
Abstract
The possibility that cortical spreading depression (CSD) may have neuroprotective action during subsequent focal cerebral ischemia was examined in rats. Three days before the imposition of focal cerebral ischemia CSDs were elicited by applying potassium chloride (KCl) for 2 h through a microdialysis probe implanted in the occipital cortex. Control animals were handled identically except that saline was infused instead of KCl. Focal ischemia was produced by the intraluminal suture method and cortical and subcortical infarct volumes were measured 7 days later. Neocortical infarct volume was reduced from 124.8 ± 49.5 mm3 in the controls to 62.9 ± 59.5 mm3 in the animals preconditioned with CSD (p = 0.012). There was no difference between the two groups in the subcortical infarct volume or in CBF, measured by the hydrogen clearance method, during or immediately after the ischemic interval. Our data indicate that preconditioning CSD applied 3 days before middle cerebral artery occlusion may increase the brain's resistance to focal ischemic damage and may be used as a model to explore the neuroprotective molecular responses of neuronal and glial cells.
Cortical spreading depression (CSD) is characterized by a propagating wave of depressed electrical activity and cell membrane depolarization. It can be elicited by a variety of means including a stab wound, ischemia, raised extracellular potassium, and strong electrical stimulation. It is also known that periinfarct depolarization occurs in cortical tissue surrounding an acute ischemic lesion (Nedergaard and Astrup, 1986). Because CSD is a process that leads to substantial activation of energy requiring metabolism (Csiba et al., 1985; Kocher, 1990; Mayevsky and Weiss, 1991), its occurrence in the peripheral zone of an occluded cerebral artery is considered harmful because it can add metabolic stress to the periinfarct region and tip the balance in favor of cell death instead of reversible injury. Recently, Mies et al. (1993) and Chen et al. (1993) have reported that the number of periinfarct depolarization events correlates with the volume of infarct.
Two lines of evidence have led us to test CSD's potential as a neuroprotective maneuver in ischemia. First, it is now clear that brief episodes of ischemic pretreatment can increase the brain's tolerance to a subsequent ischemic event (Kato et al., 1991; Kirino et al., 1991). Matsushima and Hakim (1995) have recently confirmed these findings and showed that this phenomenon referred to as ischemic preconditioning, is not associated with changes in rCBF. This suggests that the neuroprotective response is acquired through molecular upregulation of the brain's ability to resist ischemic injury. Second, we have shown that CSD is associated with a moderate increase in the binding of [3H]nimodipine to brain, consistent with the activation of voltage-sensitive calcium channels (VSCC) (Osuga et al., 1993). This activation implies limited intracellular influx of calcium ions, which act as second messengers but may be reexported after inducing a number of molecular responses (Kiessling et al., 1993). Thus, we speculated that CSD could be used as a preconditioning treatment and lead to increased resistance to ischemic damage. Recently, Kawahara et al. (1994) reported that rats subjected to CSD had significantly less damage to hippocampal CA1 neurons after cardiac arrest cerebral ischemia, but this is the first report of CSD used as a preconditioning maneuver for subsequent focal cerebral ischemia. Our data show that preconditioning with CSD could attenuate the outcome from such ischemia, and that this neuroprotective effect is not mediated through a change in rCBF during ischemia.
MATERIALS AND METHODS
Experimental design
Two groups were prepared. The “CSD + 180-min middle cerebral artery occlusion (MCAO) group” (n = 14) had left MCA occlusion lasting 180 min imposed 3 days after a 2-h episode of ipsilateral cortical spreading depression. CSDs were produced by perfusing KCl into the left occipital cortex. The other group (n = 14), labeled “sham + 180 min MCAO group,” was subjected to a similar duration of MCA occlusion after perfusion with normal saline for 2 h instead of KCl. DC potentials were recorded from two platinum electrodes implanted bilaterally in the frontal cortex during the instillation of KCl or saline as well as during the MCA occlusion. Thus, the two groups were compared for the number of CSD waves, rCBF during MCA occlusion, and histological outcome.
Surgical and experimental procedures
Twenty eight male Sprague Dawley rats weighing 260–300 g were used. Instruments used during surgery were autoclaved and all implanted probes were sterilized in 70% ethanol solution. The surgery was performed under sterile conditions. During the induction of CSD and in controls, rats were intubated and ventilated mechanically with 1% halothane in a mixture of 30% oxygen-70% nitrous oxide. The tail artery was cannulated with a polyethylene catheter for monitoring of arterial blood pressure, blood gases, and blood glucose. All procedures followed the guidelines of the Canadian Council on Animal Care (Guide to the Care and Use of Experimental Animals), and were approved by the University of Ottawa Animal Care Committee. The rats were then put in the stereotaxic frame (Kopf Instruments, Tujunga, CA, U.S.A.) and four small burr holes were made over the left frontal, frontoparietal, and occipital cortex and over the right frontal cortex. Three platinum electrodes (0.1 mm in diameter) were implanted: in the left frontoparietal cortex (bregma −1.5 mm; lateral 4.0 mm, depth 2.0 mm) for subsequent measurement of rCBF by the hydrogen clearance method, in the left frontal cortex (bregma 2.0 mm; lateral 6.0 mm; depth 2.0 mm) and the right frontal cortex (bregma 1.5 mm; lateral 5.0 mm; depth 2.0 mm) for recording of CSD and fixed with dental cement. A platinum wire reference electrode was inserted into the auricle of the right ear and both electrodes were connected to a DC amplifier and a chart recorder (WindoGraf Recorder, Gould Instruments Inc., Valleyview, OH, U.S.A.). A microdialysis probe (diameter 0.5 mm, active length 1.0 mm, CMA-12; CMA/Microdialysis AB, Sweden) was inserted stereotaxically into the left occipital cortex (bregma −7.8 mm; lateral 6.0 mm; depth 2.0 mm) and also fixed with dental cement. Accordingly, the recording of the CSD waves occurred 9.8 mm from the microdialysis instillation of KCl. Recording of CSD was started at the same time as implantation of a microdialysis probe to record any elicitation of CSD by the probe insertion itself. Recurrent waves of CSD were induced by perfusion with 3 M KCl through the microdialysis probe at the rate of 2.0 μl/min. In a separate study, using an Ag/AgCl agar bridge electrode, it was confirmed that deflections recorded by the platinum electrode corresponded to DC potential shift. After 2 h, CSD was stopped by flushing the microdialysis probe with normal saline. The rats were allowed to recover from anesthesia and returned to their cages.
Three days later, rats were again anesthetized with 1% halothane in a mixture of 30% oxygen-70% nitrous oxide. The right femoral artery was cannulated with a polyethylene catheter for monitoring of physiological parameters, and focal cerebral ischemia was induced by the intraluminal occlusion method using a nylon monofilament thread (Longa et al., 1989). Briefly, the left common, external, and internal carotid arteries were carefully exposed after a ventral midline incision in the neck. The distal portions of the left external carotid and ipsilateral common carotid arteries were ligated with 4–0 silk suture. Immediately after that, a 3–0 nylon monofilament thread was introduced through the ipsilateral common carotid artery, distal to the ligation, into the internal carotid artery. After introducing this occluder thread, the common carotid artery just distal to the point of insertion was ligated to prevent bleeding and the tip of the occluder thread was advanced 18.5 mm distal to the carotid bifurcation. The occlusion of the MCA was confirmed by measuring rCBF using the hydrogen clearance method, and the number of periinfarct depolarization waves was recorded in each animal. For measurement of rCBF, the platinum electrode implanted previously was polarized to + 500 mV relative to an Ag/AgCl reference inserted under the scalp using a H2 clearance amplifier (Computer Technology Centre, The University of Virginia). Then, ∼10% hydrogen gas was added to the inhaled gas mixture. Inhalation of hydrogen gas was stopped after reaching saturation level and the hydrogen clearance curve was recorded (WindoGraf Recorder, Gould Instruments Inc.). rCBF was measured hourly during ischemia and immediately after release of the MCA occlusion. The thread was withdrawn 3 h after induction of ischemia. The suture over the external carotid was released and reperfusion was again confirmed by measuring rCBF. The wound was then sutured with topical instillation of Xylocaine jelly, anesthesia was discontinued and the rat was allowed free access to food and water until the next procedure. During MCA occlusion, rectal and temporal muscle temperatures, MABP, arterial blood gases, plasma pH, and the plasma glucose concentration were monitored.
Seven days after MCA occlusion, the rats were decapitated while they were under the same anesthetic condition and the brains were removed and frozen in methylbutane cooled by liquid nitrogen and processed for histologic assessment. Brain sections, 20 μm thick, were stained with cresyl violet. The infarct areas in 15 brain sections that were equally spaced along the brain's entire anterior–posterior axis were measured by a microcomputer-based image display system (MCID, Imaging Research Inc., St. Catharines, Ontario, Canada) using the method described by Swanson et al. (1990). Infarct volume was calculated as the integral of the infarct areas for the cortex and the striatum separately. In measuring the latter, white matter fascicles passing through the basal ganglia were not counted.
Statistical analysis
All data were expressed as mean ± SD. Comparison of physiologic parameters, rCBF, and total infarct volumes between groups was done using independent t tests. Linear regression was used to correlate infarct volume with rCBF and the number of periinfarct depolarizations. The effect of prior conditioning with CSD on infarct volumes was assessed using analysis of covariance with rCBF as the covariate.
RESULTS
Physiological variables
During 2 h of KCl application and MCA occlusion, MABP, rectal and temporalis muscle temperatures, blood glucose concentration, hematocrit, and blood gases were not statistically different between the two groups and are given in Table 1.
Physiological variables
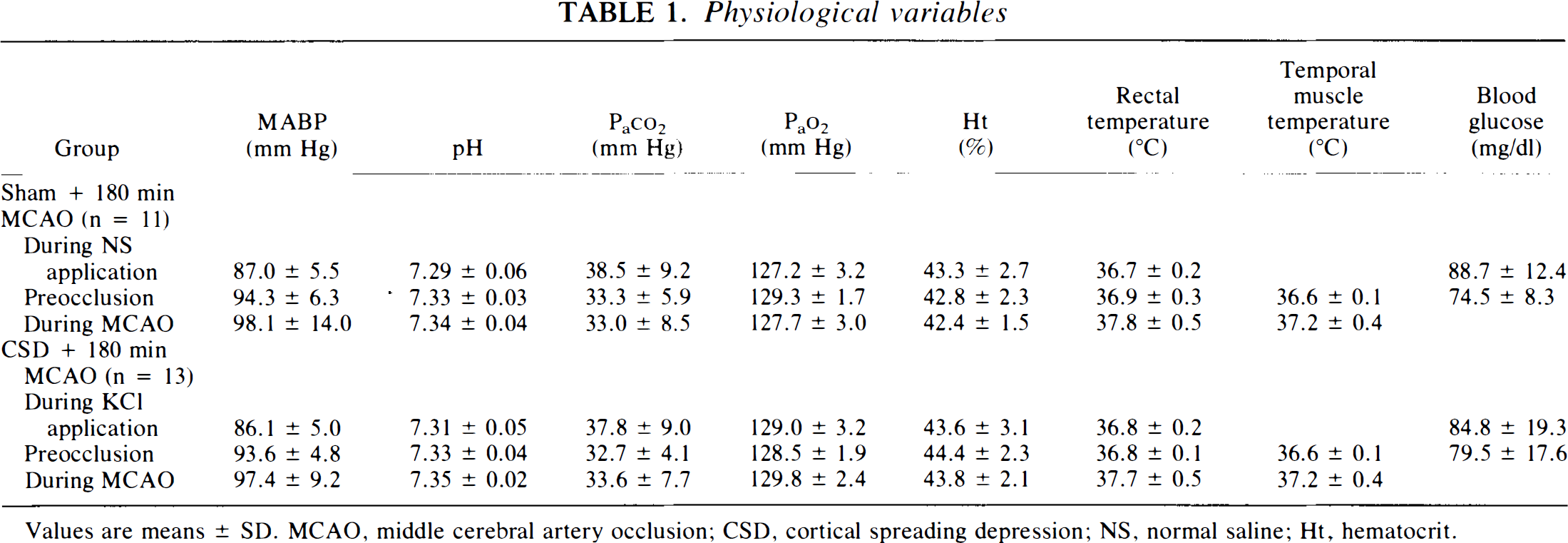
Values are means ± SD. MCAO, middle cerebral artery occlusion; CSD, cortical spreading depression; NS, normal saline; Ht, hematocrit.
KCl-elicited CSD and periinfarct depolarization
No CSD waves were observed on implantation of the microdialysis probe in the CSD + 180-min MCAO or the sham + 180-min MCAO groups. In the CSD + 180-min MCAO group, CSD waves were recorded during 2 h of KCl instillation as a transient negative shift of DC potential. The waves had an amplitude of 4.9 ± 1.8 mV and a duration of 81.7 ± 10.4 s consistent with previously reported characteristics of CSD (McLachlan, 1992; Somjen et al., 1992). The number of CSD waves recorded in the left frontal cortex of CSD + 180 min MCAO group during 2 h was 11.5 ± 4.2. In the sham + 180-min MCAO group perfused with normal saline, no CSD was observed during 2 h. No CSD waves were recorded in the contralateral frontal cortex during perfusion with KCl or normal saline. The number of periinfarct depolarization events recorded during MCA occlusion was 4.1 ± 7.9 and 1.6 ± 1.6 in the groups without and with preconditioning CSD, respectively, without significant difference. One rat in the sham + 180-min MCAO group had 27 periinfarct depolarization events during 3 h of MCA occlusion. The number of periinfarct depolarization events excluding this rat was similar in the two groups (1.8 ± 2.1 versus 1.6 ± 1.6 in the sham and CSD groups, respectively).
rCBF
rCBF during MCA occlusion showed no significant difference between the two groups (Fig. 1). Mean rCBF during MCA occlusion was 17.2 ± 2.9 and 17.0 ± 4.2 ml/100 g/min in the groups without and with preconditioning CSD, respectively. Immediately after release of MCA occlusion, mean rCBF values were also not statistically different between the two groups.
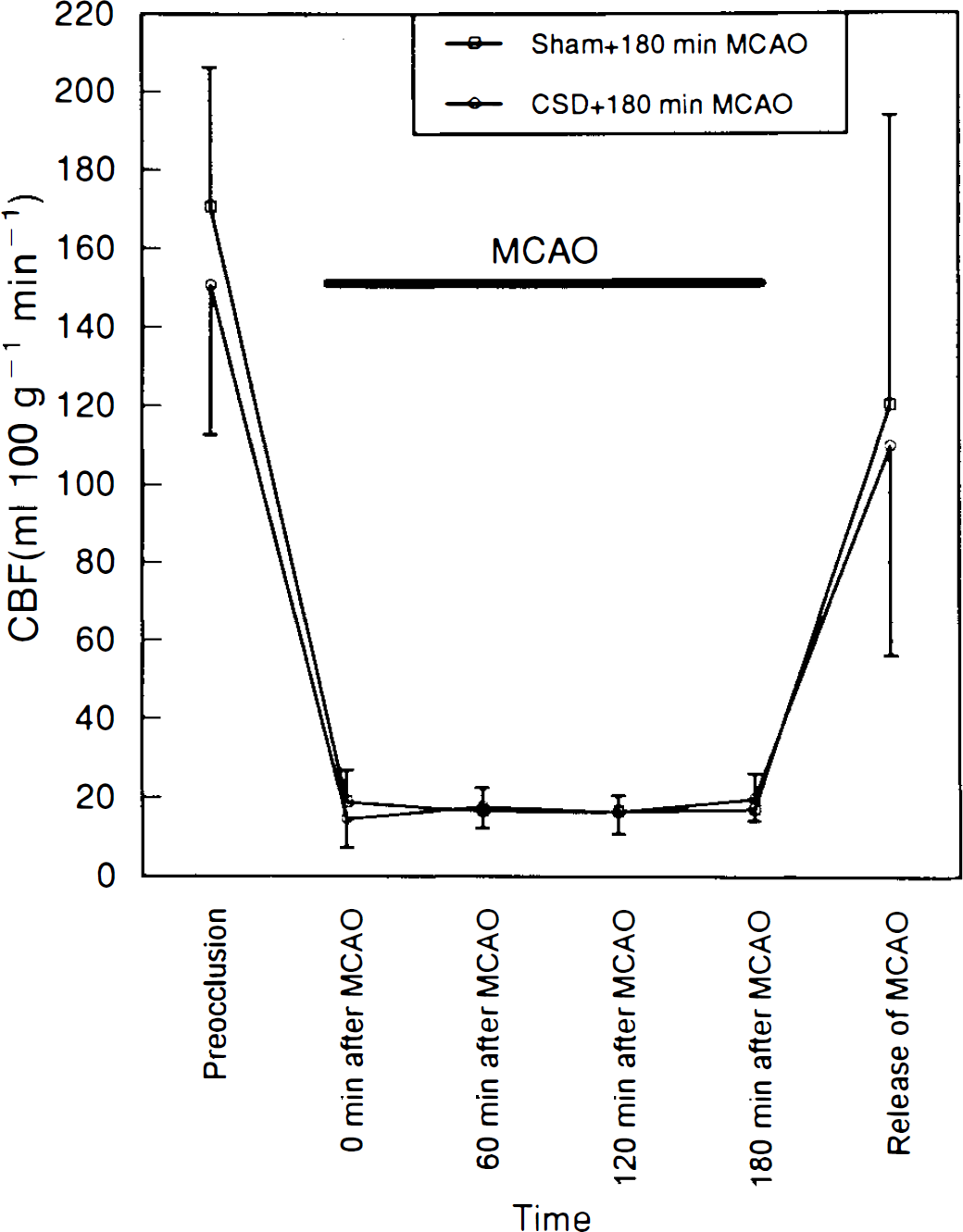
Time course of rCBF during cerebral ischemia shows the changes of rCBF in the sham + 180-min middle cerebral artery occlusion (MCAO) group (□) and in the cortical spreading depression (CSD) + 180-min MCAO group (•). Values are means ± SD.
Survival and histologic outcomes
One animal in the CSD + 180-min MCAP group died immediately after the removal of the occluder thread from a subarachnoid hemorrhage. Three animals in the sham + 180-min MCAO group also died: one at the same time point as already mentioned, and two within 2 days of MCAO from massive cerebral edema. Thus the analysis was performed on n = 13 for the CSD + 180 MCAO group and n = 11 for the sham + MCAO group.
In the CSD + 180-min MCAO group, a zone of tissue necrosis, ∼0.5 mm deep, surrounded the microdialysis probe. No such necrosis was evident in the control groups.
Infarction was noted in the neocortex and striatum in both groups (Table 2). Total cortical infarct volumes were 124.8 ± 49.5 and 62.9 ± 59.5 mm3 in the groups without and with the preconditioning CSD (p = 0.012) indicating that this maneuver significantly reduced total cortical infarct volume. Infarct volumes for the striatum were 13.7 ± 4.5 and 15.0 ± 5.5 mm3 in the groups without and with the preconditioning CSD, respectively, with no significant difference (p = 0.56). Figure 2 shows the relationship between histologic outcomes and CBF in individual animals. In the sham + 180-min MCAO group, cortical infarct volumes appeared to increase linearly with decreasing rCBF (r = 0.7, p = 0.016), whereas in the CSD + 180-min MCAO group, cortical infarct volume remained low until rCBF fell below 14 ml/100 g/min. If the infarct volume data for the CSD + 180-min MCAO group were fit linearly against rCBF, a significant correlation did result (r = 0.6, p = 0.024). The difference in these correlations was highly significant (F(1.21)) = 12.83, p = 0.0018 by one-way analysis of covariance) with CSD pretreatment resulting in reduced cortical infarct volumes over a range of rCBF values. Below an rCBF of ∼14 ml 100 g−1/min−1, both groups developed large cortical infarcts.
Infarct volumes in groups with and without CSD
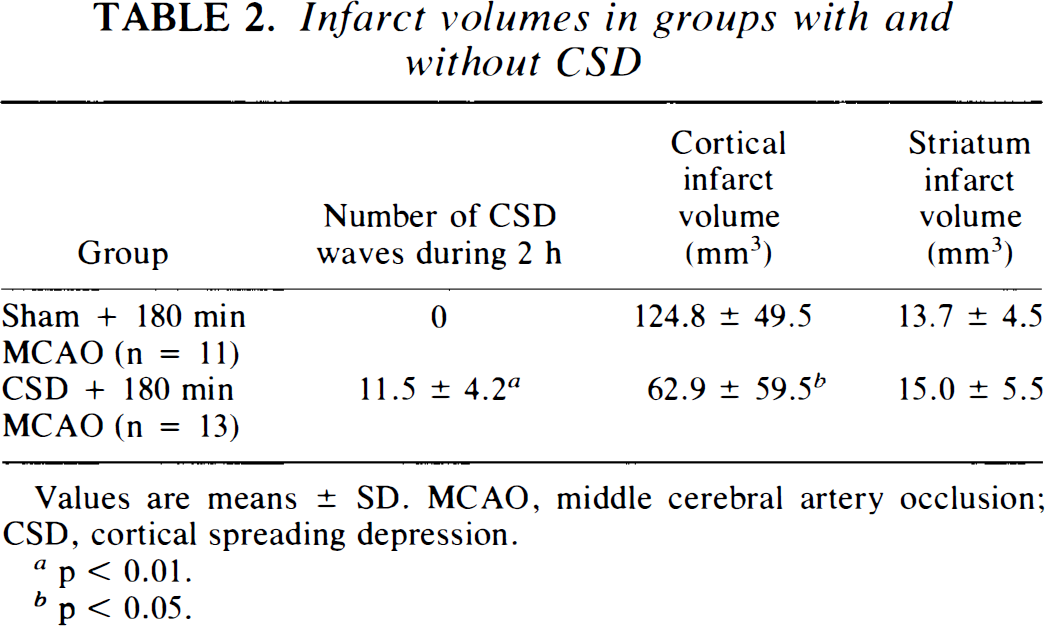
Values are means ± SD. MCAO, middle cerebral artery occlusion; CSD, cortical spreading depression.
p < 0.01.
p < 0.05.
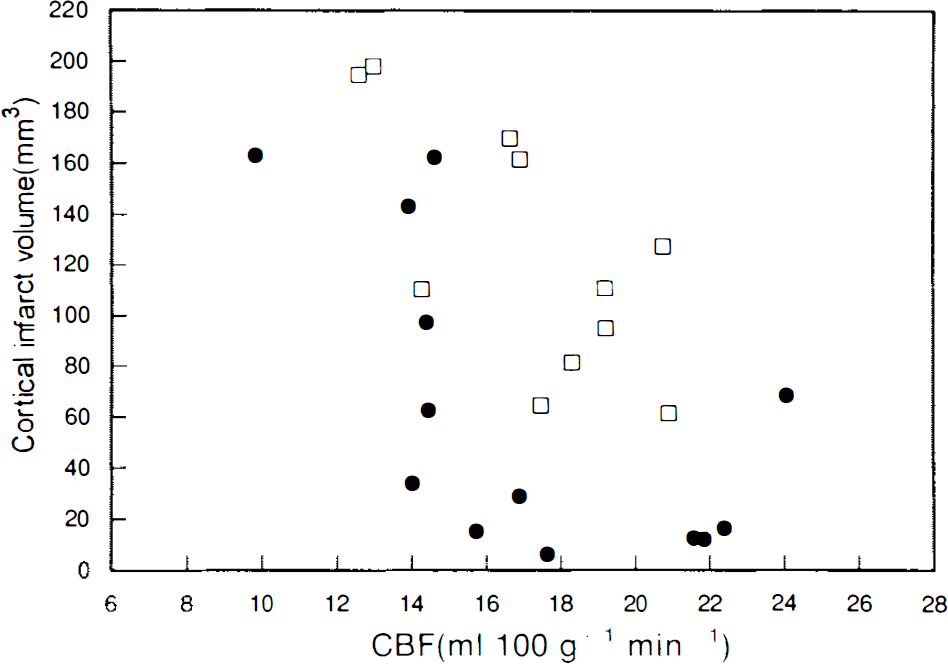
The relationship between mean rCBF during ischemia and cortical infarct volume of each animal in the sham + 180-min middle cerebral artery occlusion (MCAO) group (□) and in the cortical spreading depression (CSD) + 180-min MCAO group (•). The equations for the linear regressions shown are infarct volume = − 12 × rCBF + 33 (r = 0.7, p = 0.016) for the sham group and −8.7 × rCBF + 21 (r = 0.6, p = 0.024) for the CSD group.
Figure 3 shows that there is no correlation between the number of periinfarct depolarization waves during 3 h of MCAO and histological outcome.
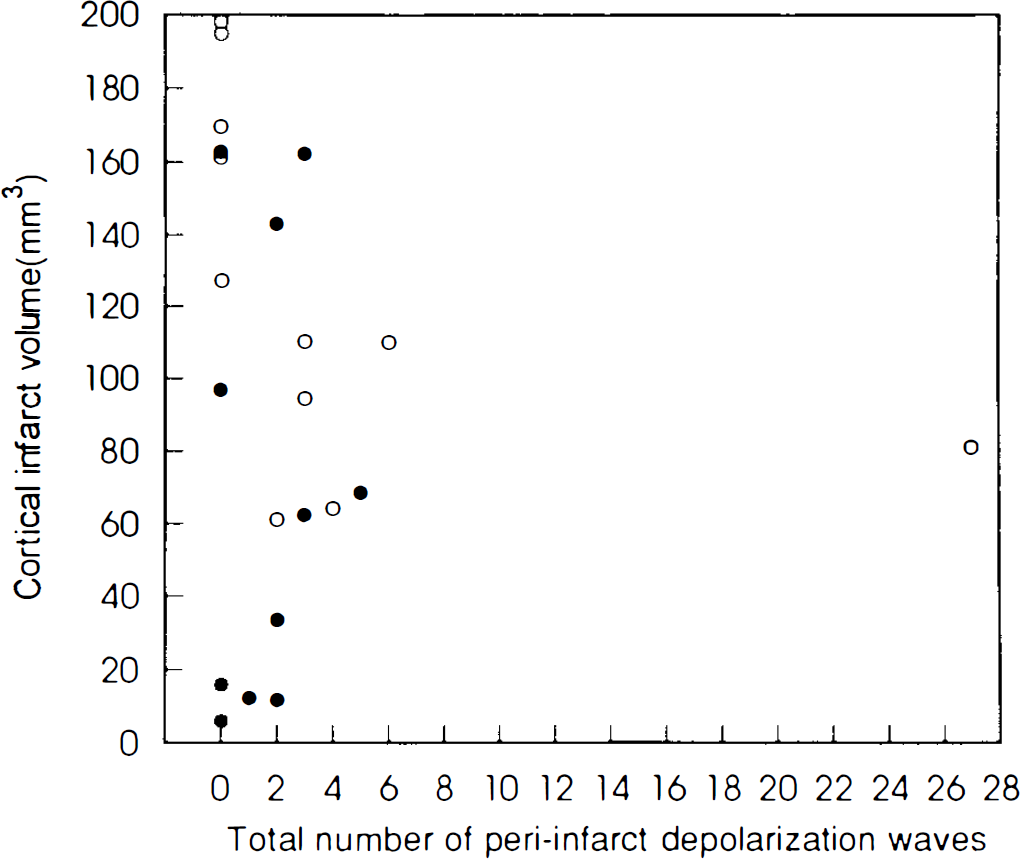
The relationship between cortical infarct volume and the number of periinfarct depolarization events during 3 h of middle cerebral artery occlusion (MCAO) of each animal in the sham + 180-min MCAO group (○) and in the cortical spreading depression + 180-min group (•).
DISCUSSION
The data presented here suggest that CSD can attenuate subsequent ischemic cerebral damage. Cerebral blood flows measured in the area surrounding the ischemic core were not significantly different between the group that had received and the one without preceding CSD, suggesting that the attenuation in ischemic damage is not related to improved perfusion. Two constraints seemed to apply: The beneficial effect of the preceeding CSD was limited to the neocortex, and rCBF had to remain >14 ml 100 g−1/min−1 for a neuroprotective effect from CSD to become evident. This would indicate that preconditioning with CSD is likely effective only in penumbral tissue. The experimental design limits our conclusions in other ways. In our experiment, only one time interval (3 days) between CSD and ischemia was used. This was chosen because ischemic preconditioning studies in global ischemia indicated that this was an effective interval (Kirino et al., 1991). The 180-min duration of MCAO was chosen because it caused both subcortical and cortical damage with minimum variability (Memezawa et al., 1992). The duration of CSD selected was sufficiently long to permit effective preconditioning, but other durations may have been more or less neuroprotective.
In this study, a platinum wire electrode was used for DC potential measurement. This electrode may not be ideal, but the waveforms recorded were consistent with previously reported characteristics of CSD (McLachlan, 1992; Somjen et al., 1992). CSD may be generated by insertion of the electrode itself (Verhaegen et al., 1992) and by the microdialysis probe (Tomida et al., 1989), but this would have affected both groups equally. In fact, no CSDs were recorded during the microdialysis probe implantation in any animals. This may be due to the fact that our recording electrode was 8.9 mm from the microdialysis probe.
In our study, the mean number of periinfarct depolarization waves was not statistically different between the two groups and no correlation between the number of periinfarct depolarization waves and infarct volume was evident (Fig. 3). Because CSD leads to activation of energy-requiring metabolism (Csiba et al., 1985; Kocher, 1990; Mayevsky and Weiss, 1991), it is supposed that CSD-like periinfarct depolarization events may lead to temporary energy deficits that endanger the brain tissue in the border zone of cerebral infarct. In this regard, Mies et al. (1993) and Chen et al. (1993) demonstrated that the number of periinfarct depolarization events correlates with the volume of infarct. Our data do not support this, which might be due to differences in the ischemic model, the duration of ischemia, or technical factors such as the sites of recording. Despite these limitations, our results suggest that decreasing cortical infarct volume is possible through preceding CSD, supporting the data of Kawahara et al. (1994), who reported that rats subjected to CSD 3 days before cardiac arrest cerebral ischemia had significantly reduced damage to CA1 neurons of the hippocampus. The combined information implies that the neuroprotective effect of prior CSD extends beyond the cortex to involve the hippocampus but does not reach the striatum.
The mechanisms of neuroprotection induced by CSD can only be speculated on. We have shown that CSD elicits the activation of VSCC, implying influx of calcium ions into neuronal cells (Osuga et al., 1993). VSCC activation mediates the induction of a variety of immediate early genes (Murphy et al., 1991) which may regulate neuronal gene expression (Dragunow et al., 1989; Hengerer et al., 1990; Sheng and Greenberg, 1990). The level of brain-derived neurotrophic factor mRNA rapidly increases in cortical neurons after CSD (Kokaia et al., 1993). Herrera and Robertson (1990) showed that the application of KCl to the brain surface elicits the induction of c-fos and nerve growth factor mRNA in the treated cerebral hemisphere. This preceded an increase in FOS immunoreactivity and nerve growth factor-like protein (Herrera et al., 1993). In addition, there is evidence that CSD can affect glial as well as neuronal gene expression. KCl application to the brain surface leads to an increase in glial fibrillary acidic protein (GFAP) mRNA level (Bonthius et al., 1993, 1994) and GFAP immunoreactivity throughout the treated cerebral cortex (Kraig et al., 1991). An increase in GFAP immunoreactivity denotes the transformation of normal astrocytes to reactive ones that can secrete neuronotrophic factors, which in turn may promote the growth and survival of injured neurons (Bignami and Dahl, 1976; Nieto-Sampedro et al., 1982; Needels et al., 1986). Kawahara et al. (1994) also observed a strong ipsilateral GFAP reaction at 3 days after CSD induction. Thus, CSD induces a number of neuronal and glial genes, which may become templates for proteins that have neuroprotective properties. The one exception to this appears to be heat shock proteins. Nowak et al. (1991) and others more recently (Ikeda et al., 1994; Kawahara et al., 1994) have shown that CSD could not induce heat shock proteins despite the suggestion that they play a role in the acquisition of ischemic tolerance through ischemic preconditioning (Kitagawa et al., 1991). In conclusion, CSDs appear to be capable of inducing many neuronal and glial neurotrophic and other mRNAs, some of which may be translated into proteins with neuroprotective properties.
It is unlikely that CSD would ever become a clinically useful means of increasing the brain's resistance to subsequent ischemic damage. Rather, CSD provides an experimental tool to understand the responses involved in neuroprotection from cerebral ischemia. Further study is needed to clarify the effective time window and intensity of CSD as well as the exact mechanism linking CSD to the neuroprotection.
Footnotes
Acknowledgments:
We thank Robin Millbank and Rose Moore for clerical help. This work was supported by a grant from the Heart and Stroke Foundation of Ontario, the Medical Research Council of Canada, and London Life Canada.