
Abstract
Cortical application of KCl has previously been shown to induce tolerance to a subsequent episode of cerebral ischemia. KCl triggers recurrent episodes of cortical spreading depression and produces a small lesion at the cortical application site. To determine whether a cortical lesion alone is sufficient to induce tolerance to ischemia, the authors used 5-mol/L NaCl to precondition rat brain 3 days before permanent occlusion of the middle cerebral artery. NaCl produced a small lesion at the application site without evoking cortical spreading depression. Preconditioning with 5-mol/L NaCl significantly attenuated the decrease in CBF after middle cerebral artery occlusion and reduced the volume of cortical infarction by 35%. The results show that a small cortical lesion, by itself, is sufficient to induce tolerance to ischemia.
Keywords
Previous studies have demonstrated that application of KCl to the cerebral cortex induces tolerance to a subsequent episode of ischemia (Kawahara et al., 1995; Kobayashi et al., 1995; Matsushima et al., 1996; Otori et al., 2003; Taga et al., 1997; Yanamoto et al., 1998). KCl-induced tolerance is believed to be mediated by cortical spreading depression (CSD) evoked at the application site. However, application of KCl also produces a small lesion at the cortical surface, which undoubtedly triggers an inflammatory response. Inflammatory cytokines, such as tumor necrosis factor-alpha (TNF-α), have previously been associated with the induction of tolerance to ischemia (Nawashiro et al., 1997; Tasaki et al., 1997). Thus, it is possible that a cortical lesion alone is sufficient to induce tolerance to ischemia via inflammatory cytokin-mediated pathways. Accordingly, the objective of the present study was to determine whether a small cortical lesion induces tolerance to ischemia in the absence of CSD. Previous work has shown that application of 5-mol/L NaCl to the cerebral cortex causes a small cortical lesion without triggering CSD (Kawahara et al., 1999). In the present study, therefore, we determined whether application of 5-mol/L NaCl is sufficient to induce tolerance to ischemia. A preliminary report of this work has been reported previously (Muramatsu and Welsh, 2003).
MATERIALS AND METHODS
Application of 5-mol/L NaCl to the cerebral cortex was performed using procedures described previously for KCl (Otori et al., 2003). All experiments were conducted in accordance with protocols approved by the institutional animal care and use committee, including the use of sterile procedures. In brief, male Sprague-Dawley rats (n = 8), weighing 281 to 363 g, were anesthetized with halothane, intubated, and ventilated mechanically with a mixture of 1% halothane, 70% nitrous oxide, and 29% oxygen. The tail artery was cannulated for measurement of arterial pressure, blood gases, and blood glucose. Core temperature was regulated at 37.5°C using a rectal thermistor and heating blanket. The head of the animal was placed in a stereotactic frame, and a 2-mm burr hole was made over frontal cortex of the left hemisphere (3 mm rostral to bregma, 2 mm lateral to the midline), leaving the dura intact. NaCl was applied to the frontal cortex using a 1-mm2 filter paper soaked in 5-mol/L NaCl, refreshed every 20 minutes. Physiologic saline (0.15-mol/L NaCl) was applied in a similar fashion in controls (n = 8). The occurrence of CSD was monitored through the thinned skull over the ipsilateral cortex (2 mm caudal to bregma, 4 mm lateral to the midline) using laser-Doppler flowmetry as described previously (Karikó et al., 1998; Rangel et al., 2001).
Three days after NaCl application, the middle cerebral artery (MCA) was permanently occluded using a silicone-coated intraluminal suture as described previously (Otori et al., 2003). In brief, animals were anesthetized with 1% halothane, 70% nitrous oxide, and 29% oxygen delivered through a nose cone. An arterial catheter and rectal temperature probe were inserted as described above. CBF was monitored in the same location described earlier using a laser-Doppler probe held in place with a tight-fitting rivet cemented to the skull. The animal was then placed in the supine position, and the MCA on the left side was permanently occluded using an intraluminal filament after ligating the external and common carotid artery. CBF was monitored continuously before and 60 minutes after MCA occlusion. At this time, the physiologic probes were disconnected, the surgical wounds closed, and the animal returned to its cage. The use of analgesics or antibiotics was not required. Animals were killed 24 hours after occlusion, and the brain was removed and sectioned at 2-mm intervals. Sections were immersed in 2% TTC (2,3,5-triphenyltetrazolium chloride) for 15 minutes at 37°C. Images of stained sections were digitized, and infarction volumes (total, cortical, and subcortical) were determined using an indirect method (Swanson et al., 1990).
Additional animals were killed 2 hours (n = 4) and 24 hours (n = 4) after application of 5-mol/L NaCl for measurement of mRNA levels indicative of CSD. In these animals, RNA was extracted from cortical samples and analyzed for c-fos mRNA and brain-derived neurotrophic factor (BDNF) mRNA using northern blots as described previously (Karikó et al., 1998). Differences in mean values between groups were tested for statistical significance using unpaired t-tests and Bonferroni corrections for multiple comparisons. Data are reported as means ± SD,
RESULTS
Application of 5-mol/L NaCl for 2 hours, as well as 2 hours thereafter, did not trigger the characteristic laser-Doppler CBF transients that are normally associated with CSD (data not shown). Cortical levels of c-fos mRNA and BDNF mRNA, which are normally increased in the ipsilateral hemisphere after CSD, remained unchanged at 2 hours and 24 hours after NaCl application (data not shown). Application of 5-mol/L NaCl for 2 hours produced a small, cone-shaped lesion (3.9 ± 1.8 mm3; n = 8) in the frontal cortex of TTC-stained sections. No lesion was detected in control animals preconditioned with physiologic saline. Arterial variables (pH, pCO2, pO2, mean pressure, and glucose) were not significantly different in animals preconditioned with 5-mol/L NaCl or physiologic saline (Table 1). Arterial pCO2 increased slightly in both groups of spontaneously respiring animals during MCA occlusion. There were also no significant differences between groups in rectal temperature or body weight (Table 1). In both groups, a 14% loss of body weight occurred 24 hours after MCA occlusion.
Physiologic variables
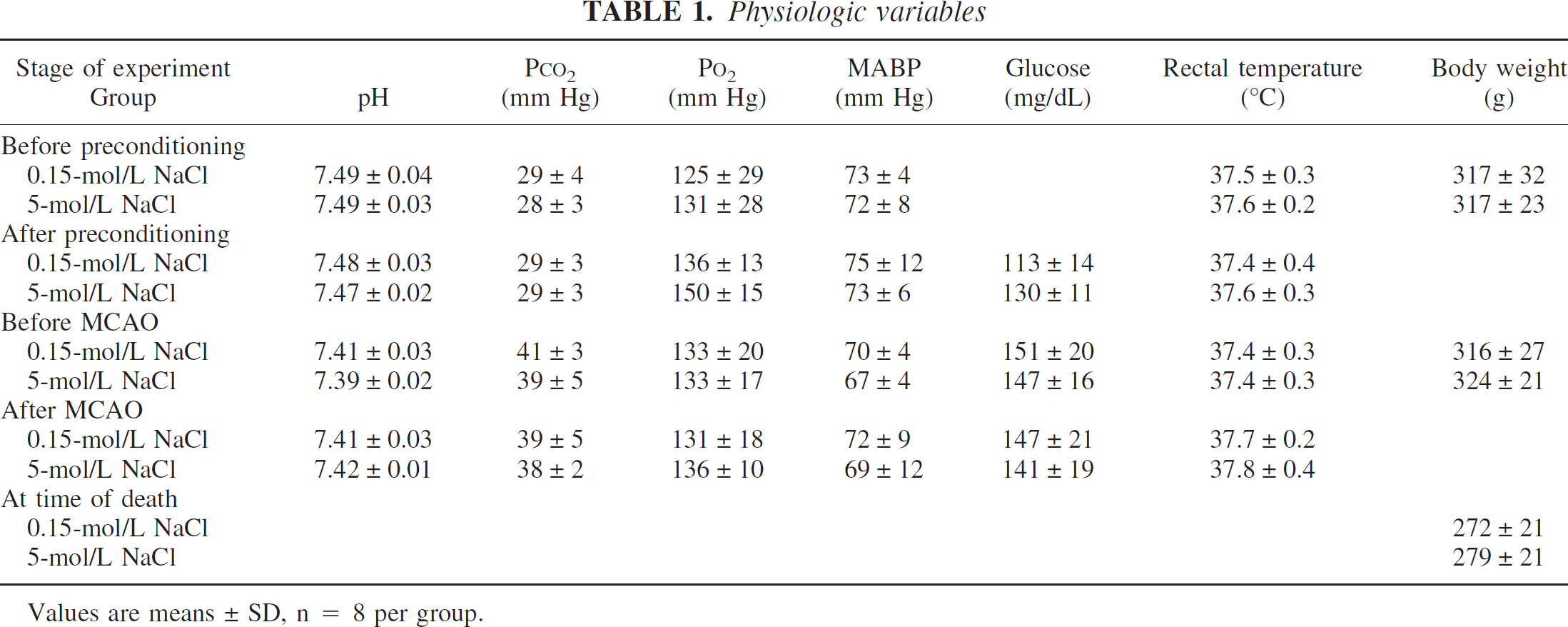
Values are means ± SD, n = 8 per group.
Preconditioning with 5-mol/L NaCl markedly reduced the volume of infarction resulting from permanent MCA occlusion (Fig. 1). The volume of infarction in the cerebral cortex was reduced from 194 ± 55 mm3 (n = 8) in saline-preconditioned controls to 127 ± 48 mm3 (n = 8) after preconditioning with 5-mol/L NaCl (P < 0.05). Analysis of individual brain sections indicated that preconditioning with 5-mol/L NaCl significantly decreased cortical infarct areas in both rostral and caudal sections of the infarct (data not shown), suggesting that neuroprotection was not limited to the cortex nearest the frontal lesion site. The volume of infarction in subcortical structures (striatum, thalamus) was not significantly different between groups. The total hemispheric volume of infarction was also reduced from 285 ± 63 mm3 to 204 ± 51 mm3 (P < 0.05) in animals preconditioned with 5-mol/L NaCl.
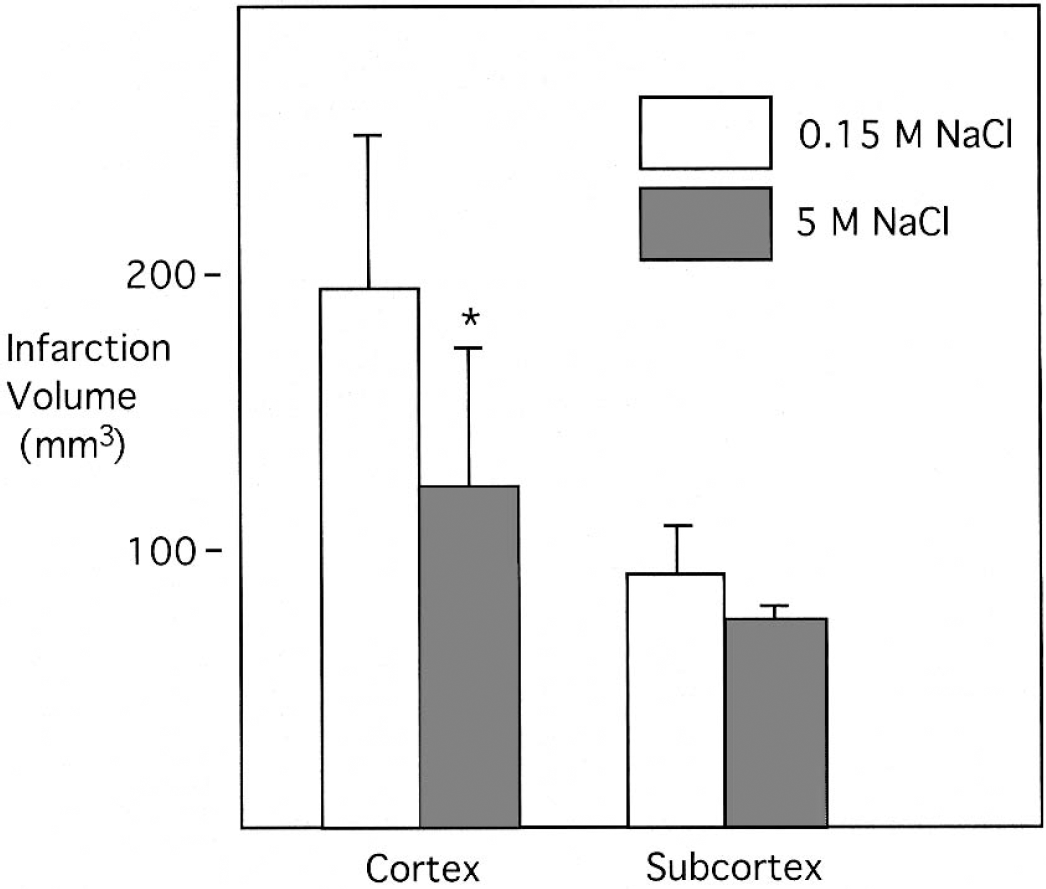
Infarction volumes after MCA occlusion in animals preconditioned with 5-mol/L NaCl or 0.15-mol/L NaCl (control). The brain was preconditioned by applying 5- or 0.15-mol/L NaCl to the intact dura for 2 hours. Three days later, the MCA was permanently occluded for 24 hours, and infarct volumes were measured in the cortex and subcortex in TTC-stained sections. Values are means ± SD, with n = 8 animals per group. *P < 0.05 compared with 0.15-mol/L NaCl (control).
Laser-Doppler flowmetry indicated that the reduction in CBF after MCA occlusion was attenuated in animals preconditioned with 5-mol/L NaCl (Fig. 2). Although the initial reduction in CBF was similar in both groups, CBF recovered by 60 minutes to 47% ± 24% of preischemic values in animals preconditioned with 5-mol/L NaCl, compared with 27% ± 12% in saline-conditioned controls (P < 0.05).
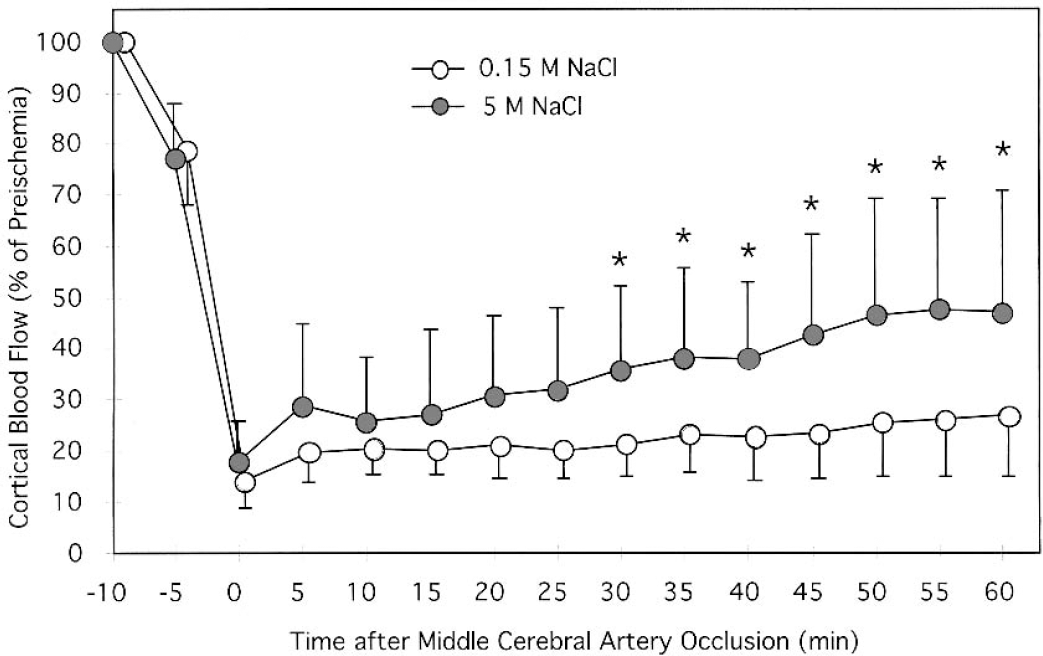
Cerebral blood flow during MCA occlusion in animals preconditioned with 5-mol/L NaCl or 0.15-mol/L NaCl. The brain was preconditioned by applying 5- or 0.15-mol/L NaCl to the intact dura for 2 hours. Three days later, the MCA was occluded, and CBF was measured in the cerebral cortex using laser-Doppler flowmetry. The initial decline (at −5 min) occurred after clamping of the common carotid artery. Values are means ± SD, with n = 8 animals per group. *P < 0.05 compared with 0.15-mol/L NaCl (control).
DISCUSSION
The central finding of the present study is that a small cortical lesion, by itself, is sufficient to induce tolerance to ischemia. In many models of preconditioning, including brief ischemia and CSD, depolarization of the tissue has been considered essential to trigger the induction of tolerance (Abe and Nowak, 2004; Glazier et al., 1994; Kobayashi et al., 1995; Otori et al., 2003). However, in these same models, a small amount of tissue injury is produced during the preconditioning stimulus. Thus, in addition to the cellular response to depolarization, there is an inflammatory cellular response to tissue injury. Because inflammatory cytokines have been implicated in the induction of tolerance to ischemia (Nawashiro et al., 1997; Tasaki et al., 1997), in the present study we asked whether tissue injury alone induces tolerance to ischemia. However, it is difficult to prevent CSD during cortical lesioning (even a pinprick to the dura normally triggers an episode of CSD). Application of 5-mol/L NaCl to the intact dura was previously reported to cause a small cortical lesion without evoking CSD (Jander et al., 2001; Kawahara et al., 1995, 1999). Thus, in the present study we used NaCl to produce a small cortical lesion in the absence of CSD.
Previous studies, including our own, have shown that CSD triggers a robust increase in CBF that is readily detected using laser-Doppler flowmetry (Karikó et al., 1998; Rangel et al., 2001). In the present study, CSD was not detected with laser-Doppler flowmetry during the 2-hour period of 5-mol/L NaCl application, nor was it detected during 2 hours of subsequent recovery. Thus, unless NaCl evokes CSD without its characteristic change in CBF, we conclude that 5-mol/L NaCl failed to trigger CSD within the first 4 hours of the onset of application. This conclusion is consistent with the previously reported absence of direct current transients after application of 5-mol/L NaCl (Jander et al., 2001; Kawahara et al., 1995, 1999). The occurrence of CSD at later times is also unlikely because of the absence of changes in gene expression that are normally associated with CSD. Previous studies have shown that CSD triggers a robust elevation of c-fos mRNA in the ipsilateral cortex (Karikó et al., 1998; Rangel et al., 2001). In the present study, changes in c-fos mRNA or BDNF mRNA levels were not detected at 2 hours or 24 hours after application of 5-mol/L NaCl. Although we cannot completely rule out the occurrence of CSD at some point during the 3-day period after 5-mol/L NaCl application, we believe the chances of undetected CSD to be exceedingly small. Thus, we conclude that the presence of a small cortical lesion is sufficient to induce tolerance to ischemia in the absence of CSD. Moreover, we suspect that the presence of tissue injury in many other models of preconditioning, including injury caused by insertion of intracerebral electrodes, may have contributed significantly to the induction of tolerance.
What mechanisms might account for the ability of a cortical lesion to induce tolerance to ischemia? The present study indicates that preconditioning with 5-mol/L NaCl attenuated the ischemic decrease in CBF, as measured using laser-Doppler flowmetry. Thus, the mechanism of induced tolerance appeared to have a vascular component, which promoted tissue perfusion during ischemia. This finding of improved perfusion, however, is in apparent conflict with previous reports that have failed to detect a beneficial effect of preconditioning with CSD on CBF during ischemia (Matsushima et al., 1996; Yanamoto et al., 1998). It should be cautioned, however, that laser-Doppler flowmetry measures changes in CBF relative to values obtained during the preischemic baseline period, and, thus, does not represent absolute CBF. In our previous study, preconditioning the brain with KCl caused an attenuation in the decrease of laser-Doppler CBF after MCA occlusion similar to that observed in the present study (Otori et al., 2003). Measurement of baseline CBF using [14C]-iodoantipyrine 3 days after KCl preconditioning indicated that the absolute value of CBF was 25% to 33% lower than that in controls conditioned with physiologic saline. It is not known whether preconditioning with 5-mol/L NaCl also results in long-lasting decrease in baseline CBF. However, in another study using [14C]-2-deoxyglucose to measure CMRglu, application of high concentrations of KCl or NaCl both caused a long-lasting decrease CMRglu (Kawahara et al., 1999). Further, comparable reductions in CMRglu have been reported after cerebral trauma caused by freeze lesions or lateral fluid percussion (Pappius, 1981; Yoshino et al., 1991). Thus, it appears that cortical injury, in general, induces a prolonged suppression of metabolic rate. Assuming that blood flow remains coupled to metabolic rate, baseline CBF will be reduced accordingly. Thus, it is likely that preconditioning with 5-mol/L NaCl also causes a long-lasting decrease in baseline CBF. If so, then preconditioning with NaCl may not increase the absolute level of CBF during MCA occlusion, but, rather, may improve in the ratio between CBF and metabolic rate, thereby lessening the degree of ischemia.
Apart from its suppression of CMRglu, the mechanisms underlying the induction of tolerance to ischemia after a small cortical lesion remain speculative. Presumably, there is a response to tissue injury that is likely to include an inflammatory component. Cortical lesions induced by application of high concentrations of electrolytes, including NaCl, trigger the expression of proinflammatory cytokines, such as TNF-α (Jander et al., 2001). At the site of electrolyte-induced lesions, lysis of cells will release intracellular constituents into the extracellular space. Thus, it is possible that any of these constituents may have a role in the induction of tolerance. Resident microglia are believed to act as sensors of such cellular damage, resulting in the expression of proinflammatory cytokines (Herx et al., 2000). Microglia possess Toll-like receptors, which specifically recognize degradation products of the extracellular matrix as well as intracellular constituents released from necrotic cells (for review, see Nguyen et al., 2002). Further, recent evidence indicates that release of ATP into the extracellular space induces the expression of TNF-α in microglia via P2X7 receptors (Suzuki et al., 2004). As noted earlier, TNF-α has been directly implicated in the mechanism of induced tolerance to ischemia, particularly in models of lipopolysaccharide preconditioning (Nawashiro et al., 1997; Tasaki et al., 1997). The means by which local expression of cytokines causes neuroprotective changes in remote regions of the brain remain unknown. However, it has been shown that local induction of cytokines in circumventricular organs after systemic administration of lipopolysaccharide is followed by delayed expression of cytokines in microglia throughout the brain parenchyma (Nadeau and Rivest, 2000). Although the route and mechanisms by which widespread cytokine expression is induced remain poorly understood, it possible that similar mechanisms may be operating after cortical lesioning. Future work will be required to clarify this intriguing phenomenon.
In conclusion, the present study shows that a small cortical lesion, by itself, induces tolerance to ischemia. It is possible that the degree of neuroprotection induced by the lesion alone is less than that induced by a lesion in combination with CSD. However, confirmation of this possibility will require contemporaneous experiments with groups of animals preconditioned with NaCl or KCl and subjected to the same model of cerebral ischemia. The present results also suggest that depolarization, which is most likely restricted to the NaCl application site, is not required for the induction of ischemic tolerance in remote regions of the cortex. Finally, the experimental model of NaCl-induced tolerance may be a convenient model to investigate neuroprotective preconditioning pathways because the molecular sequelae are likely to be less complex than those that occur after widespread depolarization.
Footnotes
Acknowledgment:
The authors thank Ann Muramatsu for her excellent technical assistance.