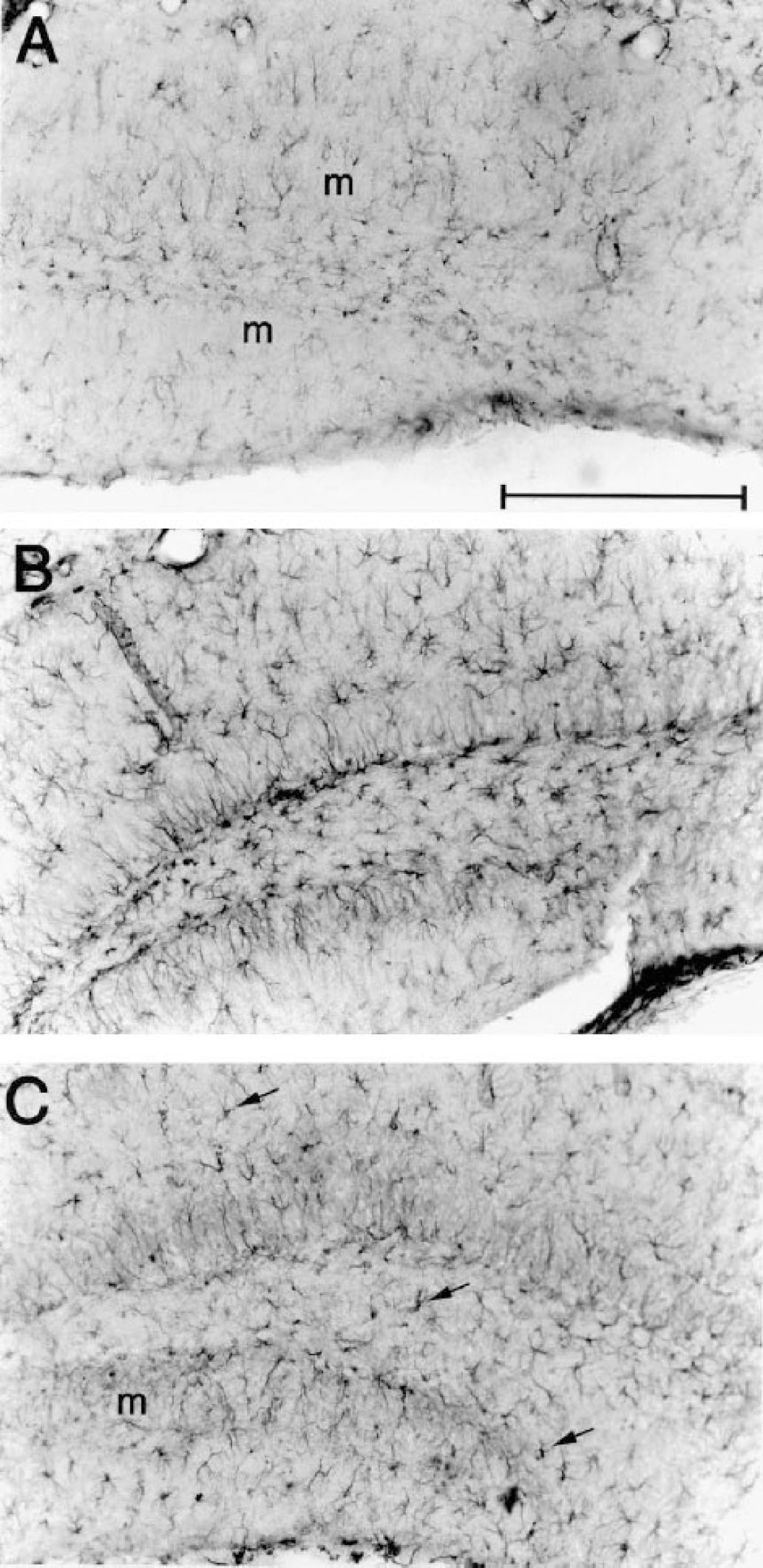
Photomicrographs of coronal brain sections (40 μm) containing the dentate gyrus of the hippocampus of a sham-operated mouse
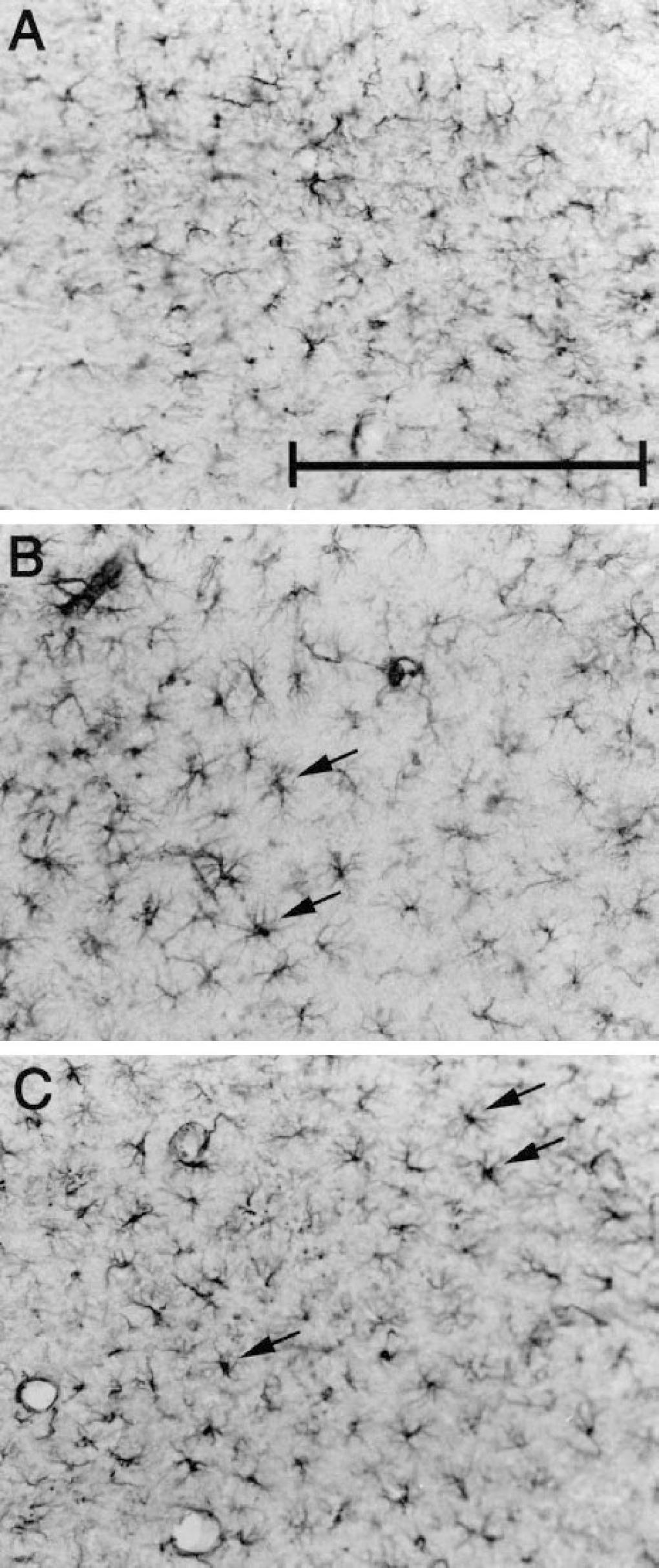
Photomicrographs of coronal brain sections (40 μm) showing a portion of stratum lacunosum moleculare of the hippocampus of a sham-operated mouse
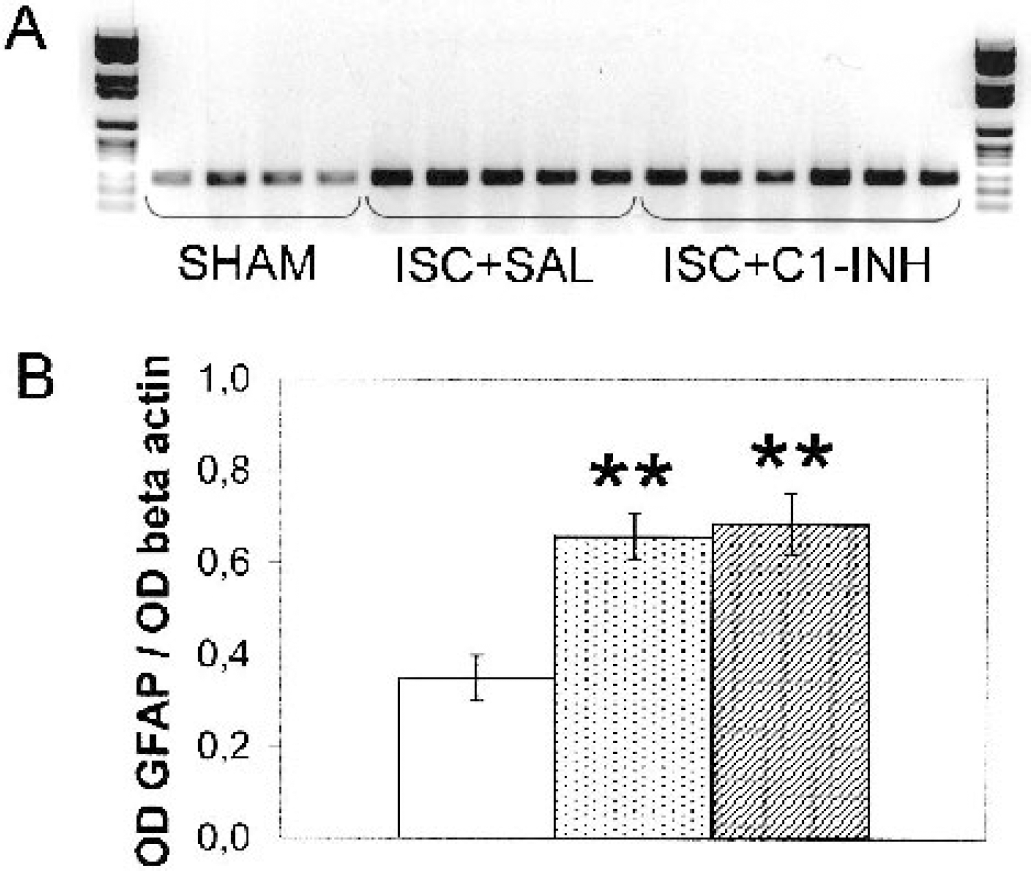
A polymerase chain reaction gel
DISCUSSION
The data obtained in the present study show that C1-INH effectively prevents neurologic deficits and tissue damage associated with reperfusion of ischemic brain. A single intravenous administration of C1-INH markedly reduced both general and focal deficits 48 hours after MCAO, inducing an improvement of the appearance and motor performance of the ischemic mice and a significant reduction in ischemic volume. A detailed examination of the neuronal damage by neutral red staining further showed the effectiveness of the treatment in reducing ischemia-induced degeneration. Furthermore, although C1-INH administration decreased the number of activated astrocytes, RT-PCR analysis of GFAP mRNA expression showed that C1-INH did not reduce their metabolic activation. Astrocytes activated during ischemia produce several molecules with neuroprotective properties including nerve growth factor, transforming growth factor β-1, and brain-derived neurotrophic factor (Culmsee et al., 1999; Gottlieb and Matute, 1999; Lee et al., 1996; Matsushima et al., 1998), suggesting that the activation of these factors may contribute to the rescue of damaged neurons and limit the spread of the injury. A protective role of reactive astrocytes in ischemia is further supported by the observation that gliosis inhibition by the gliotoxin ethidium bromide results in an increase in hippocampal injury (Louw et al., 1998). Thus, the possible beneficial effects of reactive astrocytosis are not affected by C1-INH. We found that the intravenous infusion of C1-INH had no effect on the coagulation system. This finding, in combination with previous reports that C1-INH treatment does not influence global hemodynamic parameters, blood gases, or microcirculation in animal models (Heimann et al., 1999; Horstick et al., 1997), indicates that the protective effect in our experiments can be ascribed to the antiinflammatory property of this drug.
C1-INH, the only known physiologic inhibitor of activated first component of the complement classical pathway (C1), prevents generation of the complement anaphylatoxins (C3a, C5a), opsonins (C4b, C3b) and the membrane attack complex through an irreversible binding to the C1 subcomponents, C1r and C1s. The source of complement factors in ischemic brain is not well known. A full complement cascade can be generated locally by microglia, astrocytes, and neurons when exposed to the appropriate stimuli, including the ischemic insult (Gasque et al., 2000; Huang et al., 1999). Because of the enormous difference in the concentration of complement factors in plasma (mg/mL range) and brain (μg/mL range) compartments, however, a major contribution to the overall complement action in the brain is likely due to blood in any condition involving damage to the blood—brain barrier, such as in ischemia (Stahel et al., 1998).
After an ischemic insult, complement activation seems to occur very rapidly in plasma and, at later time points, in brain parenchyma (Huang et al., 1999; Van Beek et al., 2000); however, the respective contribution to the ischemic damage of complement factors from these compartments is unknown. In the present study, because C1-INH was administered 5 minutes after the beginning of ischemia, it is not possible to determine whether the protective effect was due to complement inhibition in plasma, brain tissue, or in both. The presence, however, of marked C1-INH immunostaining in the brain capillary endothelium and surrounding tissue 30 minutes after reperfusion indicates that the inhibitor may reach compartments, different from blood stream, where it may act.
Since injured vascular endothelial cells are major activators of contact system in vivo, we cannot exclude that activation of this system could have also occurred in our model through the binding of the contact system factors (factor XII, prekallikrein, high-molecular-weight kininogen) to the ischemic vascular endothelium. A possible involvement of contact-kinin system in brain reperfusion injury has been suggested in previous studies (Huang et al., 1999; Lew et al., 1999) showing that complement inhibition at the C3 level failed to induce a significant reduction of cerebral tissue injury. If this were the case, activation of the contact—kinin system would have been prevented by treatment with C1-INH, a major inhibitor of activated factor XII and kallikrein (Mollnes et al., 2002). C1-INH forms stable bimolecular complex with these proteases, and the rate constants of interaction (plasma kallikrein, 1.7 and 4.5 × 104 mol/L−1 · sec−1; activated FXII, 3.7 and 3.1 × 103 mol/L−1 · sec−1) are in the same range as those for complex formation with C1s (2.8 × 103 mol/L−1 · sec−1) and C1r (1.2 × 104 mol/L−1 · sec−1). Whereas C1-INH is of primary importance in the control of complement classical pathway as the only known inhibitor of C1, it is more difficult to assess its physiologic role in the control of contact system because the susceptible proteases of this system may also be inactivated by one or more inhibitors (Davis, 1988). In addition, the contact and complement systems are closely linked cascades that, besides sharing a major inhibitor (C1-INH), are liable of reciprocal activation. Thus, it is difficult to delineate their specific contribution to the overall inflammatory response in vivo.
CONCLUSIONS
The present findings show that C1-INH has a strong neuroprotective action in ischemia—reperfusion brain injury. Establishing the exact mechanism of action and the optimal time-window after an ischemic event will be important in defining C1-INH potential therapeutic use in the treatment of stroke.
Footnotes
Acknowledgments
This work is dedicated to the memory of R. Samanin, former head of the Department of Neuroscience of the Mario Negri Institute. The authors thank Mr. G. Gambaro for his invaluable assistance in the EEG set-up and Veronica Marrella for her skillful contribution to the RT-PCR analysis.