
Abstract
Cortical spreading depression (CSD) has previously been shown to induce tolerance to a subsequent episode of transient cerebral ischemia. The objective of the present study was to determine whether CSD also induces tolerance to permanent focal ischemia and, if so, whether tolerance may be mediated by alterations in cerebral blood flow (CBF). Sprague-Dawley rats were preconditioned by applying potassium chloride to one hemisphere for 2 hours, evoking 19 ± 5 episodes of CSD (mean ± SD, n = 19). Three days later, the middle cerebral artery (MCA) was permanently occluded using an intraluminal suture. In a subset of animals, laser Doppler blood flow (LDF) was monitored over the parietal cortex before and during the first 2 hours of MCA occlusion. Preconditioning with CSD reduced the hemispheric volume of infarction from 248 ± 115 mm3 (n = 18) in sham-conditioned animals to 161 ± 81 mm3 (n = 19, P < 0.02). Similarly, CSD reduced the neocortical volume of infarction from 126 ± 82 mm3 to 60 ± 61 mm3 (P < 0.01). Moreover, preconditioning with CSD significantly improved LDF during MCA occlusion from 21% ± 7% (n = 9) of preischemic baseline in sham-conditioned animals to 29% ± 9% (n = 7, P < 0.02). Preconditioning with CSD therefore preserved relative levels of CBF during focal ischemia and reduced the extent of infarction resulting from permanent MCA occlusion. To determine whether CSD may have altered preischemic baseline CBF, [14C]iodoantipyrine was used in additional animals to measure CBF 3 days after CSD conditioning or sham conditioning. CSD, but not sham conditioning, significantly reduced baseline CBF in the ipsilateral neocortex to values 67% to 75% of those in the contralateral cortex. Therefore, CSD causes a long-lasting decrease in baseline CBF that is most likely related to a reduction in metabolic rate. A reduction in the rate of metabolism may contribute to the induction of tolerance to ischemia after preconditioning with CSD.
Keywords
Previous studies have demonstrated that preconditioning the brain with cortical spreading depression (CSD) reduces the extent of cellular injury resulting from a subsequent episode of ischemia (Kawahara et al., 1995; Kobayashi et al., 1995; Matsushima et al., 1996; Taga et al., 1997; Yanamoto et al., 1998). The mechanisms by which CSD induces tolerance to ischemia, however, are not well understood. To date, preconditioning with CSD has been assessed in models of transient focal and global ischemia, but not in models of permanent focal ischemia. Reperfusion after transient ischemia has been associated with excessive generation of reactive oxygen species (ROS), which are believed to contribute importantly to ischemic injury (for a recent review, see Chan, 2001). Antagonism of ROS-mediated cell damage, therefore, is one mechanism by which CSD might induce tolerance to ischemia. Experimental models of permanent focal ischemia lack a reperfusion period per se and, thus, may not benefit from preconditioning with CSD. Indeed, transgenic animals overexpressing the antioxidant enzyme, Cu, Zn-superoxide dismutase, were protected against transient focal ischemia (Yang et al., 1994), but not against permanent focal ischemia (Chan et al., 1993). Accordingly, the primary objective of the present study was to determine whether preconditioning with CSD induces tolerance to permanent occlusion of the MCA.
A second objective of the present investigation was to determine whether preconditioning with CSD alters the reduction in CBF during focal ischemia. Previous studies have failed to detect a beneficial effect of preconditioning with CSD on CBF during focal ischemia, suggesting that vascular mechanisms do not contribute to the induction of tolerance to ischemia (Matsushima et al., 1996; Yanamoto et al., 1998). It is difficult to exclude the possibility, however, that small increments in CBF during focal ischemia may have escaped detection yet contributed importantly to the reduction in ischemic injury after preconditioning with CSD. In the present study, we used LDF before and after MCA occlusion to determine whether preconditioning with CSD improves CBF during focal ischemia. Because LDF is a relative measure of CBF, additional animals were studied using IAP to measure the absolute level of baseline CBF 3 days after CSD conditioning or sham conditioning. Preliminary results of this study have been reported previously (Otori et al., 2001).
MATERIALS AND METHODS
All procedures were performed in strict accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania.
Cortical spreading depression
Cortical spreading depression was induced in one hemisphere of male Sprague-Dawley rats using modifications of previously described methods (Kobayashi et al., 1995). In brief, animals weighing 270 to 350 g were anesthetized with halothane, intubated, and ventilated mechanically with a mixture of 1% halothane, 30% oxygen, and a balance of nitrous oxide. The tail artery was catheterized for measurement of arterial pressure, blood gases, and blood glucose. A thermistor was inserted into the rectum, and core temperature was regulated at 37.0°C using a heating pad. The head of the animal was placed into a stereotaxic frame, and a 2-mm burr-hole was made over the frontal cortex of the left hemisphere (3 mm rostral to bregma, 2 mm lateral to the midline), leaving the dura intact. Cortical spreading depression was evoked by placing a 1-mm2 filter paper soaked in 2 mol/L potassium chloride onto the dura, refreshing the filter paper every 20 minutes for 2 hours. Filter paper soaked in physiologic saline was used in sham-conditioned controls. Episodes of CSD were monitored through the thinned skull over the ipsilateral parietal cortex (2 mm caudal to bregma, 4 mm lateral) using LDF as described previously (Karikó et al., 1998). After CSD conditioning or sham conditioning, the surgical sites were sutured, and the animal was returned to his cage.
Permanent occlusion of the middle cerebral artery
Three days after CSD conditioning or sham conditioning, the MCA was permanently occluded using a silicone-coated intraluminal filament and modifications of methods described previously (Koizumi et al., 1986; Mori et al., 2000). In brief, animals were anesthetized with 1% halothane, 30% oxygen, and a balance of nitrous oxide, delivered through a nose cone. An arterial catheter and rectal temperature probe were inserted as previously described, and needle thermistors were placed bilaterally in the temporalis muscle to monitor the temperature of the head. The animal was placed in the supine position, and the common carotid artery on the left side was isolated and dissected to the carotid bifurcation. A silicone-coated nylon filament was prepared from 4–0 monofilament, the tip of which was rounded and enlarged to 0.35 mm using heat. The filament was coated with a silicone polymer (Konishi, Osaka, Japan), yielding an outer diameter of 0.35 mm for a distance of 5 mm from the tip. To occlude the MCA, the external and common carotid arteries were ligated, and the filament was inserted into the internal carotid artery and advanced until resistance was encountered (≉18 mm). The filament was then fastened securely, the surgical sites were sutured, and the animal was returned to its cage. In a subset of animals, CBF was monitored in the ipsilateral parietal cortex using LDF. In these animals, an aluminum cylinder was cemented to the thinned skull over the parietal cortex (2 mm caudal to bregma, 4 mm lateral) for insertion of the LDF probe. The animal was then placed in the supine position on an elevated platform, allowing for clearance of the probe. Rectal and head temperatures were monitored as previously described and regulated at 37.0°C using a heating lamp. Laser Doppler flow was monitored before MCA occlusion and for 2 hours afterward. The aluminum cylinder and LDF probe were removed, the surgical sites sutured, and the animal was returned to its cage.
Measurement of infarct volumes
Animals were killed 24 hours after permanent occlusion of the MCA. The brain was removed from the cranium and the underside of the brain was examined for location of the tip of the intraluminal suture. The brain was then sectioned in the coronal plane at 2-mm intervals, and the sections were examined for subarachnoid hemorrhage. Animals exhibiting significant hemorrhage (usually associated with suture advancement past the origin of the MCA) were excluded from the study. Brain sections were immersed in 2% 2,3,5-triphenyltetrazolium chloride and incubated for 15 minutes at 37°C. Images of the stained sections were digitized, and infarction volumes were determined with image-analysis software (NIH Image) using an indirect method (Swanson et al., 1990). In each brain section, the area of red-staining tissue in the ipsilateral hemisphere was subtracted from that in the contralateral hemisphere to obtain the total area of hemispheric infarction. Similar measurements were made in hemispheric subdivisions, including neocortex, subcortex, and caudoputamen (Fig. 1).
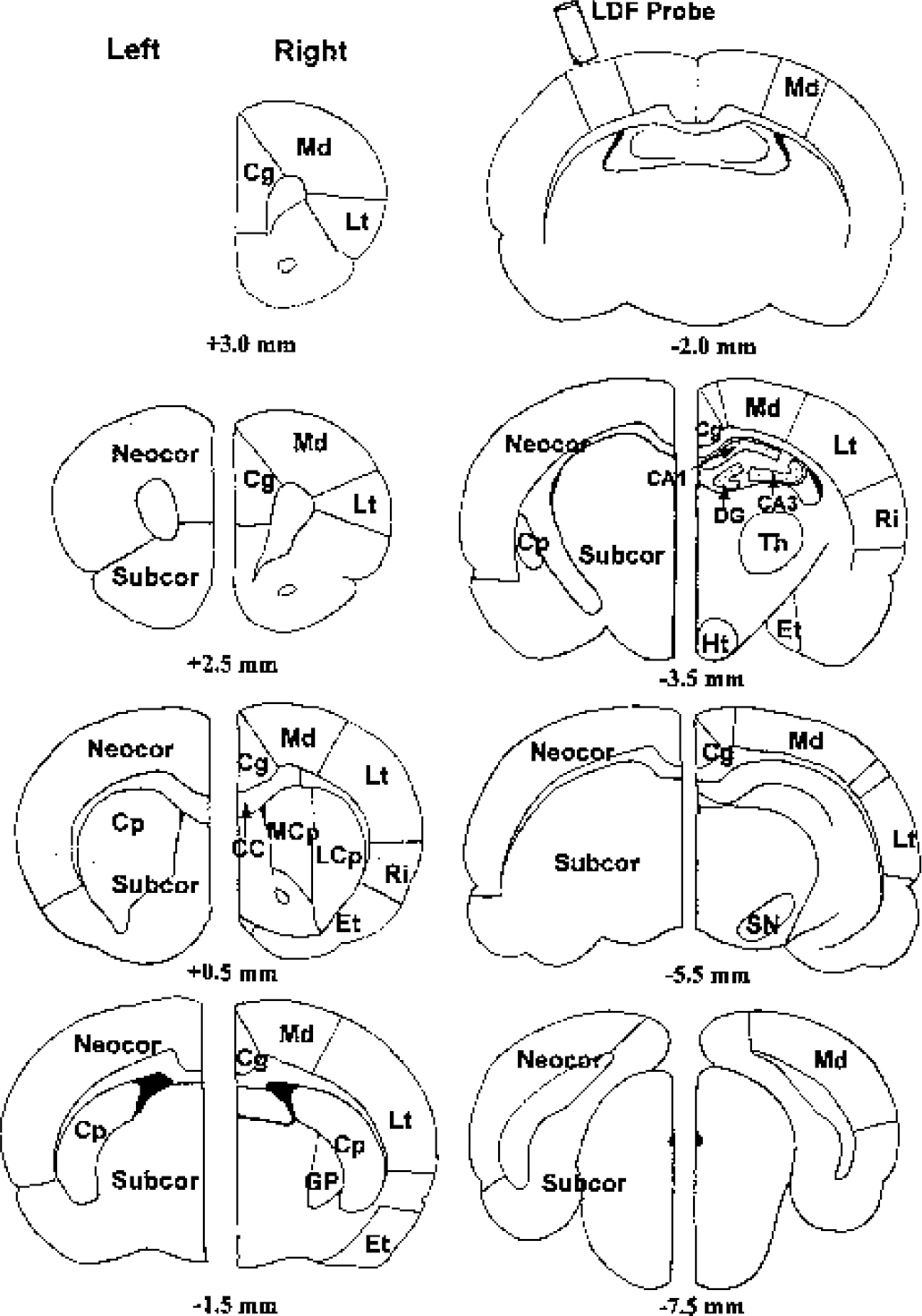
Anatomic location of regions analyzed for infarction volume and cerebral blood flow (CBF). Brain levels are indicated by distance from bregma. Infarction areas were measured using 2,3,5-triphenyltetrazolium chloride in neocortex (Neocor), subcortex (Subcor), and caudoputamen (Cp), as shown in the diagram of the left hemisphere. CBF was measured using [14C]iodoantipyrine autoradiography in selected areas, as shown in the diagram of the right hemisphere. The areas analyzed for CBF are listed in Table 2. The location of the laser Doppler blood flow (LDF) probe is also illustrated. Cg, cingulate; Md, medial; Lt, lateral; Ri, rhinal; Et, entorhinal; CC, corpus callosum; MCp, medial caudoputamen; LCp, lateral caudoputamen; GP, globus pallidus; CA1, CA1 hippocampus; CA3, CA3 hippocampus; DG, dentate gyrus; Th, thalamus; Ht, hypothalamus; SN, substantia nigra.
Regional cerebral blood flow: iodoantipyrine autoradiography
Regional CBF was measured 3 days after CSD conditioning or sham conditioning using [14C]-IAP autoradiography (Reivich et al., 1969; Sakurada et al., 1978). Rats were anesthetized as described previously and catheters (PE50) were placed into both femoral arteries and one femoral vein. One femoral artery was used to monitor arterial blood pressure and to obtain blood gas samples, whereas the other was used to sample blood for [14C]IAP concentration. Twenty microcuries of 4-iodo[N-methyl-14C]-antipyrine (NEN Research Products, Boston, MA, U.S.A.) was infused at a linearly increasing rate into the femoral vein, whereas arterial blood samples were obtained every 3 to 4 seconds by touching 20-μL pipettes (Unopette; Becton Dickinson, Franklin Lakes, NJ, U.S.A.) to the arterial catheter that was cut close to its entrance into the femoral artery. Sixty seconds after the start of the infusion, the animal was decapitated, and the brain was quickly removed, frozen in isopentane cooled to −50°C with dry ice, and stored at −80°C until processing. The arterial blood samples were spotted onto filter paper and allowed to dry overnight. The blood spots were placed into liquid scintillation vials with scintillation fluid and counted (LS6000IC; Beckman Instruments, Fullerton, CA, U.S.A.) for quantification of the time course of the arterial blood IAP concentration. The brain was sectioned coronally in a cryostat (Hacker Instruments, Inc., Fairfield, NJ, U.S.A.) at a thickness of 20 μm, and triplicate sections taken at intervals of 500 μm were placed onto x-ray film (BioMax MR; Eastman Kodak, Rochester, NY, U.S.A.). The autoradiographic images were digitized and blood flow was measured in predetermined regions of cerebral cortex, subcortical white matter, striatum, and hippocampus (Fig. 1) using AIS/C software (Imaging Research, St. Catharines, Ontario, Canada).
Animal groups
Experiments were performed in a pairwise fashion, matching one CSD-preconditioned animal with one sham-conditioned animal. A total of 21 pairs of animals were preconditioned 3 days prior to permanent occlusion of the MCA. Three of the sham-conditioned animals exhibited subarachnoid hemorrhage at the time of brain dissection and were excluded from the study. In a subset of nine pairs of animals, LDF was monitored before and during 2 hours of MCA occlusion. One of these CSD-preconditioned animals experienced a seizure several hours after occlusion of the MCA and another did not survive the required 24-hour interval after occlusion. These animals were also excluded from the analysis. In two additional groups of animals, CBF was measured using [14C]-IAP 3 days after CSD conditioning (n = 8) or sham conditioning (n = 5).
Statistical analysis
Differences between CSD conditioning and sham-conditioned groups were tested for statistical significance using one-way analysis of variance followed by a post hoc Tukey test. A P value less than 0.05 was considered statistically significant. Correlations between the number of episodes of CSD and infarction volumes, and between LDF and infarction volumes, were tested for statistical significance using Pearson product-moment coefficients and bivariate regression.
RESULTS
Physiologic variables
Application of potassium chloride for 2 hours evoked 19 ± 5 episodes of CSD (mean ± SD, n = 19). Cortical spreading depression was not detected in sham-conditioned animals. There were no significant differences in physiologic variables between sham- and CSD-conditioned groups (Table 1). In both groups, mild respiratory alkalosis occurred during preconditioning as a result of mechanical ventilation. Blood glucose was slightly elevated before MCA occlusion, but glucose concentrations were not significantly different between groups. Body and head temperatures remained near 37.0°C before and after MCA occlusion.
Physiologic variables
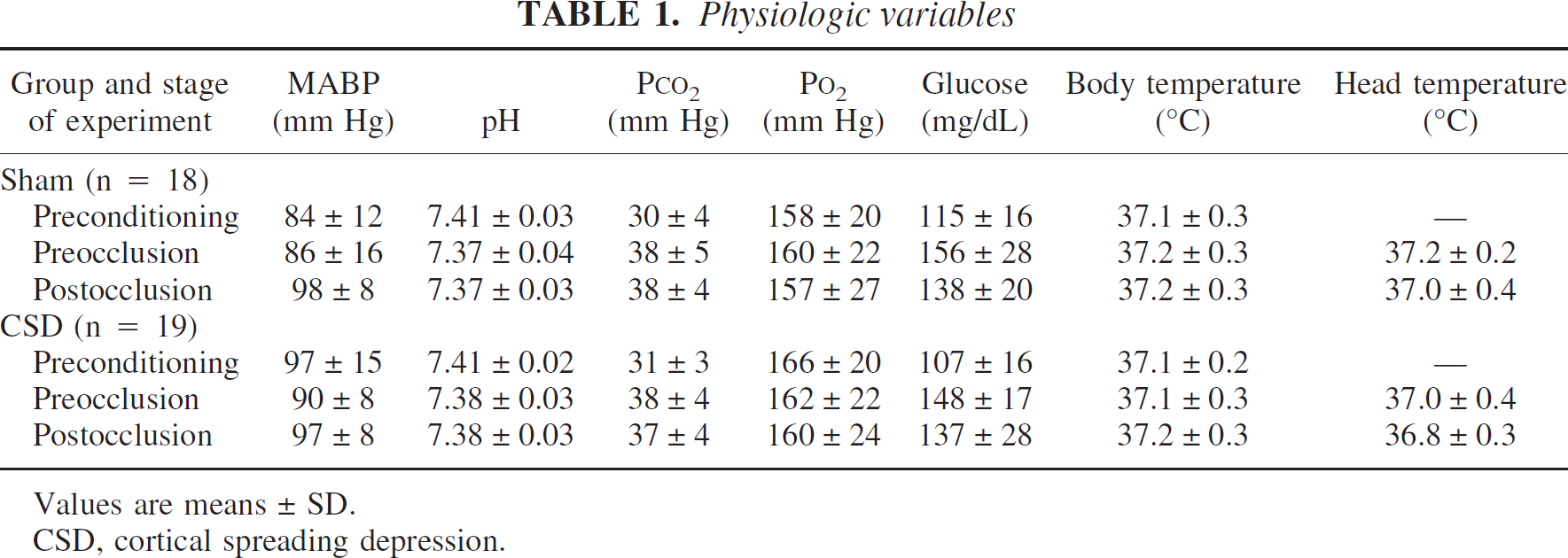
Values are means ± SD.
CSD, cortical spreading depression.
Cerebral infarction
Preconditioning with CSD decreased the hemispheric volume of infarction by 35% and the neocortical volume of infarction by 52%, relative to infarction volumes in sham-conditioned animals (Fig. 2). Significant reductions in infarct areas occurred in all brain sections, indicating that tolerance was induced throughout the distribution of the MCA. Preconditioning with CSD did not cause a significant reduction in the volume of infarction in the caudoputamen (data not shown). Within the CSD-preconditioned group, the number of episodes of CSD correlated inversely with hemispheric and neocortical volumes of infarction, indicating that the degree of induced tolerance was directly related to the number of episodes of CSD (Fig. 3).
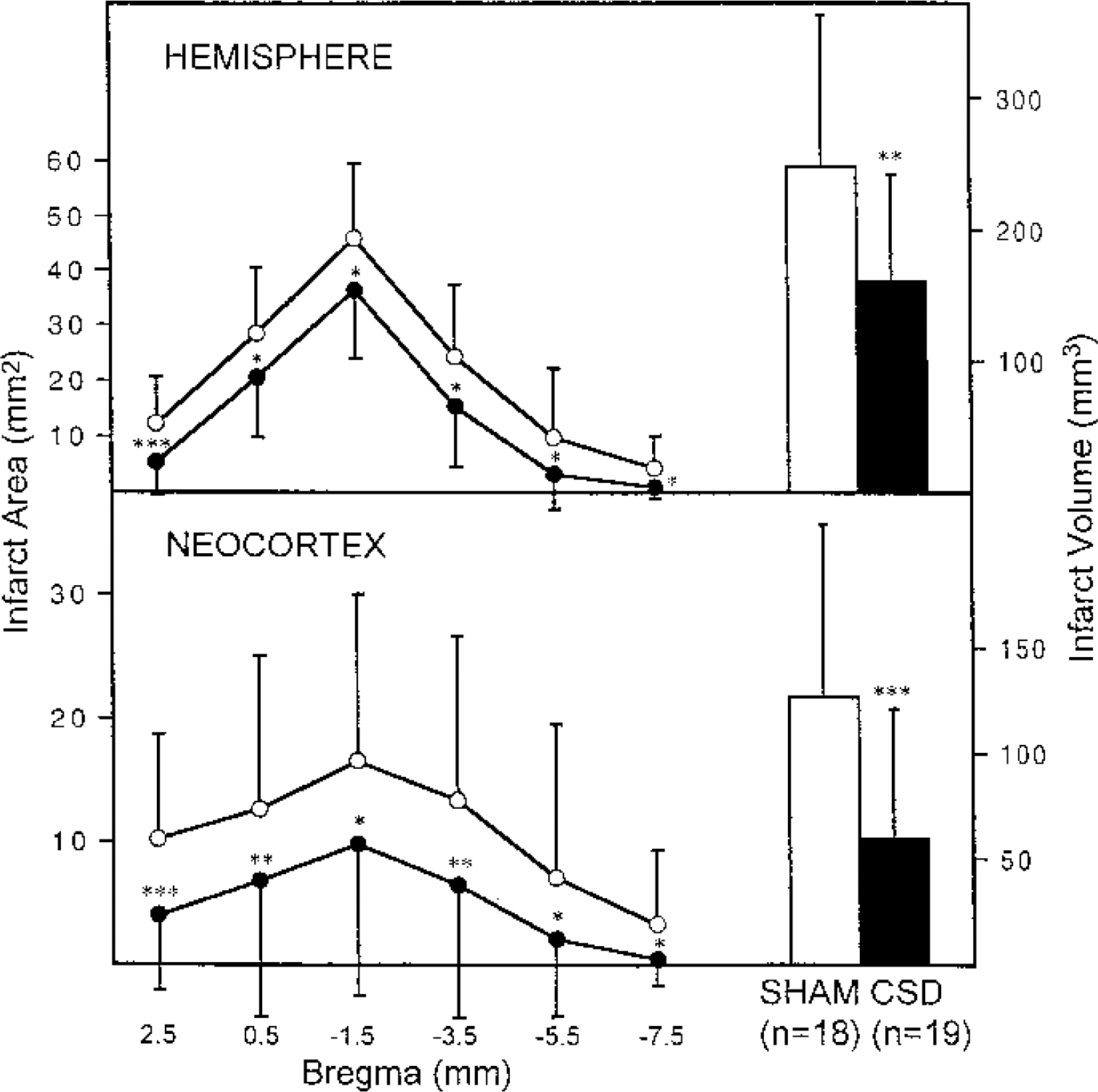
Effect of preconditioning with cortical spreading depression (CSD) on cerebral infarction. Areas of infarction in the hemisphere and neocortex were measured in brain sections indicated by distance from bregma. Values are means ± SD, with open circles representing sham-conditioned animals and closed circles representing CSD-preconditioned animals. Volumes of infarction are indicated in the adjacent bars. *P < 0.05; **P < 0.02; ***P < 0.01.
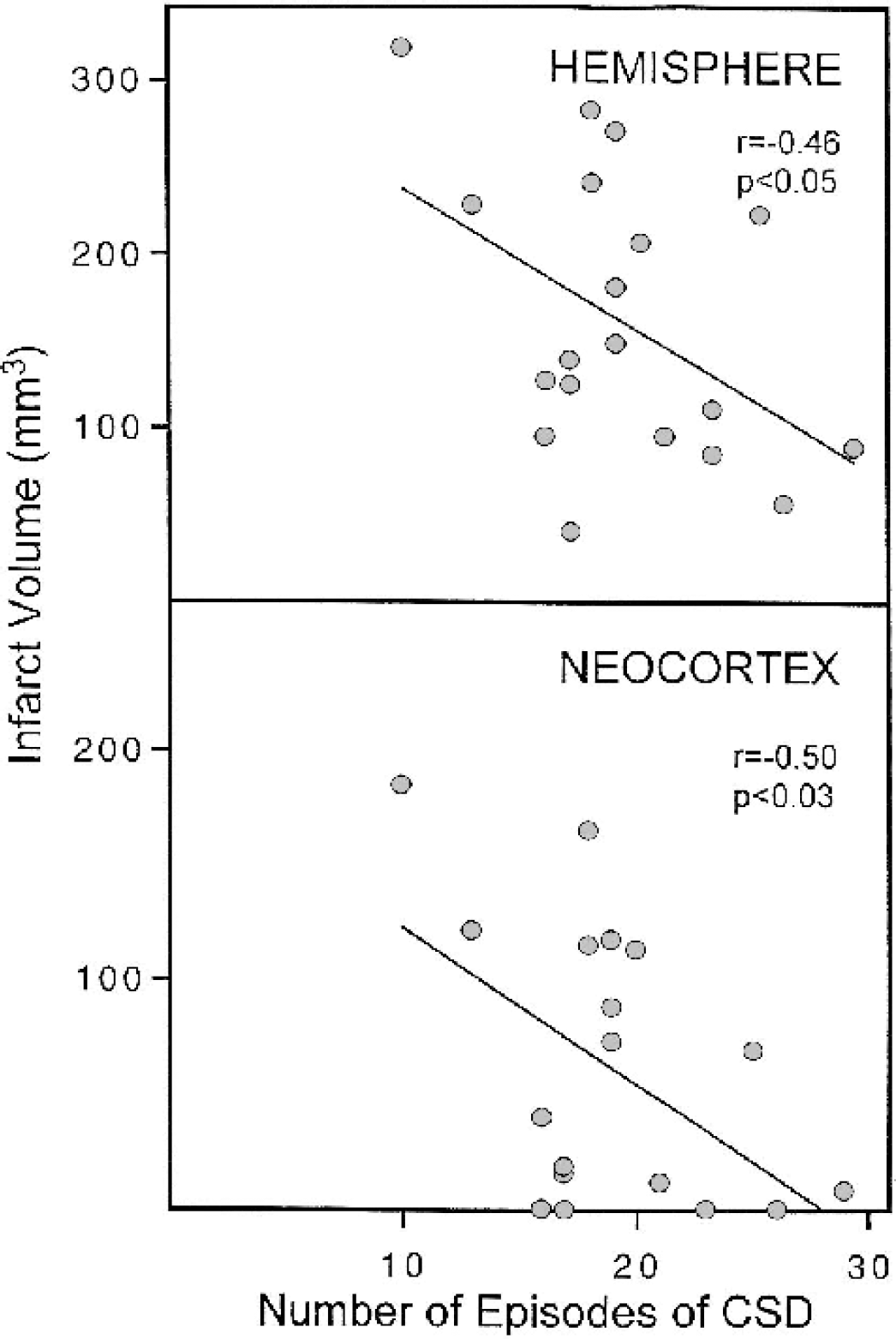
Correlation between number of episodes of cortical spreading depression (CSD) and infarction volume. For each CSD-preconditioned animal, the number of episodes of CSD detected during the preconditioning period was plotted against the volumes of infarction in the hemisphere and neocortex. There was a significant inverse correlation between the number of episodes of CSD and hemispheric infarct volume (r = −0.46, P < 0.05) and neocortical infarct volume (r = −0.50, P < 0.03).
Laser Doppler flowmetry
Permanent occlusion of the MCA caused a rapid decrease in LDF in both CSD-conditioned and sham-conditioned animals (Fig. 4). The percentage remaining LDF, however, was significantly greater in CSD-preconditioned animals than in sham-conditioned animals at nearly all times during the first 2 hours after MCA occlusion. In both groups, LDF tended to increase during the first 2 hours of occlusion, but the increase was not statistically significant in either group. The average LDF during the first 2 hours after MCA occlusion was significantly higher in animals preconditioned with CSD (29% ± 9%) than in sham-conditioned animals (21% ± 7%, P < 0.02). In both groups combined, there was an inverse correlation between the average LDF during the first 2 hours after MCA occlusion and the hemispheric and neocortical volumes of infarction, indicating that the extent of ischemic injury was related to LDF during focal ischemia (Fig. 5).
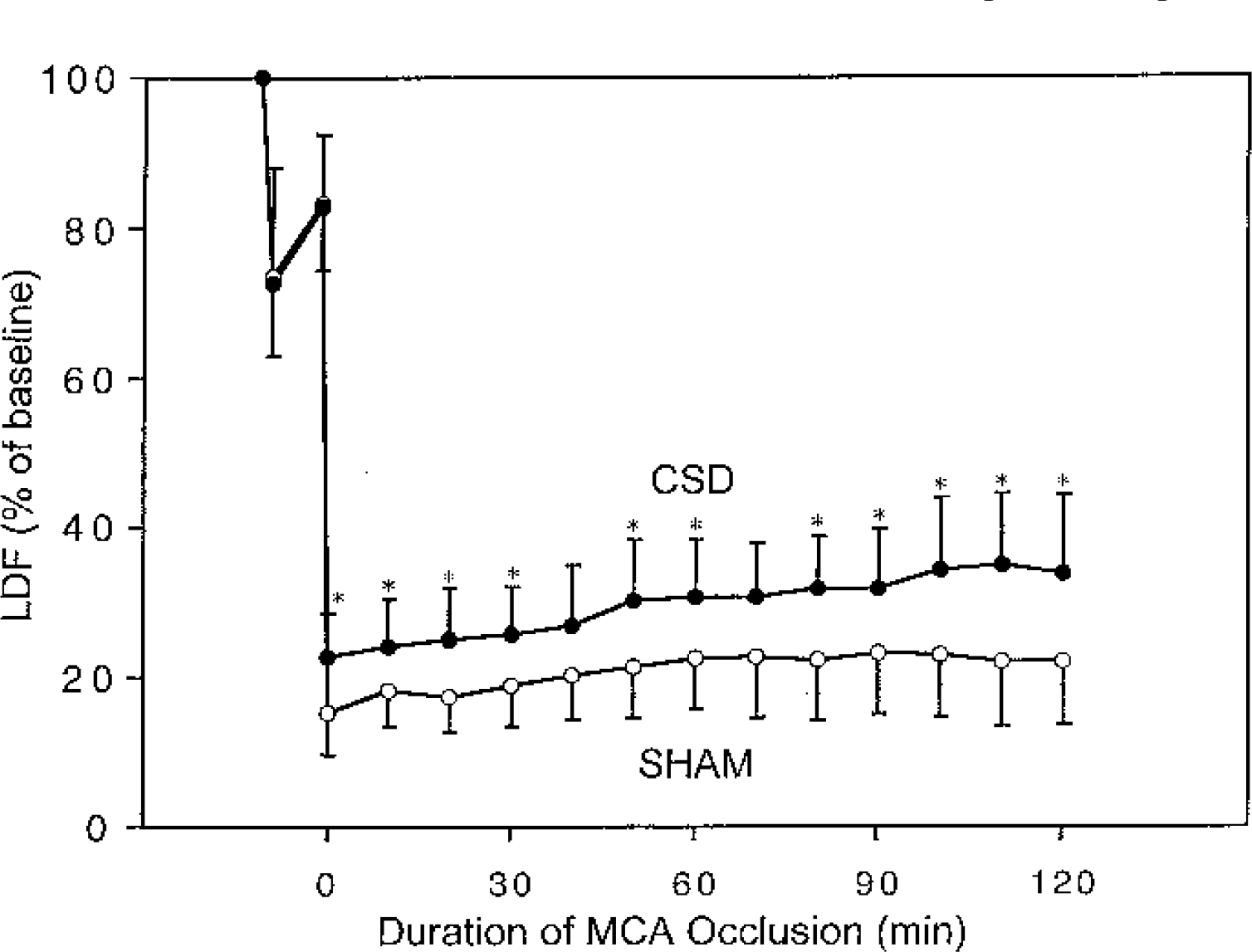
Effect of preconditioning with cortical spreading depression on laser Doppler blood flow (LDF) after middle cerebral artery (MCA) occlusion. LDF was measured continuously for 2 hours after MCA occlusion and was averaged in 10-minute intervals and expressed as a percentage of preischemic baseline LDF. *Significantly higher than LDF in sham-conditioned animals, P < 0.05.
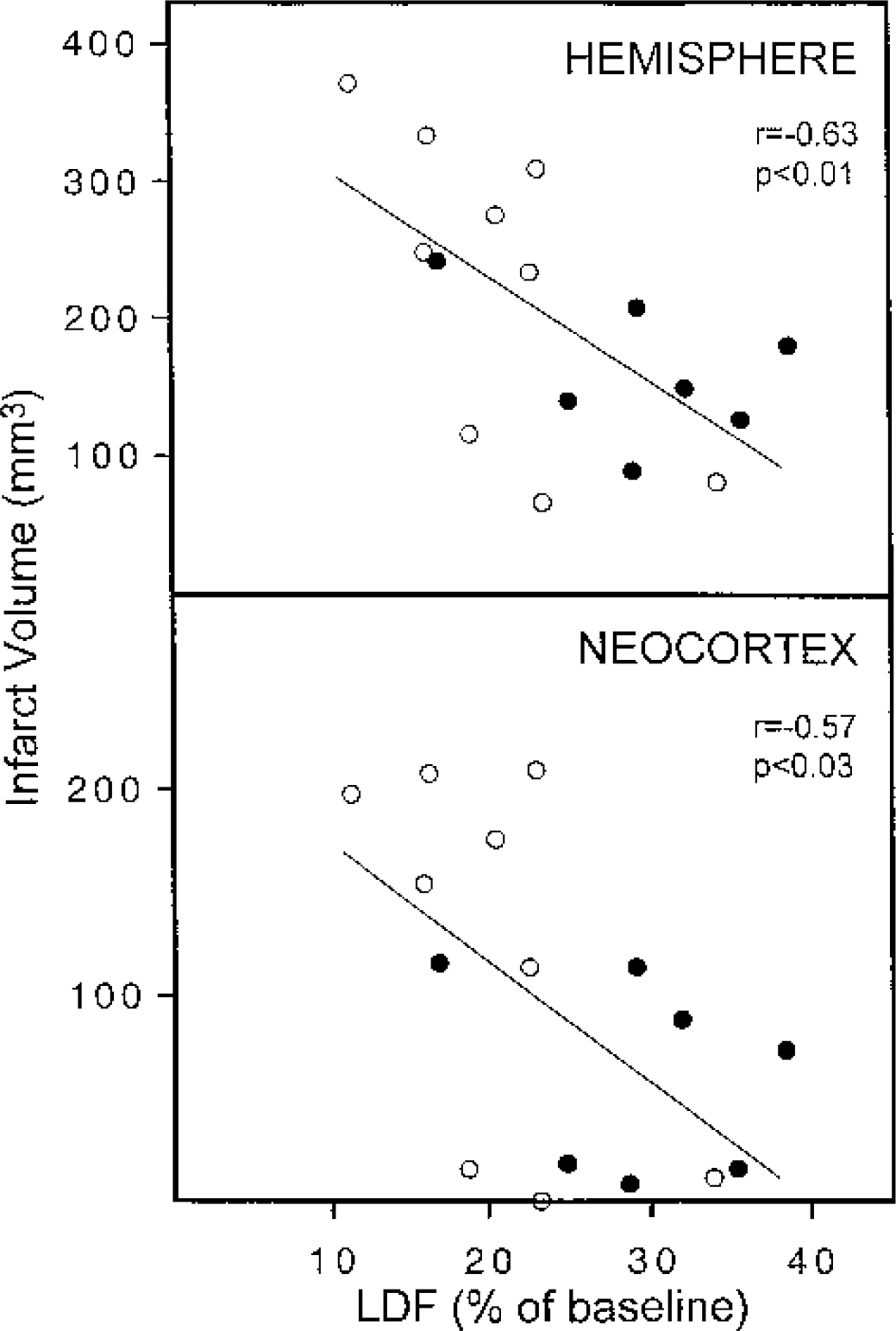
Correlation between laser Doppler blood flow (LDF) and infarction volumes. For each animal (sham-conditioned, open circles; cortical spreading depression-preconditioned, closed circles), the average LDF during the first 2 hours after MCA occlusion was plotted against hemispheric and neocortical infarction volumes. There was an inverse correlation between LDF and hemispheric infarction volume (r = −0.63, P < 0.01) and neocortical infarction volume (r = −0.57, P < 0.03).
Iodoantipyrine autoradiography
Because LDF is a relative measure of CBF, [14C]IAP autoradiography was used to determine the absolute level of baseline CBF 3 days after CSD preconditioning or sham preconditioning (Table 2). Preconditioning with CSD caused a widespread decrease in CBF throughout the ipsilateral cerebral cortex, relative to that in the contralateral cortex. In posterior portions of the cingulate cortex and in subcortical structures, regions not directly involved in CSD, CBF was not significantly different between hemispheres. In sham-conditioned animals, there were also intrahemispheric differences in CBF in a few cortical and subcortical regions, but the differences were significantly smaller than those in CSD-preconditioned animals. Because of the wide variation in CBF between animals, comparison of ipsilateral CBF between CSD-conditioned and sham-conditioned animals reached statistical significance in one region only (Table 2, medial +0.5). Expression of CBF in the ipsilateral cortex as a percentage of that in the contralateral cortex indicated that the percentage reduction of CBF in CSD-preconditioned animals was significantly greater than that in sham-conditioned animals in regions of the cerebral cortex associated with CSD (Fig. 6). These results indicate that CSD caused a depression of baseline CBF 3 days after preconditioning.
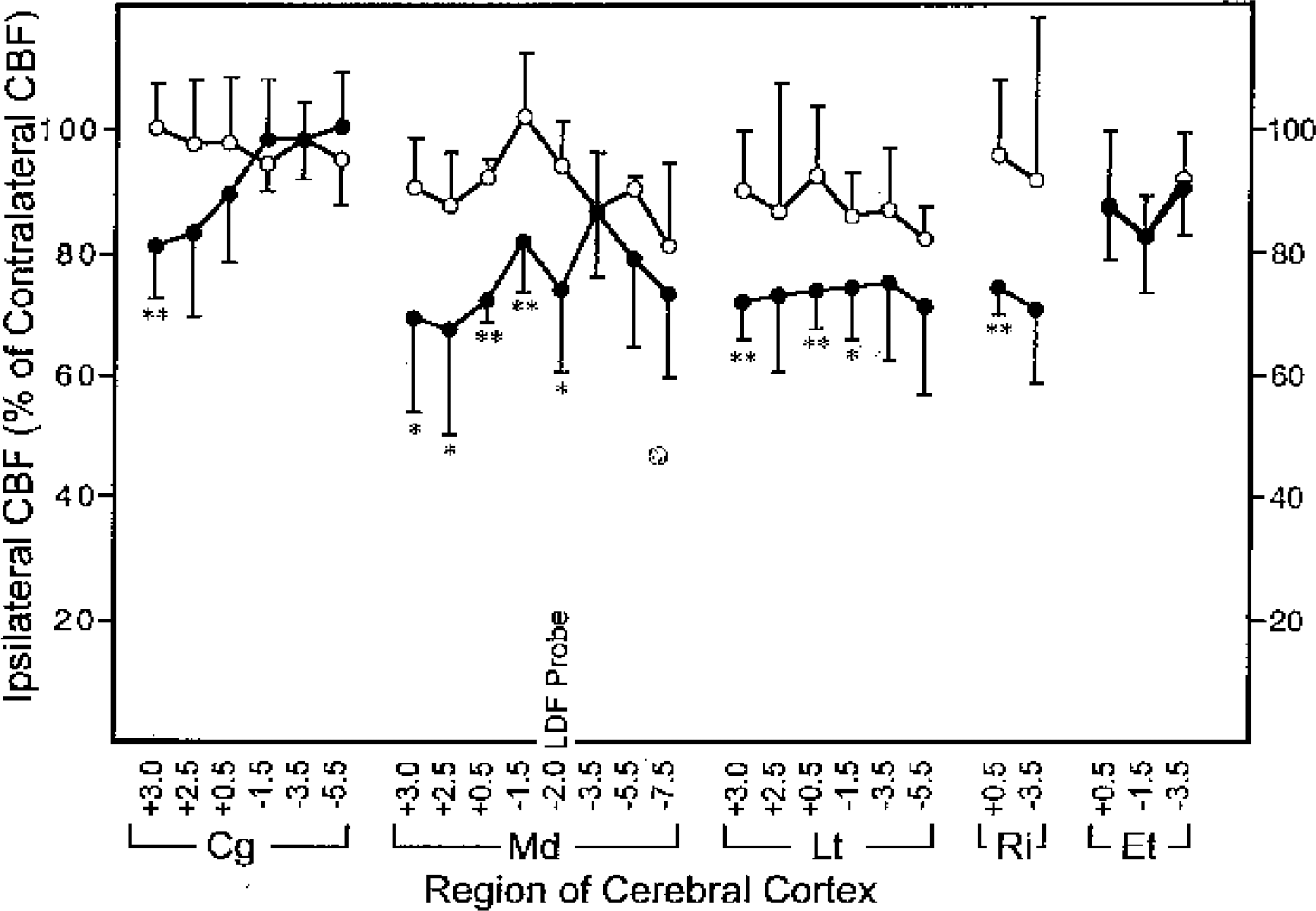
Depression of baseline cerebral blood flow (CBF) in the ipsilateral cortex 3 days after cortical spreading depression (CSD). CBF was measured using [14C]iodoantipyrine 3 days after CSD conditioning (n = 8) or sham conditioning (n = 5). Each value represents the mean ipsilateral CBF (± SD) expressed as a percentage of contralateral CBF for CSD-conditioned (closed circles) and sham-conditioned animals (open circles). Regions of cortex include Cg (cingulate), Md (medial), Lt (lateral), Ri (rhinal), and Et (entorhinal) in various brain sections indicated by distance from bregma. See Fig. 1 for location of regions. *Significantly different from sham, P < 0.05. **P < 0.02.
Regional cerebral blood flow 3 days after cortical spreading depression or sham preconditioning
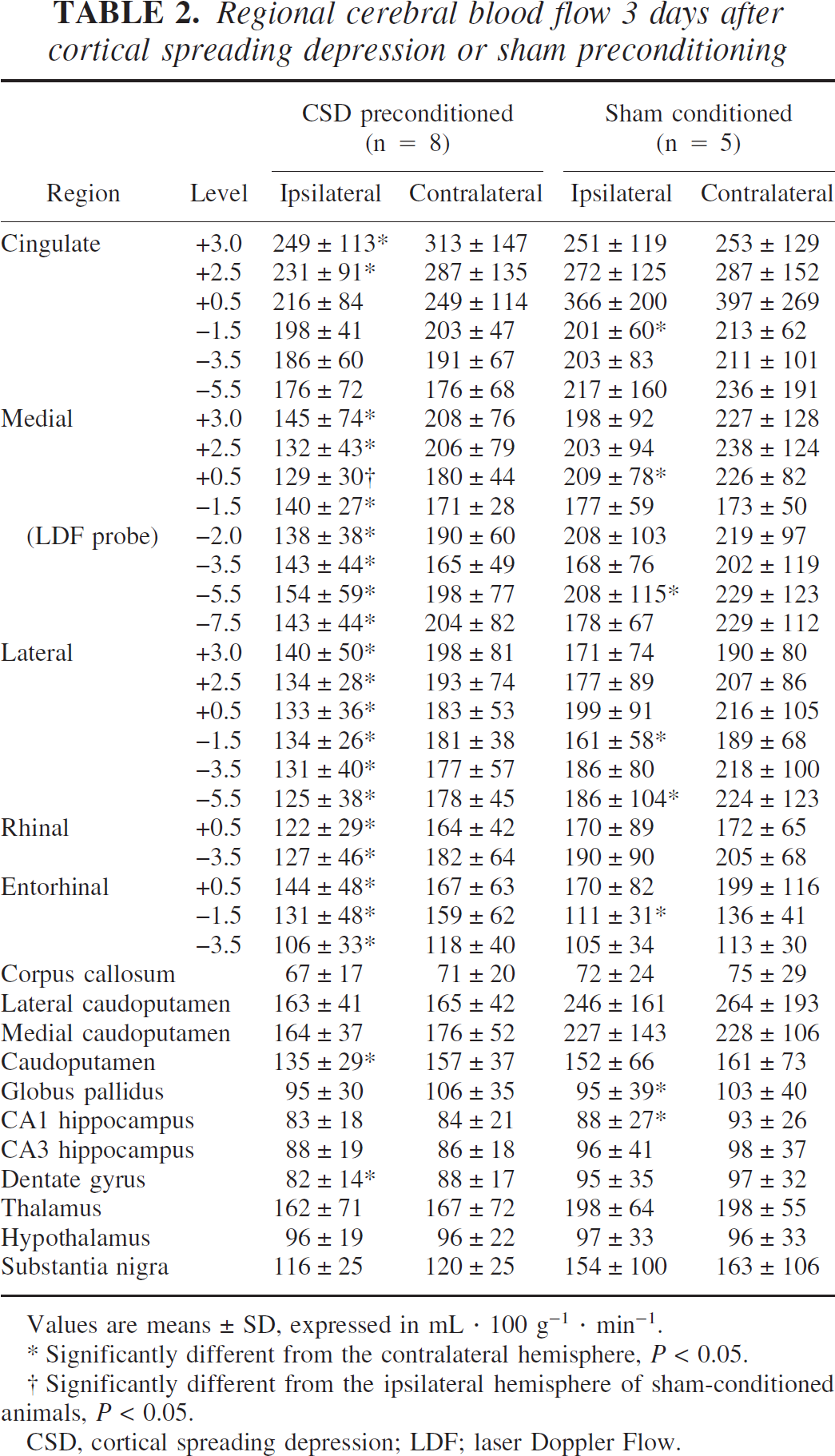
Values are means ± SD, expressed in mL · 100 g−1 · min−1.
Significantly different from the contralateral hemisphere, P < 0.05.
Significantly different from the ipsilateral hemisphere of sham-conditioned animals, P < 0.05.
CSD, cortical spreading depression; LDF; laser Doppler Flow.
DISCUSSION
The primary objective of the present study was to determine whether CSD induces tolerance to permanent focal ischemia. The results demonstrate that preconditioning with CSD decreased the extent of hemispheric and neocortical infarction resulting from permanent occlusion of the MCA. These results are significant because they indicate that reperfusion is not required to reap the benefits of preconditioning with CSD. Previous studies have demonstrated that CSD induces tolerance in models of transient cerebral ischemia, both focal and global (Kawahara et al., 1995; Kobayashi et al., 1995; Matsushima et al., 1996; Taga et al., 1997; Yanamoto et al., 1998). In these models of transient ischemia, the induction of tolerance may depend on mechanisms that antagonize the deleterious effects of excessive ROS generated during postischemic reperfusion. Permanent occlusion of the MCA, however, does not permit a reperfusion period per se and, therefore, may not generate excessive amounts of ROS. It is, therefore, likely that mechanisms other than antagonism of reperfusion-generated ROS are responsible for the induction of tolerance to permanent focal ischemia in CSD-preconditioned animals.
In the present study, preconditioning with CSD significantly improved LDF during occlusion of the MCA. Improvement in CBF during permanent focal ischemia would be expected to diminish the volume of infarction by decreasing the volume of tissue in which CBF falls below the threshold for viability. In contrast to the present results, however, previous studies have failed to detect a beneficial effect of preconditioning with CSD on CBF during focal ischemia (Matsushima et al., 1996; Yanamoto et al., 1998). In the present study, CBF during MCA occlusion was measured using LDF and expressed as a percentage of preischemic baseline LDF in the same animal. This method has the advantage of minimizing the variability in CBF between animals and thus increasing the sensitivity to detect small changes in CBF. Because LDF is a relative measure, however, it was important to determine whether preischemic baseline CBF was altered by preconditioning with CSD. Accordingly, we used [14C]IAP autoradiography to measure the absolute value of CBF 3 days after CSD conditioning or sham conditioning. Somewhat to our surprise, baseline CBF in most regions of the ipsilateral cortex was 20% to 30% lower than that in corresponding regions of the contralateral cortex. This reduction of CBF was most prominent in the frontal and parietal neocortex, but was not evident in posterior cingulate and entorhinal cortex. This regional pattern of hypoperfusion matches closely the regional pattern of expression of c-fos mRNA previously demonstrated in the model of preconditioning used in the present study (Kobayashi et al., 1995). In subcortical structures, such as striatum, hippocampus, and white matter, CBF was not significantly different between hemispheres. Therefore, the reduction of CBF in the ipsilateral hemisphere is restricted to regions of cortex preconditioned with CSD. In sham-conditioned animals, CBF was also decreased in several regions of the ipsilateral cortex, but the reductions were significantly smaller than those in CSD-preconditioned animals. In sum, these results indicate that preconditioning with CSD causes a long-lasting reduction in baseline CBF in the ipsilateral cortex.
The reduction in baseline CBF after CSD has an important consequence with respect to the interpretation of LDF measurements made during focal ischemia. Preconditioning with CSD increased LDF during MCA occlusion from 21% of baseline to 29% of baseline. If, however, preconditioning with CSD lowered baseline CBF to 70% to 80% (73% at the LDF site), then the absolute level of CBF after MCA occlusion would be estimated at 21% (73% of 29%) of the original baseline, a value identical to that measured in sham-conditioned animals. It is therefore likely that preconditioning with CSD does not significantly alter the absolute level of CBF during focal ischemia.
The finding that CSD causes a long-lasting decrease in baseline CBF may have important implications with respect to mechanisms of induced tolerance to ischemia. To our knowledge, the present results are the first to demonstrate a long-lasting reduction in CBF after CSD. Previous studies have shown that CBF is reduced to 60% to 80% of control levels for several hours after CSD (Lauritzen et al., 1982; Duckrow, 1991; Piper et al. 1991). Prolonged reductions in baseline CBF after CSD, however, have not been previously reported. There are two distinct mechanisms by which CSD may cause a long-lasting decrease in CBF. First, it is conceivable that regulation of CBF is chronically impaired after CSD, resulting in prolonged vasoconstriction. This explanation, however, is not consistent with the decreased vulnerability to ischemia of the CSD-preconditioned brain. Chronic vasoconstriction would be expected to exacerbate tissue injury resulting from a subsequent episode of ischemia. An alternative explanation for the decreased CBF after CSD is a reduction in metabolic rate and unimpaired coupling between CBF and metabolic rate. A previous study demonstrated a profound reduction in the rate of glucose utilization in the ipsilateral cortex 3 days after CSD (Kawahara et al., 1999). In this study, the reduction in the rate of glucose utilization was decreased by as much as 50% throughout the ipsilateral cerebral cortex. These results indicate that CSD causes a long-lasting reduction in the rate of metabolism, which may underlie the reduction of CBF observed in the present study.
Importantly, a reduction in the rate of metabolism after CSD may contribute to the induction of tolerance to ischemia by lowering the energy demands of the tissue. Other agents that lower the cerebral metabolic rate, such as hypothermia and barbiturate anesthesia, are known to protect the brain against ischemic injury. A decrease in the rate of energy consumption would be expected to lower the CBF threshold for energy failure and subsequent infarction (Mies et al., 1991). Therefore, levels of CBF that would normally cause infarction may be tolerated in preconditioned regions with reduced metabolic rate. Although the degree to which the CSD-induced reduction in baseline metabolic rate is maintained during ischemia is not known, the ratio between CBF and metabolic rate during ischemia is likely to be higher in the cortex of CSD-preconditioned animals than in sham-conditioned controls because the residual CBF during ischemia (relative to baseline) was greater in CSD-preconditioned animals. An improved ratio between CBF and metabolism during ischemia would be expected to diminish the extent of resulting infarction observed in CSD-preconditioned animals.
In summary, the present results demonstrate that preconditioning with CSD (1) caused a prolonged decrease in baseline CBF, (2) diminished the percentage reduction in LDF after occlusion of the MCA, and (3) reduced the extent of infarction resulting from permanent occlusion. We conclude that the long-lasting decrease in CBF after CSD is most likely caused by a reduction in the rate of metabolism, which, in turn, may contribute to the tolerance to ischemia. Finally, a word of caution should be made regarding the use of LDF to measure changes in CBF in cases in which baseline CBF may have been altered. For example, in genetically modified animals, it would be prudent to measure baseline CBF using alternative methods for absolute levels of CBF in order to determine whether the genetic modification might have altered baseline CBF.