
Abstract
Complement component C4 mediates C3-dependent tissue damage after systemic ischemia—reperfusion injury. Activation of C3 also contributes to the pathogenesis of experimental and human traumatic brain injury (TBI); however, few data exist regarding the specific pathways (classic, alternative, and lectin) involved. Using complement knockout mice and a controlled cortical impact (CCI) model, we tested the hypothesis that the classic pathway mediates secondary damage after TBI. After CCI, C4c and C3d immunostaining were detected in cortical vascular endothelial cells in wild-type (WT) mice; however, C4c and C3d immunostaining were also detected in C1q−1/− mice, and C3d immunostaining was detected in C4−/− mice. After CCI, WT and C1q−1/− mice had similar motor deficits, Morris water maze performance, and brain lesion size. Naive C4−/− and WT mice did not differ in baseline motor performance, but C4−/− mice had reduced postinjury motor deficits (days 1 to 7, P < 0.05) and decreased brain tissue damage (days 14 and 35, P < 0.05) versus WT. Reconstitution of C4−/− mice with human C4 (hC4) reversed their protection against postinjury motor deficits (P < 0.05 versus vehicle), but administration of hC4 did not impair postinjury motor performance (versus vehicle) in WT mice. The protective effects of C4−/− were functionally distinct from the classic pathway and terminal complement, as C1q−/− and C3−/− mice had postinjury tissue damage and motor dysfunction similar to WT. Thus, C4 contributes to motor deficits and brain tissue damage after CCI by mechanism(s) fundamentally different from those involved in experimental systemic ischemia-reperfusion injury.
Introduction
Traumatic brain injury induces an acute inflammatory response that contributes to posttraumatic cell death and functional neurologic deficits (Schmidt et al, 2005). Activation of complement amplifies local inflammation and exacerbates tissue damage and organ dysfunction after systemic and central nervous system ischemia-reperfusion injury (Chan et al, 2003; del Zoppo, 1999; Zhou et al, 2000), and studies in humans and in experimental animal models provide convincing evidence that complement is activated in brain after traumatic brain injury (TBI) (Schmidt et al, 2005; van Beek et al, 2003). Activated complement components, including the terminal membrane attack complex and proinflammatory anaphylatoxins, are detectable in cerebrospinal fluid and on brain parenchymal cells in patients with severe TBI (Bellander et al, 2001; Kossmann et al, 1997; Stahel et al, 2001), and experimental TBI induces intrathecal synthesis of complement proteins and their receptors on neurons, microglia, and astrocytes (Keeling et al, 2000; Stahel et al, 2000). Complement signaling in injured brain parenchyma may also be contributed by serum components that enter the brain through a compromised blood-brain barrier.
To date, only four published studies have examined a role for complement in traumatic cerebral contusion models. The investigators showed beneficial effects on histopathologic or functional outcome of pharmacologic C3 convertase inhibitors (Kaczorowski et al, 1995; Leinhase et al, 2006b), genetic inhibition of factor B (a component of the alternative pathway) (Leinhase et al, 2006a), or of astrocyte-specific overexpression of an endogenous C3 convertase inhibitor (Rancan et al, 2003). These studies suggested that terminal complement activation may induce secondary injury after TBI; however, the pathway(s) that initiate complement activation after TBI have not been completely defined (Leinhase et al, 2006a).
C1q is an essential component of the classic pathway, whereas C4 is essential to both the classic and lectin complement pathways. The classic pathway is initiated when C1q interacts with natural antibody bound to neoepitopes present on injured tissue, whereas the lectin pathway is initiated by a complex formed by mannose-binding lectin (MBL) or ficolins, immunoglobulins, and MBL-associated serine proteases. Both activated C1 complex and MBL-MASP complex may cleave C4 to C4a, C4b, and C4c. C4b and C2 react to form C4b2a, a C3 convertase that cleaves and activates C3, the convergence point for all complement pathways. The alternative pathway requires factor B to assemble a C3 convertase and is independent of C4 and C2. Complement anaphylatoxins (C3a and C5a), which amplify local and systemic inflammation, and the membrane attack complex (C5b-9), which is responsible for cell lysis, are produced from C3 cleavage products C3a and C3b.
Several studies have shown an association between C1q or C4 and acute and chronic central nervous system disorders, including central nervous system sequelae of group A beta-hemolytic streptococcus infection (Hoffman et al, 2004) and multiple sclerosis (Schwab and McGeer, 2002); however, C4 is not required to produce experimental allergic encephalomyelitis in mice (Boos et al, 2005). Increased cerebrospinal fluid C4a was associated with delayed ischemic neurologic deterioration in patients with subarachnoid hemorrhage (Kasuya and Shimizu, 1989). C4 mRNA and protein were induced in periinfarct tissue after permanent focal stroke in mice (Van Beek et al, 2000), and neonatal mice deficient in C1q had reduced cerebral ischemic tissue damage compared with wild type (WT) (Ten et al, 2005). C1q gene expression was increased in contused rat brain after experimental TBI (von Gertten et al, 2005). These studies suggest that the classic pathway might be involved in acute and chronic brain injury; however, no published studies have examined a functional role for the classic pathway in a TBI model.
Here, we tested the hypothesis that the classic complement pathway contributes to tissue injury and neurologic outcome after TBI. We used a controlled cortical impact (CCI) model and knockout mice deficient in C1q (the initiator of classic pathway), C4 (the mediator of the classic and the lectin pathways), or C3 (required for all complement pathways). The data suggest that C4 contributes to motor dysfunction and tissue damage after CCI, by mechanism(s) independent of the classic complement pathway and terminal complement activation.
Materials and methods
For all studies, investigators were masked to mouse genotype during surgery, data acquisition, and analysis. The CCI protocol was approved by the Massachusetts General Hospital Institutional Animal Care and Use Committee and complied with the NIH Guide for the Care and Use of Laboratory Animals.
Controlled Cortical Impact
The CCI model was used as described previously (Bermpohl et al, 2006). Because significant intralaboratory variability in brain lesion size can occur using the CCI model (Clark et al, 2000), mutant and WT mice were always injured concomitantly in all studies. Mice (8 to 16 weeks of age) were anesthetized with 4% isoflurane (Anaquest, Memphis, TN) in 70% N2O and 30% O2 using a Fluotec 3 vaporizer (Colonial Medical, Amherst, NH) and positioned in a stereotaxic frame. Anesthesia was maintained using 2% to 3% isoflurane. A 5 mm craniotomy was made using a portable drill and trephine over the left parieto-temporal cortex, and the bone flap was removed and discarded. Mice were then subjected to CCI using a pneumatic cylinder with a 3-mm flat-tip impounder, velocity of 6 m/sec, depth of 0.6 mm, and 100 ms of impact duration. The scalp was then sutured closed. The mice were returned to their cages to recover from anesthesia, and were observed for several hours after surgery. Mice could usually ambulate within 15 min of discontinuation of anesthesia.
Mice
Mice deficient in C3 (C3−/−) (Wessels et al, 1995) or C2/factor B (C2/fB−/−) (Taylor et al, 1998) were generated as described previously and backcrossed 10 generations into a C57Bl/6 background. C2/fB−/− mice do not express factor B or C2 protein, and are therefore deficient in both the alternative and classic pathways of complement activation. Mice deficient in C1q (C1q−/−) were generated as described previously (Botto, 1998) and backcrossed into C57Bl/6 at least 10 generations. C1q−/− mice are deficient in the classic complement activation pathway. Mice deficient in C4 (C4−/−) were generated as described previously (Fischer et al, 1996) and backcrossed 10 generations in to C57Bl/6. Wild-type C57Bl/6 mice obtained from Jackson Laboratories (Bar Harbor, ME) were used as controls. Male and female mice matched for gender, age (6 to 16 weeks), and weight (20 to 30 g) were used for experiments.
Quantitative C4 Real-Time Reverse Transcriptase Polymerase Chain Reaction
Brain tissue from contused cortex (12 h after CCI) or left hemispheric cortical tissue from sham-injured animals was obtained and total RNA extracted with Trizol® (Invitrogen, Carlsbad, CA, USA). cDNA was synthesized using the SuperScript® III First-Strand Synthesis System for RT-PCR (Invitrogen). Real-time quantitative polymerase chain reaction analysis of C4 gene expression was performed on ABI PRISM® 7000 Sequence Detection System using TaqMan® Gene Expression Assays (Mm00437890_m1 for C4 gene and Hs99999901_s1 for 18S rRNA) (Applied Biosystems, Foster City, CA, USA).
Administration of Purified Human C4 Protein
In reconstitution experiments, purified human C4 (hC4, 4 μg in 4 μL) (Quidel, San Diego, CA, USA) or vehicle (phosphate-buffered saline (PBS), 4 μL) was administered intracerebroventricularly by hand injection to mice immediately before CCI. For immunochemistry studies, hC4 was labeled with DyLight™ 547 Fluor (excitation/emission: 557/570 nm) using DyLight™ 547 protein labeling kit (Pierce, Rockford, IL, USA) according to the manufacturer's instructions.
Immunohistochemistry
At various times after CCI mice were anaesthetized with isoflurane and the brain was rapidly removed intact and frozen in nitrogen vapor and stored at −80°C. Coronal brain sections (12 mm) were placed on poly-
Assessment of Lesion Volume and Brain Tissue Loss
Morphometric image analysis (Imaging Research Inc., St Catherines, Ontario, Canada) was used to determine lesion volume (n = 10 to 21/group for 14 days) and brain tissue loss (n = 13 to 14/group for 35 days) after CCI. Mice were deeply anesthetized with isoflurane, decapitated, and brains were removed and frozen in nitrogen vapor. Coronal brain sections (12 mm) were cut on a cryostat at 0.5 mm intervals from anterior to posterior and mounted on poly-
Evaluation of Motor and Morris Water Maze Performance
Vestibulomotor function was assessed using a wire-grip test (Bermpohl et al, 2006). Mice were placed on a metal wire (45 cm long) suspended 45 cm above a foam pad and were allowed to traverse the wire for 60 secs. The latency that a mouse remained on the wire within a 60 secs interval was measured, and wire-grip scores were quantitated using a five-point scale. A score of one point was given if the mouse failed to hold on to the wire with both sets of forepaws and hind paws together; two points were given if the mice held on to the wire with both forepaws and hind paws but not the tail; three points were given if the mouse used its tail along with both forepaws and both hind paws; four points were given if the mouse moved along the wire on all four paws plus tail; and five points were given if mice that scored four points also ambulated down one of the posts used to support the wire. Mice that were unable to remain on the wire for less than 30 secs were given a score of zero. The wire-grip test was performed in triplicate and an average value calculated for each mouse on each day of testing.
The Morris water maze (MWM) task was used to evaluate spatial memory performance as described previously (Bermpohl et al, 2006). The apparatus consisted of a white pool (90 cm diameter, 60 cm deep) filled with water to 29 cm depth with several highly visible cues located on the walls of each of the four quadrants. Water temperature was maintained 21°C to 25°C. A clear plexiglass goal platform 5 cm in diameter was positioned 0.5 cm below the water's surface approximately 15 cm from the southwest wall. Each mouse was subjected to a series of 4 to 8 trials per day. For each trial, mice were randomized to one of four starting locations (north, south, east, or west) and placed in the pool facing the wall. Mice were given a maximum of 60 to find the submerged platform. If the mouse failed to reach the platform by the allotted time, it was placed on the platform by the experimenter and allowed to remain there for 10 secs. Mice were placed in a warming chamber for at least 4 min between trials. To control for possible differences in visual acuity or sensorimotor function between groups, two trials were performed using a visible platform raised 0.5 cm above the surface of the water. Performance in the MWM was quantitated by latency to find the platform.
To minimize potential variability in performance due to daily environmental differences, WT and mutant mice were always tested concomitantly in motor and MWM tasks.
Assessment of Brain Edema
Brain edema was assessed by measuring brain water content of injured and uninjured brain hemispheres using the (wet—dry)/wet brain weight method. Mice were subjected to CCI and killed at 24 h. Brains were removed and bisected at the midline to separate right and left hemispheres. The cerebellum and brain stem were removed. Uninjured brain tissue anterior and posterior to the contusion was removed by sectioning in the coronal plane. The wet weight of the remaining injured and uninjured hemispheres was obtained. Brain tissue was dried at 99°C for 48 h and dry hemispheric weights were obtained. Brain water content was expressed as (wet weight—dry weight)/wet weight × 100%.
Statistical Analyses
Data are mean ± s.e.m. Reverse transciptase polymerase chain reaction data were analyzed by rank sum test. Brain neutrophil counts and edema data were from experiments using multiple complement knockout strains and a single WT control group, therefore these data were analyzed by analysis of variance followed by comparisons between mutant strains and WT using the appropriate post hoc test. Lesion volume and brain tissue loss data were from experiments involving a single complement knockout strain versus a WT control group, therefore these data were analyzed by t-test. Motor and MWM test data were analyzed by two-factor repeated measures analysis of variance (group × time) for mutant strains versus their respective WT control groups. For all comparisons, P < 0.05 was regarded as significant.
Results
All mice survived CCI and appeared healthy during the experimental period. The lesion produced by our CCI model had similar histopathologic characteristics in complement knockout mice and WT including immediate brain swelling and hemorrhage, progressive cell death in cortical and hippocampal brain regions (1 to 72 h), increased brain edema in injured hemispheres (24 h), and robust leukocyte accumulation (24 h) in injured cortex. Gross brain anatomy was similar in C4−/−, C3−/−, C1q−/−, and WT mice. With the exception of partial pressure of oxygen in C3−/− mice, baseline blood pressure and blood gases under anesthesia did not differ between C4−/− and WT or between C3−/−and WT mice; no mice were hypoxic during the experimental monitoring period (Table 1).
Baseline physiologic measurements of WT and mutant mice
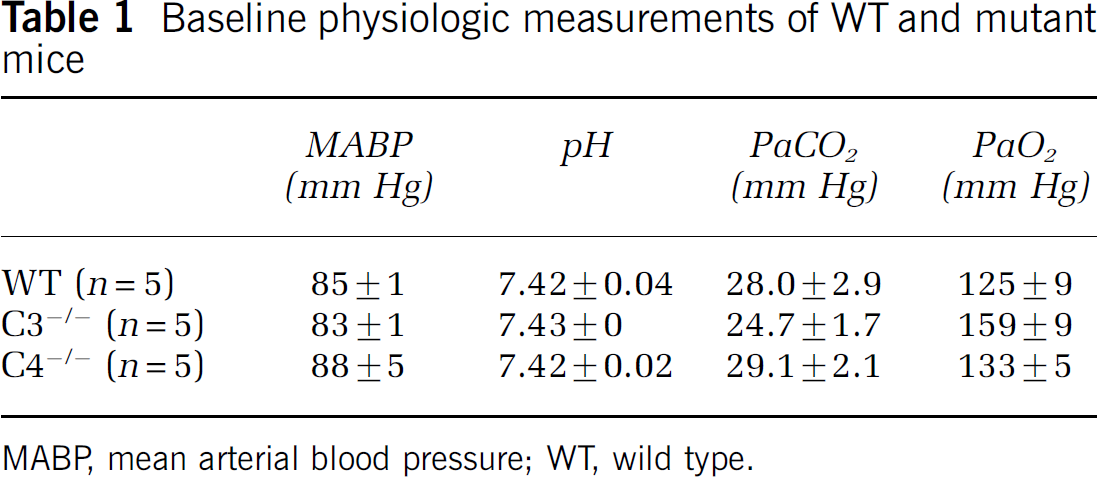
MABP, mean arterial blood pressure; WT, wild type.
Figure 1 shows the results of immunohistochemical detection of C4c in brain after CCI. Robust C4c immunostaining was detected in a subset of cortical blood vessels in injured and uninjured hemispheres, as early as 5 min and as late as 72 days after CCI (Figure 1). No staining was observed in cortices from naive (uninjured) mice or from mice at 1 min after CCI. C4c immunostaining was not detected in cell types other than vascular endothelium or choroid plexus epithelium in WT mice, and no reactivity for C4c was observed in C4−/− mice. To validate further the immunohistochemistry data, we injected Dy-Light™ 547 Fluor-labeled hC4 (75 μg) intravenously into WT mice immediately before CCI, and examined frozen brain sections using fluorescence microscopy at 1 h after injury. Similar to results using immunohistochemistry, robust hC4-DyLight™ 547 Fluor labeling was detected in a subset of cerebral microvessels in injured and uninjured cortex (Figure 1).
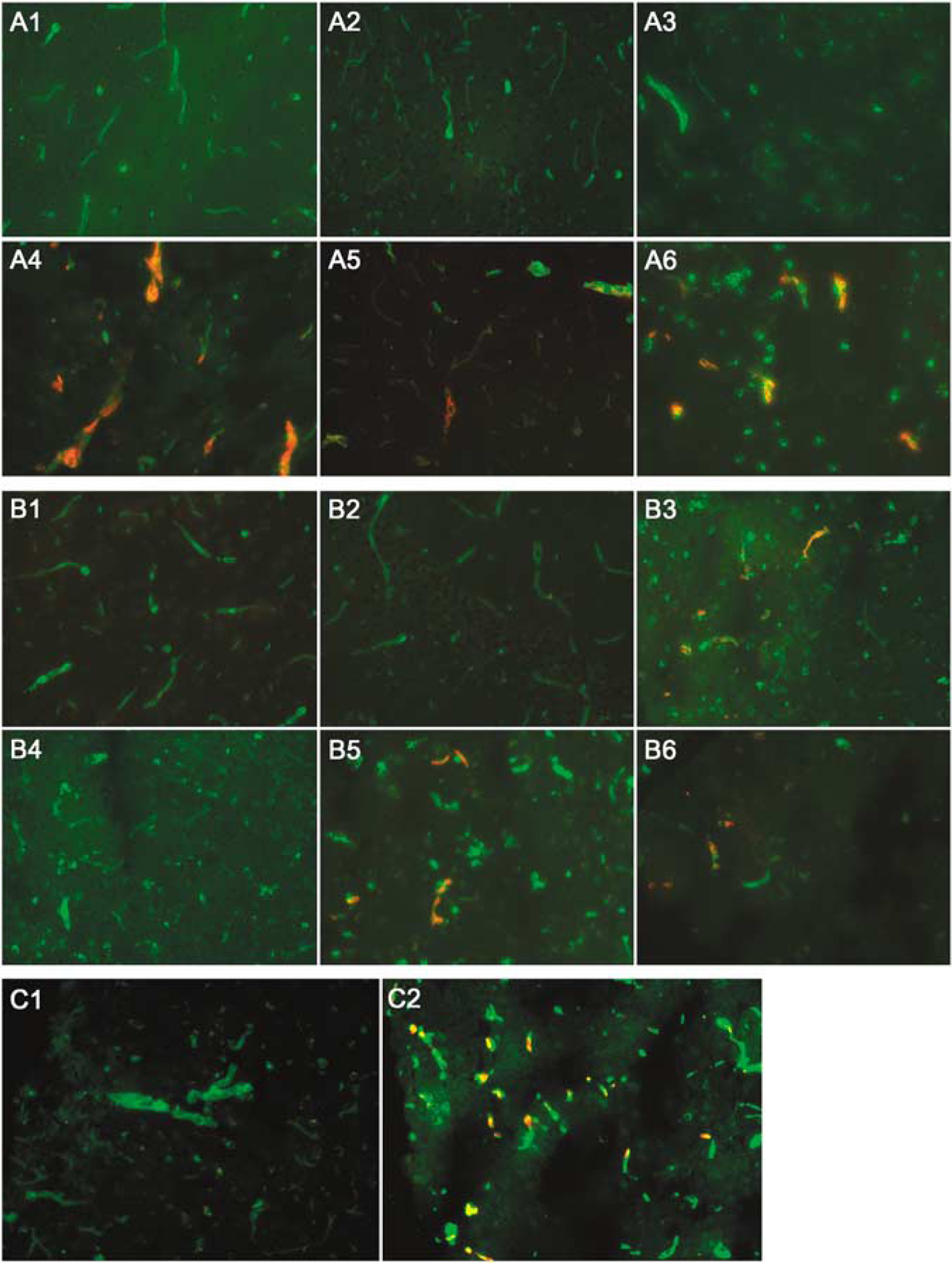
Detection of C4c and C3d in WT mouse brain after CCI. (A) Immunochemical staining of left hemispheric mouse cortex using rabbit anti-C4c (red) and FITC-labeled Griffonia simplicifolia lectin I (green) for blood vessels. Co-labeled blood vessels appear yellow. (A1) Naive WT; (A2) C4−/− at 24 h postinjury; (A3 to A5) WT at 1 min, 5 min, and 72 days postinjury, respectively; (A6) C1q−/− at 24 h after injury. (B) Immunohistochemical staining of left hemispheric mouse cortex using anti-C3d (red) and Griffonia simplicifolia lectin I-FITC (green) for blood vessels. Co-labeled cells appear yellow. (B1) Naïve WT; (B2 to B6) C3−/−, WT, C2/fB−/−, C1q−/−, and C4−/− mice at 24 h postinjury, respectively. C3d was detected in WT, C1q−/−, and C4−/− mice but not in C3−/− or C2/fB−/− mice. (
To assess whether the classic pathway is required for C3 activation after CCI, we used immunohistochemistry and an antibody recognizing C3d in mice. Robust C3d immunostaining was observed in injured cortical and subcortical microvessels and in choroid plexus at 24 h after CCI in WT and C1q−/−but not C3−/− or C2/fB−/− mice (Figure 1). The absence of staining in C2/fB−/− mice strongly suggests that this antibody preferentially reacts with C3d under the conditions used for immunohistochemistry, as full-length C3 is expressed normally in serum of C2/fB−/− mice (data not shown). We next asked whether C3 cleavage to C3d occurs in the absence of C4, by examining brain sections from C4−/− mice using the anti-C3d antibody. Figure 1 shows robust immunostaining for C3d in cortical blood vessels at 24 h after CCI in C4−/− and WT mice. These data suggest that C4 and the classic pathway are not required for activation of terminal complement after CCI.
We next examined the functional role(s) of C4 in recovery of motor and cognitive function after CCI. Figure 2 shows the results of motor testing in WT and C4−/− mice before and after CCI. Naive C4−/−and WT mice did not differ in wire-grip test performance, and motor performance was impaired after CCI in both C4−/− and WT mice (P < 0.05 versus naive mice, Figure 2A). Compared with WT, wire-grip test performance was significantly improved in C4−/− mice over the experimental period (P < 0.05 group effect). Compared with vehicle controls, pretrauma administration of hC4 intracerebroventricularly to C4−/− mice resulted in impaired postinjury motor scores (P < 0.05 versus vehicle, group effect). However, administration of hC4 to WT mice did not impair motor recovery compared with vehicle control (Figures 2B and 2C). Thus, improved motor recovery in C4−/− mice is a function of genotype and not background strain effects, and reversal of protection by hC4 in C4−/− mice is not explained by a generalized toxic effect of exogenously administered hC4.
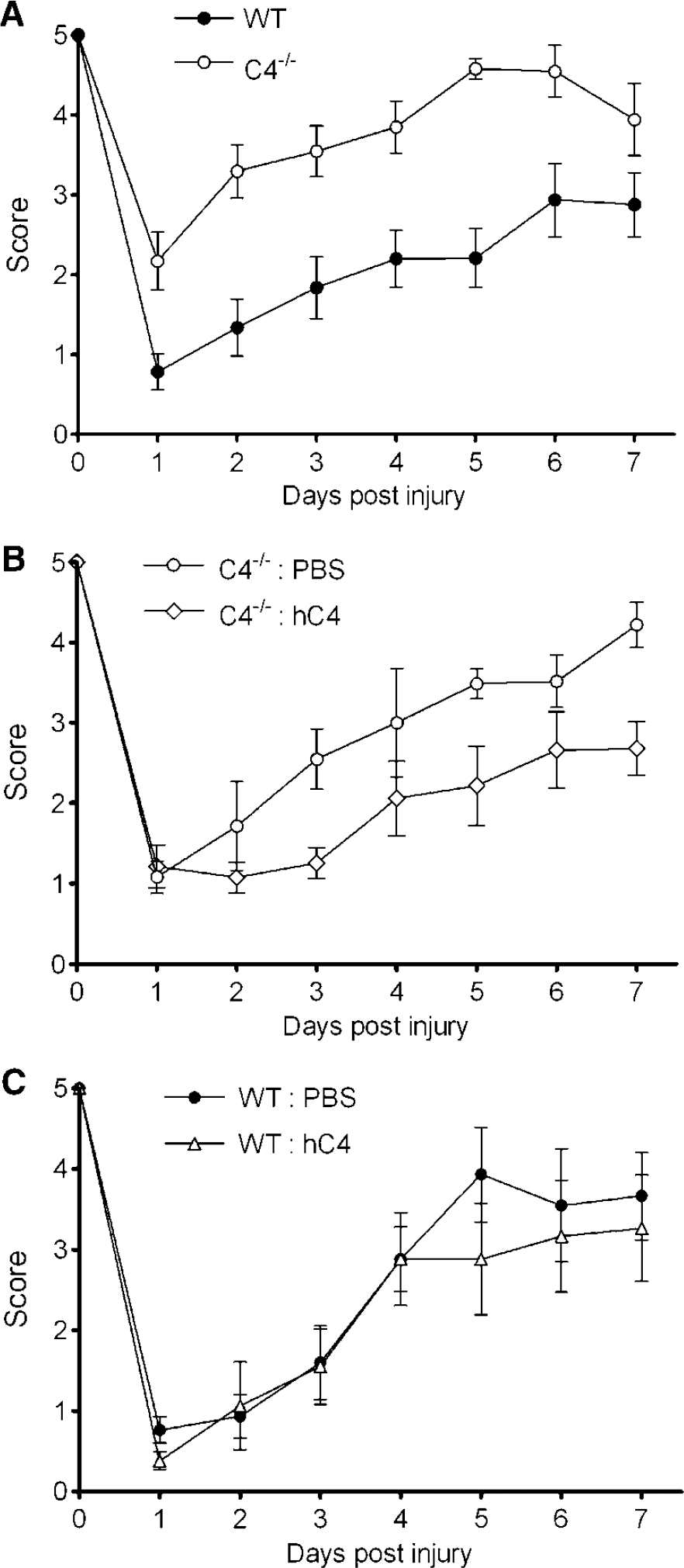
Reduced motor deficits after CCI in C4−/− mice. Vestibulo-motor function was assessed by a wire-grip test before CCI and at 1 to 7 days after CCI in (
Having shown that hC4 reverses the motor recovery phenotype in C4−/− mice, we next sought to determine whether vascular endothelium is required for C4 signal transduction, because C4 may react directly with injured brain parenchymal cells via passage into the brain across the damaged blood-brain barrier. Mice (n = 3) were injected with hC4 intracerebroventricularly, subjected to CCI, and brains examined at 3 or 24 h for hC4-DyLight fluorescence. In contrast to intravascular administration, intracerebroventricularly administered hC4 did not label vascular endothelium in injured or uninjured cortices at any time point examined (Figure 1). These data suggest that brain cells other than vascular endothelium may mediate the effects of C4 on posttraumatic motor dysfunction and tissue damage.
Figure 3 shows results of MWM testing in WT and C4−/− mice. Latency to the hidden and visible platform in the MWM paradigm was similar between groups of naive mice (Figure 3A). Both C4−/− and WT mice performed worse in the MWM after CCI compared with naive mice (P < 0.05), but no significant group differences were observed after CCI between C4−/− and WT mice in hidden or visible platform trials (Figure 3B).
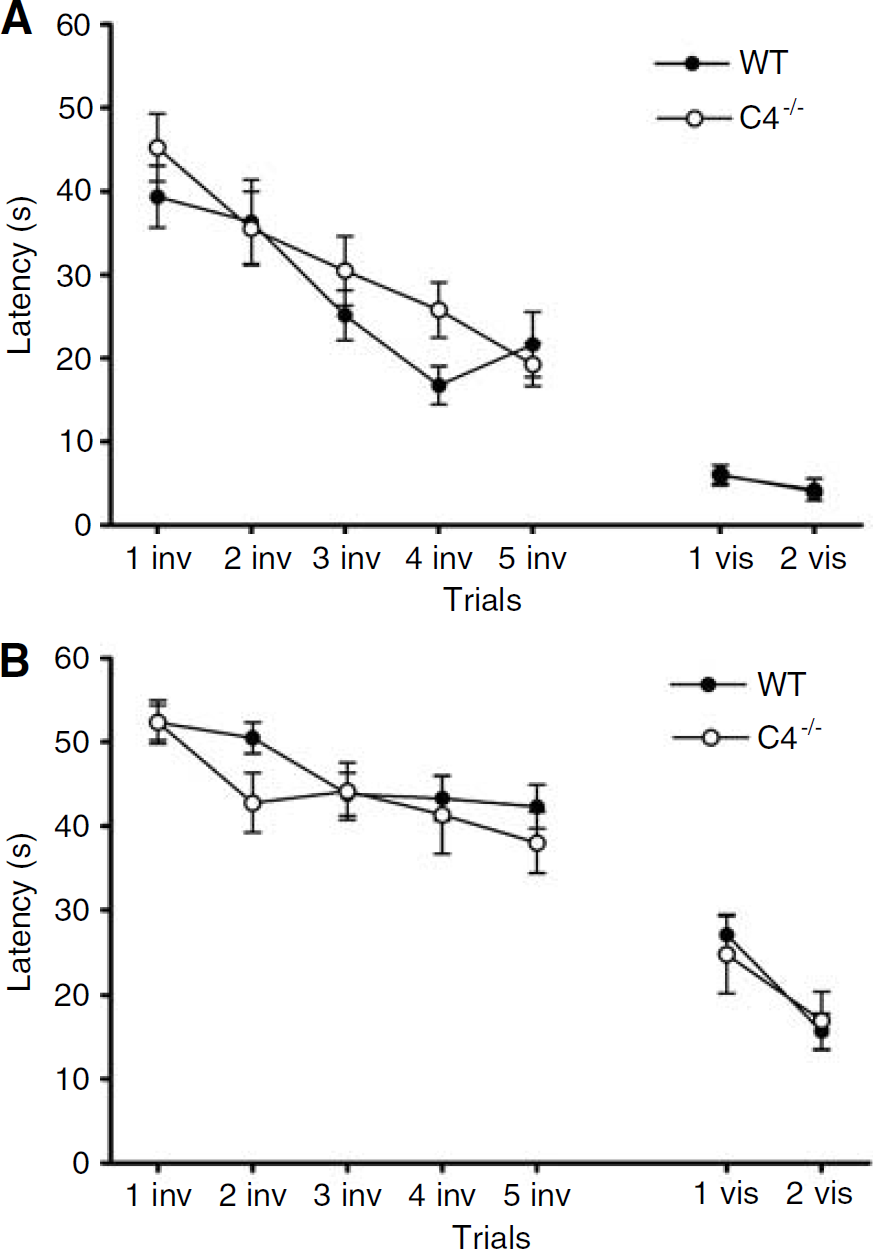
Results of MWM testing in C4−/− and WT mice. No difference in performance was observed between naive C4−/−(n = 11) and WT (n = 10) mice in hidden (inv) or visible (vis) platform trials. Compared with naive mice, injured C4−/−(n = 12) and WT (n = 20) mice exhibited impaired performance (P < 0.05), but no between-group differences in MWM performance were observed between injured C4−/− and WT mice in hidden or visible platform trials.
We next assessed the effect of C4 knockout on acute and long-term lesion size after CCI. Compared with WT, lesion volume at 14 or 35 days was modestly but significantly reduced in C4−/− versus WT mice (P < 0.05, Figure 4). To assess whether the C4−/− phenotype depends on initiation of the classic complement pathway, we performed CCI in C1q−/−and WT mice. Compared with WT, mice deficient in C1q did not differ with respect to pre- or postinjury motor or MWM performance, or posttraumatic lesion volume (Figure 5).
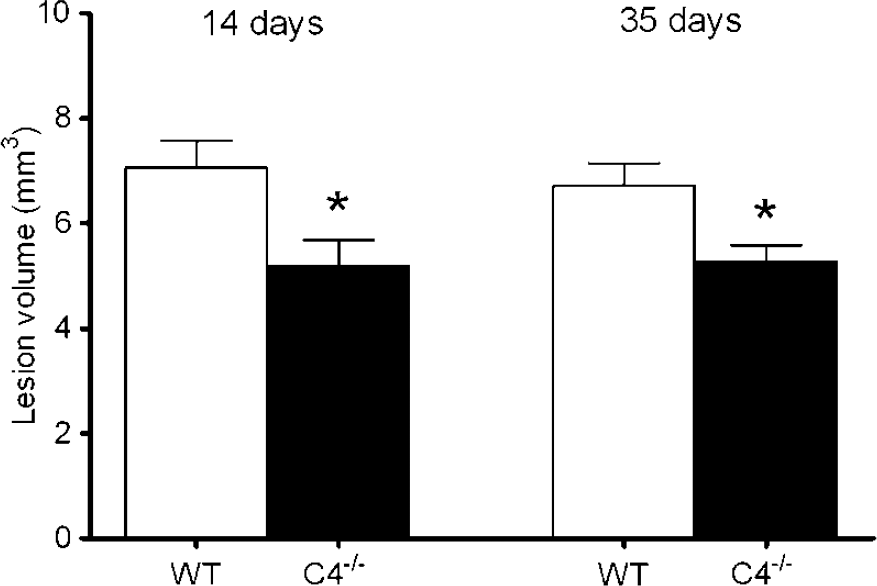
Reduced lesion size after CCI in C4−/− versus WT mice. At 14 days, posttraumatic brain lesion size was reduced in C4−/− (n = 12) versus WT (n = 20) mice (*P < 0.05). At 35 days, reduced posttraumatic brain lesion size was maintained in C4−/− (n = 10) versus WT (n = 9) mice (*P < 0.05).
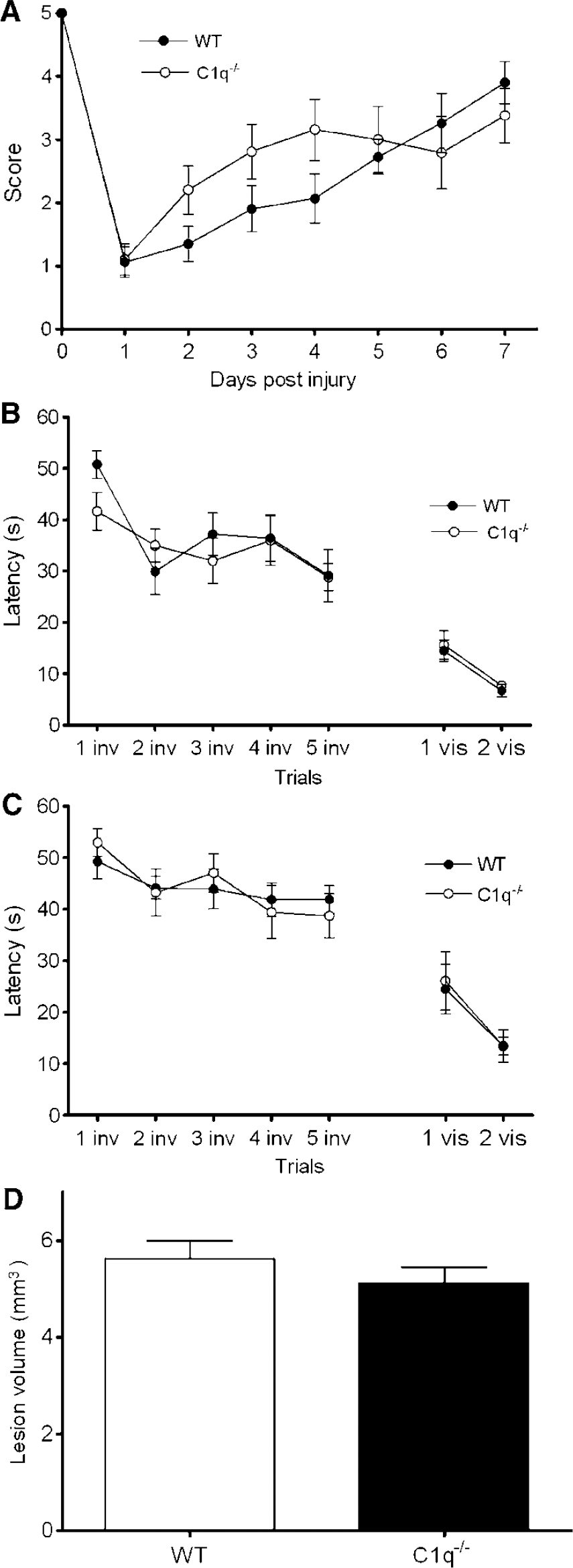
Results of behavioral and histopathologic outcome after CCI in C1q-deficient (C1q−/−) versus WT mice. Motor function was assessed by the wire-grip test at days 1 to 7 after CCI, and a MWM paradigm was used to assess cognitive function at days 8 to 14. Brain lesion size was assessed at day 30. (
To determine whether the C4−/− phenotype depends on terminal complement activation, we examined outcome after CCI in WT versus C3−/−mice. Mice deficient in C3 did not differ from WT with respect to pre- or postinjury motor or MWM performance, or brain lesion size after CCI (Figure 6). The data suggest that C4 mediates tissue damage and motor recovery independently of both the classic complement pathway and of terminal complement activation in the CCI model.
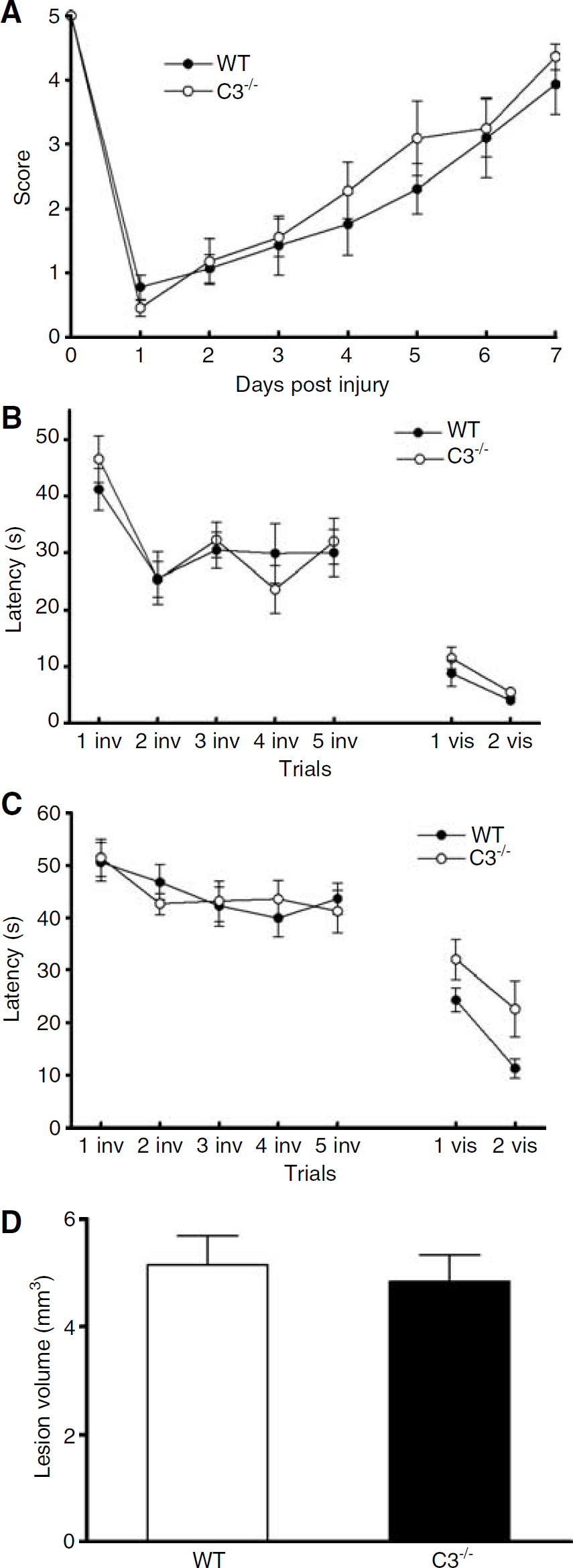
Results of behavioral and histopathologic outcome after CCI in C3-deficient (C3−/−) versus WT mice. Motor function was assessed by the wire-grip test at days 1 to 7 after CCI, and a MWM paradigm was used to assess cognitive function at days 8 to 14. Lesion size was evaluated at day 14. (
We next asked whether the C4−/− phenotype is associated with decreased neuroinflammation, assessed by posttraumatic brain neutrophil accumulation at 24 h (the time of peak neutrophil accumulation after CCI), and with decreased brain edema (24 h, the time of peak increase in brain water content in the CCI model). No differences were observed in postinjury brain neutrophil counts between C4−/− (39 ± 3, n = 7) and WT (39 ± 6, n = 9) mice. In contrast, brain neutrophils were decreased by approximately 50% in mice deficient in C3, (P < 0.05, versus WT, Figure 7). Brain edema, assessed by brain water content difference ((left hemisphere—right hemisphere)/left hemisphere) did not differ between C4−/− (2.1 ± 0.3, n = 6) and WT (2.0 ± 0.3, n = 6), or C3−/− (2.3 ± 0.1, n = 6) and WT (2.3 ± 0.1, n = 6) mice.
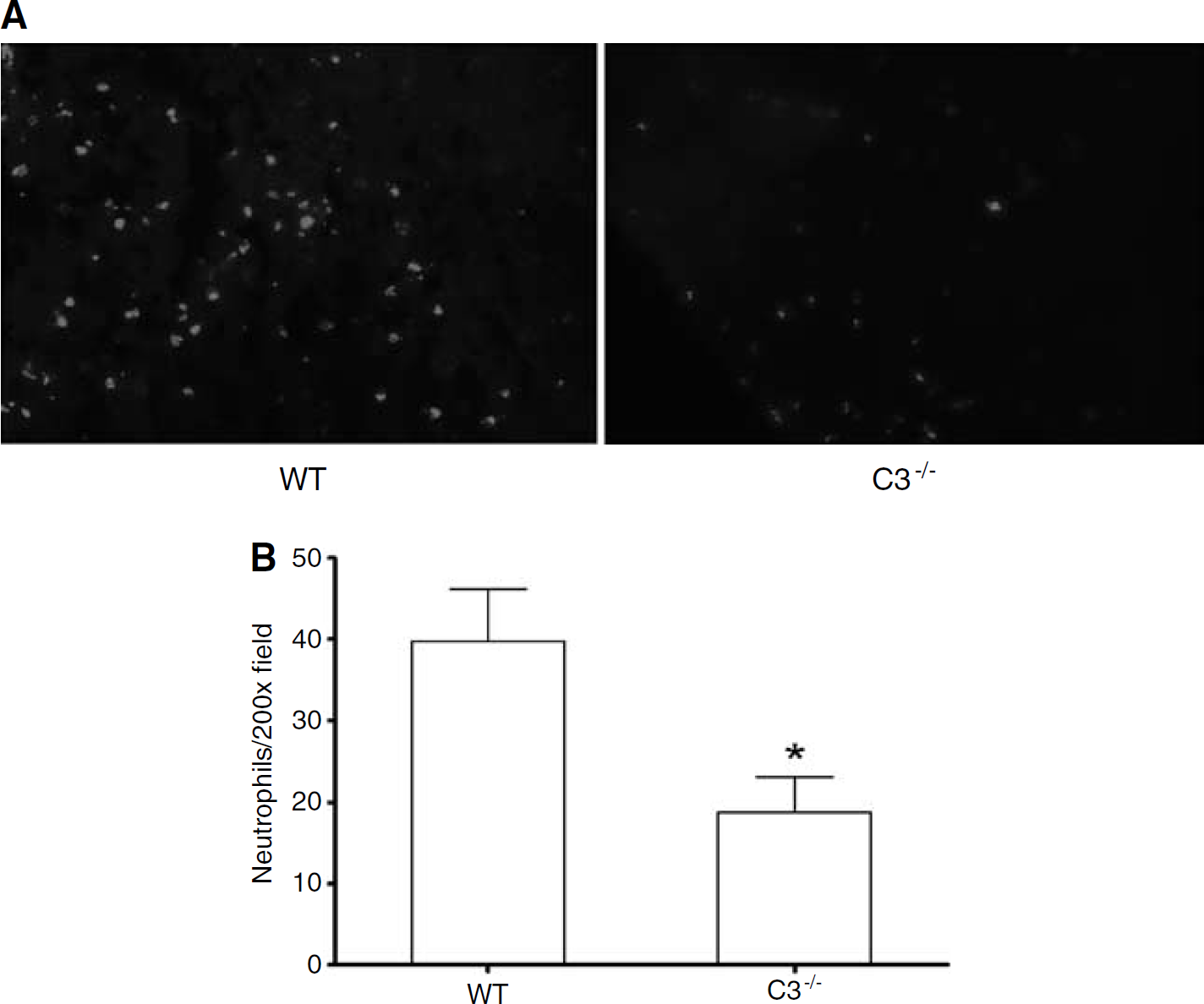
Quantification of brain neutrophils in injured cortex at 24 h after CCI. (
To determine whether C4 gene expression is induced early after TBI, C4 gene expression was analyzed in sham-operated and injured WT mice at 12 h after CCI using quantitative real-time reverse transcriptase polymerase chain reaction. No difference in C4 mRNA was observed between sham-injured (1.04 × 10−5 ± 2.9 × 10−6, n = 4) and injured (1.2 × 10−5 ± 3.8 × 10−6, n = 4) mice.
Discussion
The major finding of this study is that C4 mediates motor dysfunction and brain tissue damage independent of C3 after CCI in mice. Reconstitution experiments using hC4 ruled out the possibility that motor protection was related to potential confounding effects of strain differences between C4−/− and WT mice. Although extensive characterization would be required to examine dose-response and therapeutic window of hC4 replacement, our data strongly suggest that the C4−/− genotype accounts for the improved motor function (and probably decreased tissue damage) after CCI in C4−/− mice.
Several lines of evidence suggest that the protective effects of C4−/− on posttraumatic motor dysfunction are independent of the classic complement pathway. First, anti-C3d antibody reacted with cerebral vascular endothelium and choroid plexus epithelium after CCI in C1q−/−, C4−/−, and WT mice, suggesting that the classic pathway is dispensable for terminal complement activation after CCI. Secondly, posttraumatic motor deficits and tissue damage were not improved in C1q−/− mice versus WT, as would have been expected if the classic pathway mediated these C4−/− phenotypes. Although the lectin pathway is also inhibited in C4−/− mice, our preliminary findings with MBL−/−mice do not support a link between the lectin pathway and motor deficits or tissue damage after CCI (Yager et al, 2005). In support of our findings, Leinhase et al (2006a) recently reported a predominant role for the alternative pathway in a closed head cerebral contusion model.
In addition to ruling out the classic pathway, our data suggest that the effect of C4 on postinjury tissue damage and motor dysfunction is also independent of terminal complement activation. The presence of C3d immunostaining in vascular endothelium of C4−/− mice suggests that C4 is dispensable for cleavage and activation of C3. Furthermore, C3−/− mice did not have reduced postinjury motor deficits or tissue damage, as would have been expected if the protective effect of C4−/− were related to inhibition of C3. In addition, posttraumatic brain neutrophil accumulation was not decreased in C4−/− mice compared with WT. We and others found that brain neutrophil accumulation after cerebral contusion in rodents is C3-dependent, as shown in rats using a pharmacologic C3 convertase inhibitor (Kaczorowski et al, 1995) and in mice deficient in C3 in this study. Collectively, these findings imply that C3-independent mechanism(s) mediate the biologic effects of C4 in our CCI model.
In gastrointestinal and skeletal muscle ischemia-reperfusion models, C4-mediated tissue damage is dependent on activation of C3 (Weiser et al, 1996; Williams et al, 1999). However, C4 also has biologic effects independent of terminal complement activation. Activated products of C4 are ligands for complement receptors CR1 and CR2, which function to enhance antibody production in B lymphocytes after an immune response. Thus, C4 may play a role in adaptive immunity by shaping the host antibody repertoire (Carroll, 1998). In addition, C4 inhibits systemic autoimmunity by suppressing self-reactive antibody production through (unknown) mechanism(s) independent of CR1, CR2, and C3 (Chen et al, 2000; Cook and Botto, 2006). Whether these mechanisms are relevant to the acute (motor dysfunction) or chronic (tissue damage) effects of the C4−/−phenotype in our CCI model remains to be determined. Nonetheless, our data strongly suggest that the biologic activity of C4 in the CCI model is independent of C3.
Immunohistochemical data and studies using fluorescent-labeled hC4 suggested that after TBI, C4 signaling might involve vascular endothelium. However, blood-derived C4 could enter the brain early after CCI via a damaged blood-brain barrier and react directly with parenchymal cells. To differentiate between these two possibilities, we administered DyLight™ 547 Fluor-labeled hC4 intracerebroventricularly before CCI and examined for blood vessel labeling after injury. In contrast to systemically administered hC4-DyLight™ 547, intracerebroventricularly administered hC4-DyLight™ 547 did not label vascular endothelium at 3 or 24 h after CCI, suggesting that vascular signaling may not be required for the detrimental effects of C4 after CCI. Our inability to detect C4 or C4c on brain parenchymal cells may have been due to relative insensitivity of the immunohistochemical method/reagents used. Taken together with the lack of increase in C4 mRNA after CCI in WT mice, the hC4 reconstitution experiments also show that blood C4 is sufficient, and that novel C4 gene expression is not required, to mediate the effects of C4 in traumatically injured brain.
With the exception of C4, genetic deletion of most of the complement components studied influenced surprisingly little secondary injury in our CCI model. In contrast, C1q, C3, and other complement components contributed to brain edema and histopathology in models of ischemic and hemorrhagic brain injury (Ten et al, 2005; Yang et al, 2006). In a closed head TBI model, Rancan et al (2003) reported that astrocyte overexpression of soluble complement receptor protein y (sCrry), a mouse-specific C3 convertase inhibitor, reduced posttraumatic blood-brain barrier damage and improved neurologic severity scores (including motor function, alertness, and physiologic behavior). Subsequently, Leinhase et al (2006b) reported that systemic administration of chimeric immunoglobulin G1-Crry (Ig-Crry) fusion protein also improved neurologic severity scores, and reduced hippocampal CA3/CA4 cell loss.
The divergent results of our study and studies using complement inhibitors may be attributable to model-specific differences, as well as the use of functional outcome studies in CCI, which may be less manipulable than those reported in other TBI models. However, histopathology, brain edema, and functional outcome tests used in this study can be influenced after CCI by targeting inflammation (Sinz et al, 1999; Whalen et al, 2000). Alternatively, off-target effects of Ig-Crry fusion protein on neuroprotective gene expression may have mitigated cell death and improved functional outcome in previous reports (Leinhase et al, 2006b).
Previous studies linking inflammatory mechanisms with learning and memory deficits suggested that complement activation might influence spatial learning deficits after CCI (Biegon et al, 2002, 2004; Sinz et al, 1999). However, we found no effect of inhibition of C1q, C4, or C3 on postinjury MWM performance. The lack of an association between reduced tissue damage and MWM performance in C4−/− mice is not surprising, as gross histopathology and functional outcome often do not correlate in experimental TBI (Bermpohl et al, 2006; Clark et al, 2007). The present data suggest that C1q, C4, and C3 do not influence MWM performance in our CCI model; however, other complement components may influence postinjury cognitive brain function, as suggested by preliminary findings in our CCI model using mice deficient in MBL (Yager et al, 2005).
We conclude that C4 impairs recovery of posttraumatic motor deficits, and contributes to overall brain tissue damage after CCI, through mechanisms independent of C3 activation. In this regard, the mechanism(s) of C4 in TBI appear to be fundamentally different from those that mediate systemic ischemia-reperfusion injury. Possible mechanisms may include activation of CR1/CR2, a direct effect of C4 or its cleavage products on injured brain cells, or C4 signaling via an unidentified receptor in the brain. On the basis of current findings, we propose a novel function for C4 in the pathogenesis of TBI.
Footnotes
Acknowledgements
We thank Marina Botto (Imperial college, London, England) for generating the C1q−/− and C2/fB−/− mice.