
Abstract
Tissue-type plasminogen activator (tPA) is available for the treatment of thromboembolic stroke in humans. However, adverse effects of tPA have been observed in animal models of ischemic brain injuries. In the present study, we have used a synthetic tPA inhibitor, named 2,7-bis-(4-amidinobenzylidene)-cycloheptan-1-one dihydrochloride (tPA stop), to investigate the role of endogenous tPA in the cerebral parenchyma. In mouse cortical cell cultures, we observed that although tPA stop reduced N-methyl-
tPA is involved in many physiological cerebral processes, both in the developing and the adult brain (Seeds et al., 1990; Frey et al., 1996). For instance, in the adult brain, tPA may contribute to the late phase of long term potentiation and to the subsequent synaptic growth in the hippocampal mossy fiber pathway (Frey et al., 1996; Huang et al., 1996; Baranes et al., 1998). The role of tPA has also been investigated in pathological conditions such as stroke. In fact, tPA is the only treatment of ischemic stroke approved by the Food and Drug Administration (NINDS, 1995). Nevertheless, although its thrombolytic activity seems to be beneficial in the acute treatment of stroke, recent studies have suggested that tPA could also play deleterious effects in the cerebral parenchyma. This hypothesis is based on several experimental results: (1) tPA-deficient mice are resistant to excitotoxic neuronal death induced by the intrahippocampal injection of kainate (Tsirka et al., 1995); (2) the infarct volume induced by middle cerebral artery occlusion is reduced in tPA knock-out mice (Wang et al., 1998); and (3) the intravenous injection of tPA is able to potentiate the infarct volume in animals subjected to middle cerebral artery occlusion (Wang et al., 1998). This deleterious effect of tPA has been suggested to result from a proteolytic-dependent potentiation of N-methyl-
MATERIALS AND METHODS
Materials
Dulbecco's Modified Eagle Medium (DMEM), cytosine β-
tPA activity assay
Twenty-five microliters of human recombinant tPA (final concentration: 2 μg/mL) were incubated in the presence of 50 μL of tPA substrate (final concentration: 1.65 μmol/L) alone or in the presence of 25 μL of tPA stop (0.01 μmol/L). tPA activity was estimated by using a specific chromogenic substrate for t-PA, spectrozyme XF444 (excitation: 360, emission: 440 nm). The Ki of tPA stop for the proteolytic activity of tPA is 0.082 μmol/L. Its Ki for other trypsin-like serine proteases is 0.096 μmol/L for trypsin, 0.32 for thrombin μmol/L, 6.4 μmol/L for plasmin, and 3.4 μmol/L for urokinase.
Sodium dodecyl sulfate polyacrylamide gel electrophoresis zymography assay for t-PA
For each treatment, an equal amount of protein contained in the conditioned media was subjected to electrophoresis in a 15% sodium dodecyl sulfate polyacrylamide gel containing 1 mg/mL of casein and 4.5 μg/mL of plasminogen. After removal of SDS by washing with buffer containing Triton X-100 2.5%, the gels were incubated in a glycine-EDTA buffer (pH = 8.3) at 37°C for 2 hours. Caseinolytic bands indicating proteolytic activities were visualized as clear bands after Coomassie staining.
Neuronal cultures
Neuronal cortical cultures were prepared from fetal mice E15-E16 (Rose et al., 1993). Cortices were dissected in DMEM, dissociated in the same medium, and plated on 24-well plates coated with poly-
Estimation of tPA release
Cells were treated as described in the corresponding figure (Fig. 1) and were conditioned media harvested. Then, sodium dodecyl sulphate polyacrylamide gel electrophoresis was performed and revealed Coomassie staining or immunoblotting performed with an antibody raised against tPA.
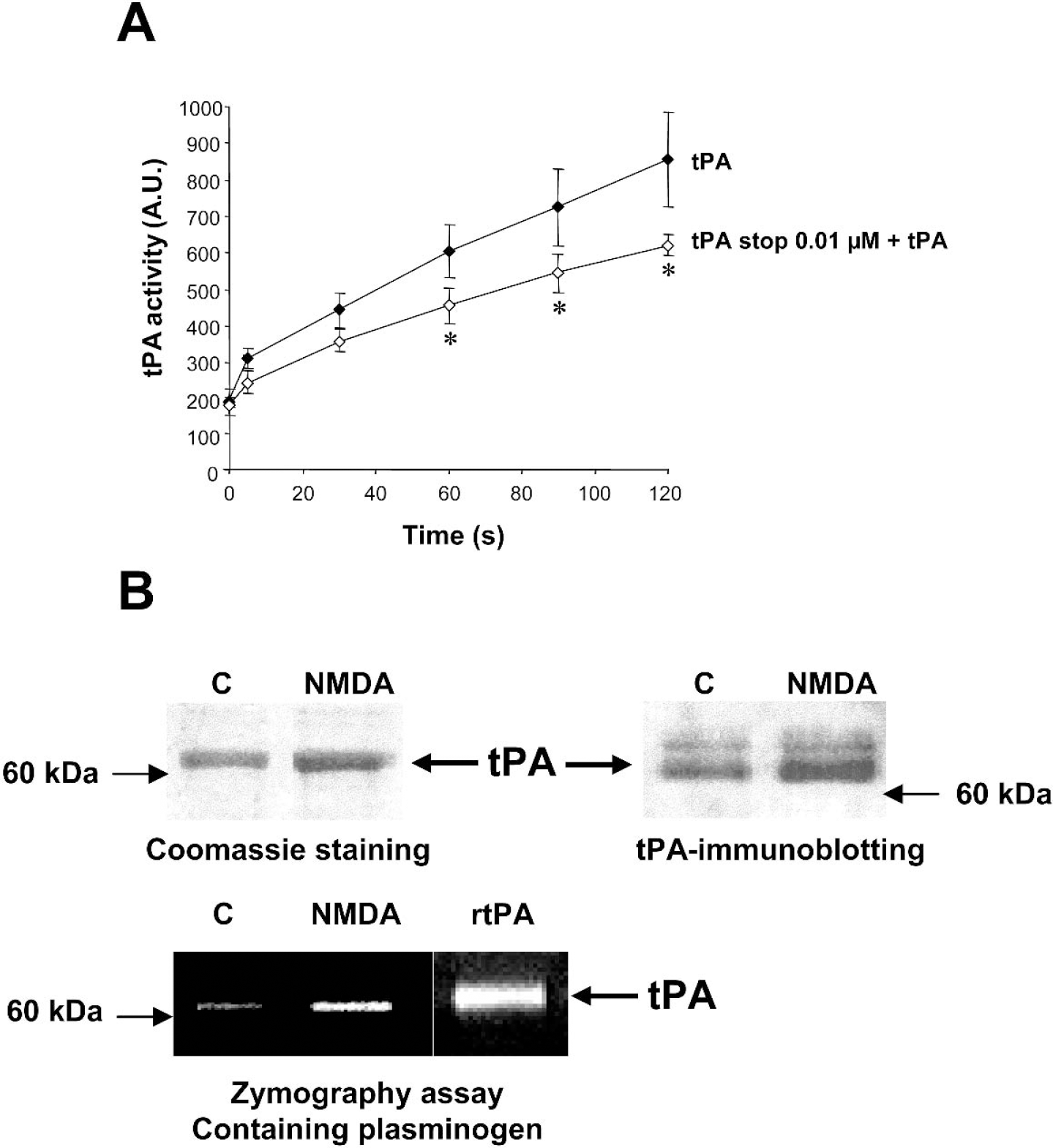
Tissue-type plasminogen activator (tPA) activity is blocked by tPA stop and endogenous tPA is released from cortical neurons after N-methyl-
Induction of excitotoxicity
Excitotoxicity was induced by a 24-hour exposure to NMDA (12.5 μmol/L), AMPA (10 μmol/L), or kainate (50 μmol/L) in serum-free DMEM (Medium Solution supplemented with 10 μmol/L of glycine). MK-801 (10 μmol/L) was added concurrently with AMPA and kainate to block secondary NMDA receptor activation. Increasing concentrations of tPA or tPA stop or combination of one dose of each compound were coapplied with the excitotoxin and left in the bathing media for 24 hours. Neuronal death was quantified by measurement of lactate dehydrogenase release from damaged cells into the bathing medium (Koh and Choi, 1987)
Calcium imaging
Cell cultures were loaded with Fura-2 (45 min, 37°C) in 5 μmol/L Fura-2 AM plus 0.1% pluronic F-127. Experiments were performed at room temperature, on the stage of a Nikon Eclipse (Paris, France) inverted microscope equipped with a 75-W xenon lamp and a Nikon 40x, 1.3 numerical aperture epifluorescence oil immersion objective. Fura-2 (excitation: 340, 380 nm, emission: 510 nm) ratio images were acquired with a CDD camera (Princeton Instrument, Trenton, NJ, U.S.A.), and digitized (256 × 512 pixels) using Metafluor 4.11 software (Universal Imaging Corporation, Chester, PA, U.S.A.).
Excitotoxic lesions
All experiments were carried out in the framework of the French national legislation that governs animal experimentation. The research protocols were approved by the Scientific Council of our institution. Male Sprague-Dawley rats weighing 280 to 320 g (CERJ, Paris, France) were anesthetized with chloral hydrate (300 mg/kg, IP) and received an unilateral injection into the left striatum: coordinates 0.2 mm posterior, 3.5 mm lateral, 5.5 mm ventral to the bregma of either NMDA (75 nmol in phosphate buffered saline, pH 7.4, n = 5), NMDA + tPA stop (1.5 nmol, n = 5), AMPA (6 nmol in phosphate buffered saline, pH 7.4, n = 5), or AMPA + tPA stop (1.5 nmol, n = 5) in a total volume of 3 μL. For AMPA-induced lesion, MK-801 (6 nmol) was co-injected. Five minutes after the insertion of the needle, the solution was injected using a Hamilton syringe pump at a rate of 0.5 μL/min. The needle was removed 5 minutes later.
Histological analysis
After 24 hours for NMDA experiments and 48 hour for AMPA injections, rats were killed and the brains were removed and frozen in isopentane for histological analysis. Cryostat-cut coronal brain sections (20 μm) were stained with cresyl violet and analyzed by using an image analyzer. For volume analysis, one section of 20 μm of 20 was stained and analyzed (total of 12 sections, covering the whole lesion). Regions of interest were determined through the use of a stereotaxic atlas for the rat and an image analysis system (BIOCOM RAG 200, Paris, France) was used to measure the lesioned area.
Statistical analysis
Results are expressed as the mean ± SD. Statistical analysis were performed by one-way or by a multiparametric analysis of variance followed by Fisher's or Bonferroni Dunn's test as indicated in the corresponding legend of figures.
RESULTS
tPA potentiates NMDA-mediated neuronal death
It has been reported that tPA could influence neuronal death induced by acute brain injury (Wang et al., 1998). In this situation, death occurs at least in part by excitotoxicity. To test whether tPA or tPA inhibitors are able to modulate excitotoxic neuronal death, we determined the effect of the recombinant tPA and of a synthetic reversible competitive tPA inhibitor (tPA stop) on murine cortical cell cultures exposed to stimuli that specifically induce excitotoxicity. tPA stop was previously used to block the proteolytic activity of tPA both in vitro and in vivo (Baranes et al., 1998; Flavin et al., 2000; Pawlak et al., 2002). First, we verified that tPA stop is able to inhibit tPA activity. As shown in Fig. 1A, the activity of tPA was reduced by the addition of tPA stop (0.01 μmol/L). To further complete the characterization of our model, we have investigated the release of endogenous tPA in the culture media of cortical neurons treated in the presence of NMDA. As shown in Fig. 1B, NMDA exposure led to a release of tPA in the extracellular space as estimated by Coomassie staining and Western blotting. The release of tPA after NMDA exposure in cultured cortical neurons was confirmed by zymography assay in casein and plasminogen-containing gels (Fig. 1B). Recombinant tPA was loaded as a control. No proteolytic activity was detected in gels without plasminogen, confirming that the proteolytic band detected around 69 kDa is tPA (data not shown). Then, necrosis was induced by the following ionotropic glutamatergic agonists: NMDA (12.5 μmol/L), AMPA (10 μmol/L), or kainate (50 μmol/L). The exposure of cortical neuronal cultures (DIV14) to NMDA produced acute swelling of neuronal cell bodies, followed by widespread neuronal degeneration. As shown in Fig. 2A, NMDA alone induced approximately 45% of neuronal death as estimated by lactate dehydrogenase release in the bathing media. Co-incubation with increasing concentrations of tPA promoted NMDA-induced neuronal death in a dose-dependent manner, with a significant effect at 20 μg/mL, a dose that alone was not toxic to neurons. Conversely, application of the tPA inhibitor, tPA stop, reduced NMDA-induced neuronal death in a dose-dependent manner, with a significant effect at the concentration of 0.1 μmol/L (Fig. 2B). In order to further investigate the neuroprotective activity of the tPA inhibitor against excitotoxicity, tPA stop was tested in the presence of tPA. Although tPA (20 μg/mL) potentiated the NMDA-induced neuronal death (+40%), tPA stop at 0.01 μmol/L (a dose that was without any effect on NMDA alone (Fig. 2B) was able to prevent tPA-enhanced NMDA-mediated neuronal death (Fig. 3). We have also induced excitotoxicity by using the glutamatergic agonists AMPA and kainate. In contrast to the effects observed in the presence of NMDA, tPA (20 μg/mL) and the tPA inhibitor, tPA stop (0.1 μmol/L), (Figs. 4A and 4B) failed to influence such paradigms of neuronal death.
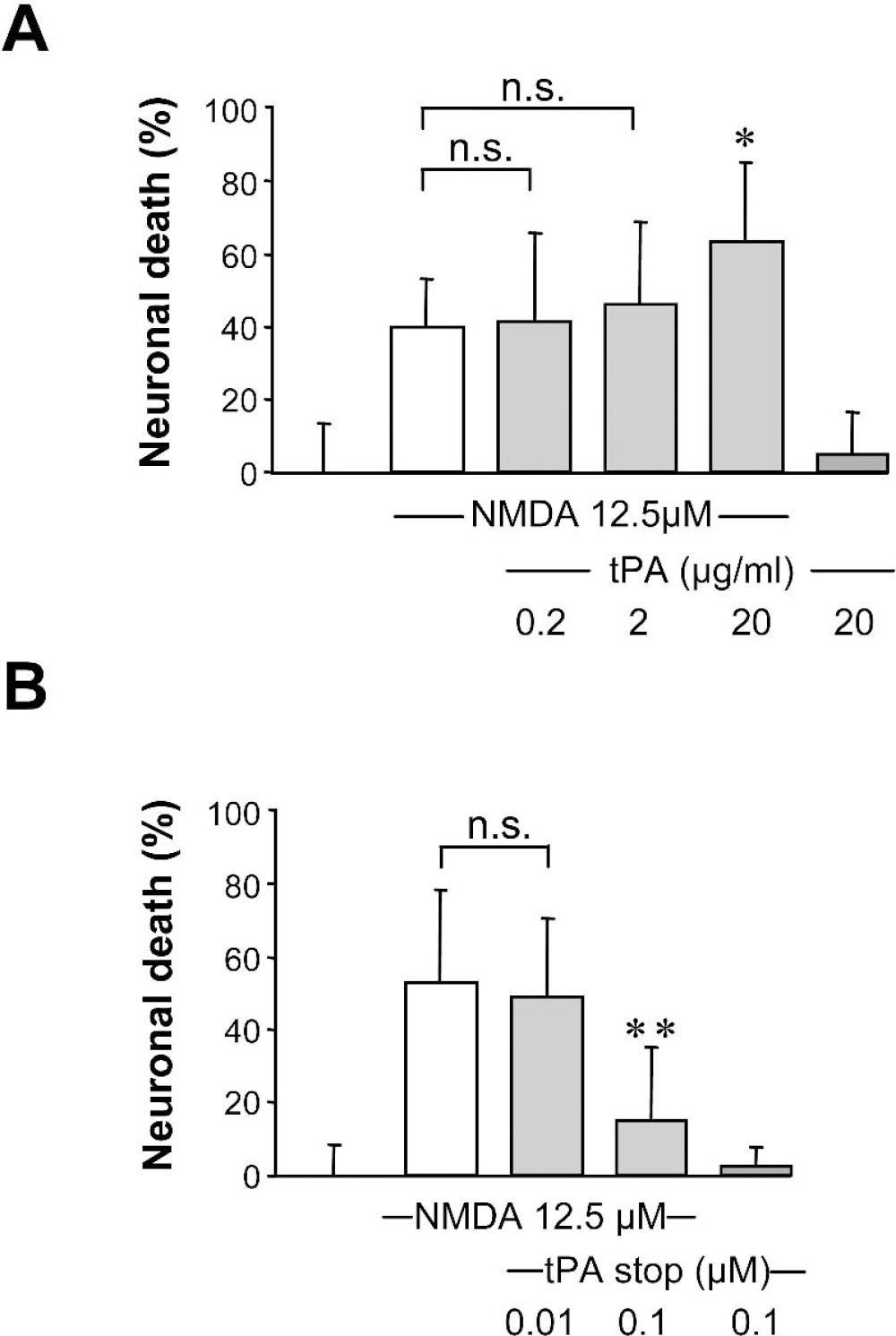
Tissue-type plasminogen activator (tPA) potentiates N-methyl-
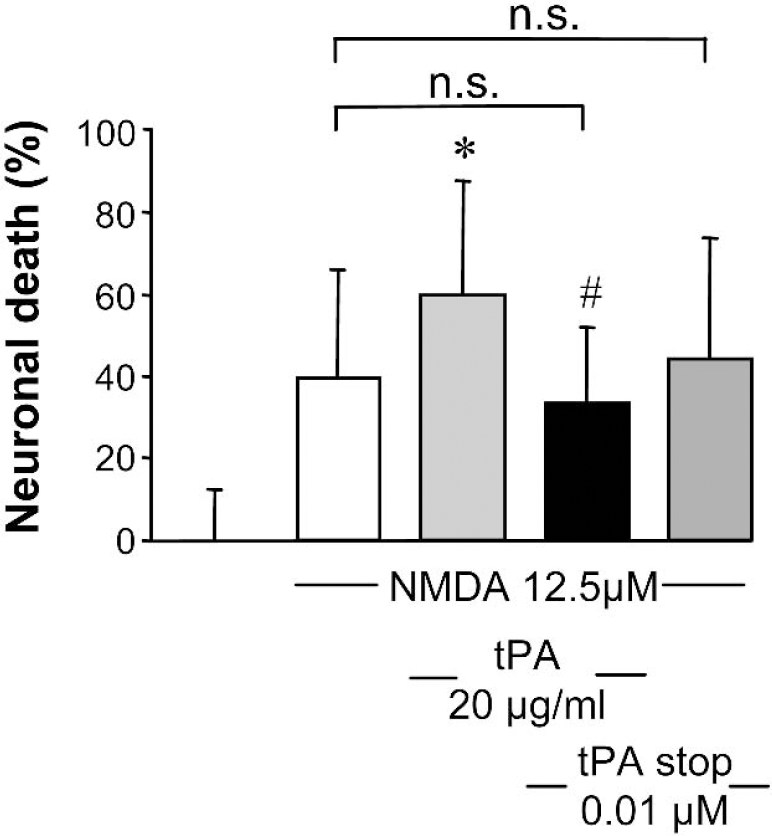
Tissue-type plasminogen activator (tPA) stop reverses the potentiating effect of exogenous tPA on N-methyl-
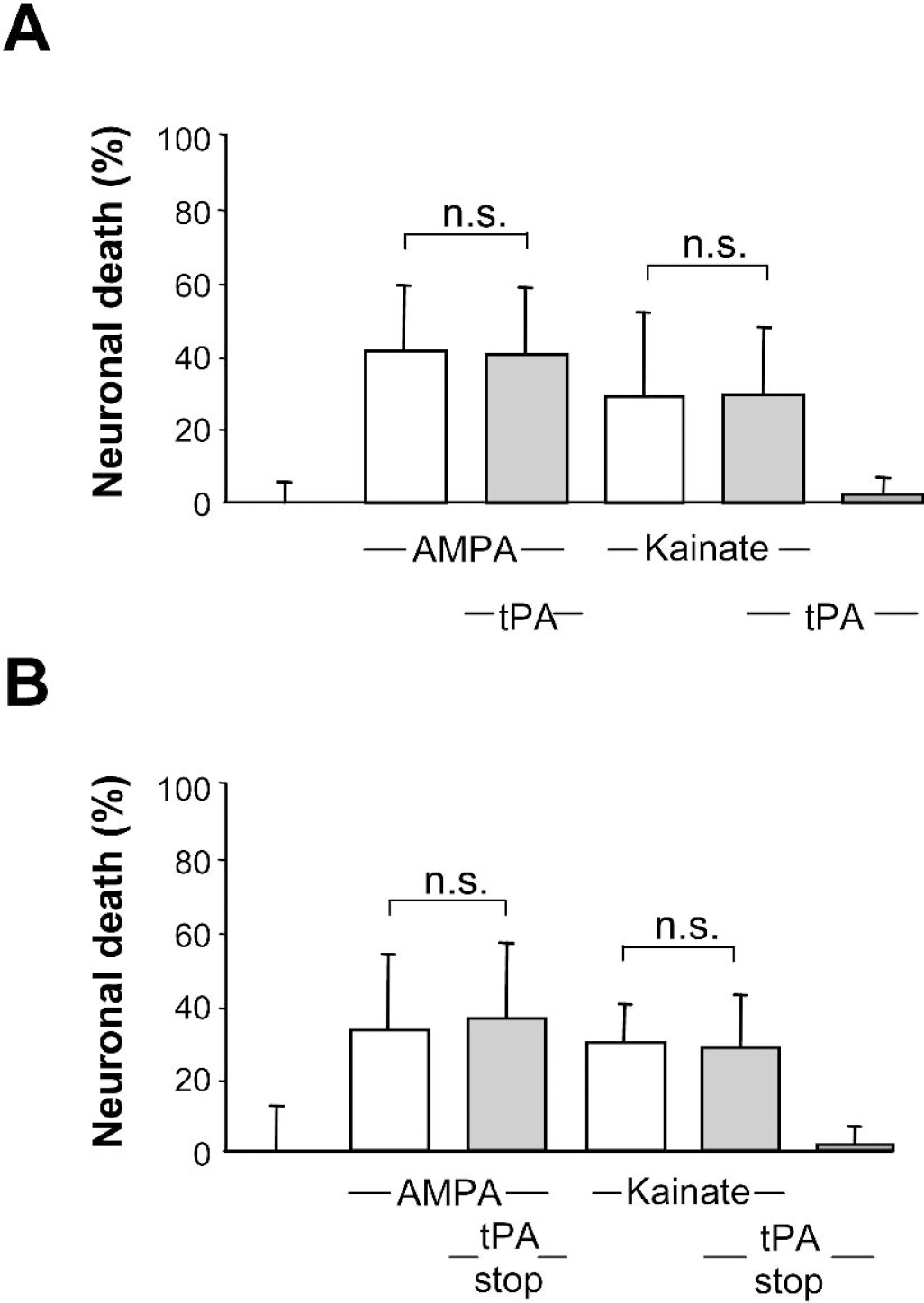
Tissue-type plasminogen activator (tPA) and tPA stop fail to modulate α-amino-2,3-dihydro-5-methyl-3-oxo-4-isoxazole propanoic acid (AMPA)/kainate-mediated neuronal death. Pure cultures of cortical neurons were exposed to AMPA (10 μmol/L) or kainate (50 μmol/L) alone or in the presence of either t-PA (20 μg/mL)
The proteolytic activity of tPA controls NMDA receptor signaling
We have previously demonstrated that tPA was able to increase NMDA receptor-dependent calcium influx (Nicole et al., 2001). Thus, we have investigated whether tPA stop could prevent tPA-induced potentiation of NMDA signaling, by using fura-2 fluorescence videomicroscopy. For these experiments, we chose a concentration of NMDA that allowed observation of a response that could be modulated (25 μmol/L of NMDA in order to observe a potentiation and 50 μmol/L in order to observe an inhibition). In cortical cell cultures, treatment with tPA stop (0.01 or 0.1 μmol/L) did not influence NMDA-induced calcium influx (Figs. 5A and 5B). Because these experiments were performed under perfusion allowing washout of endogenous tPA, these data are evidence that tPA stop does not influence NMDA signaling by itself at the doses of 0.01 and 0.1 μmol/L. In addition, as shown in Figs. 6A–C, although treatment with tPA enhanced the NMDA-induced calcium influx in cortical neurons, a co-treatment with tPA stop (0.01 μmol/L) prevented this effect of exogenous tPA.
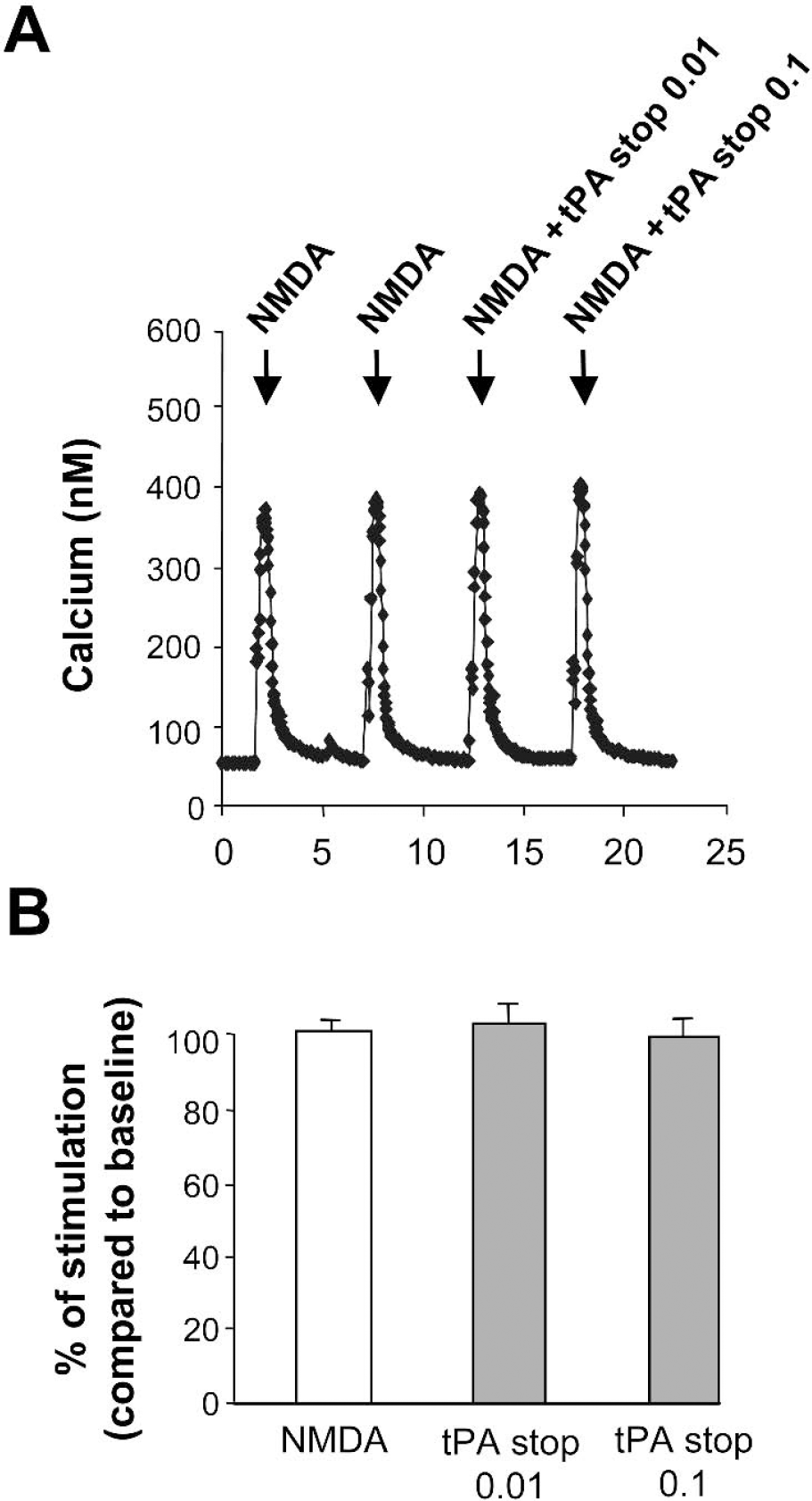
Tissue-type plasminogen activator (tPA) stop does not influence N-methyl-
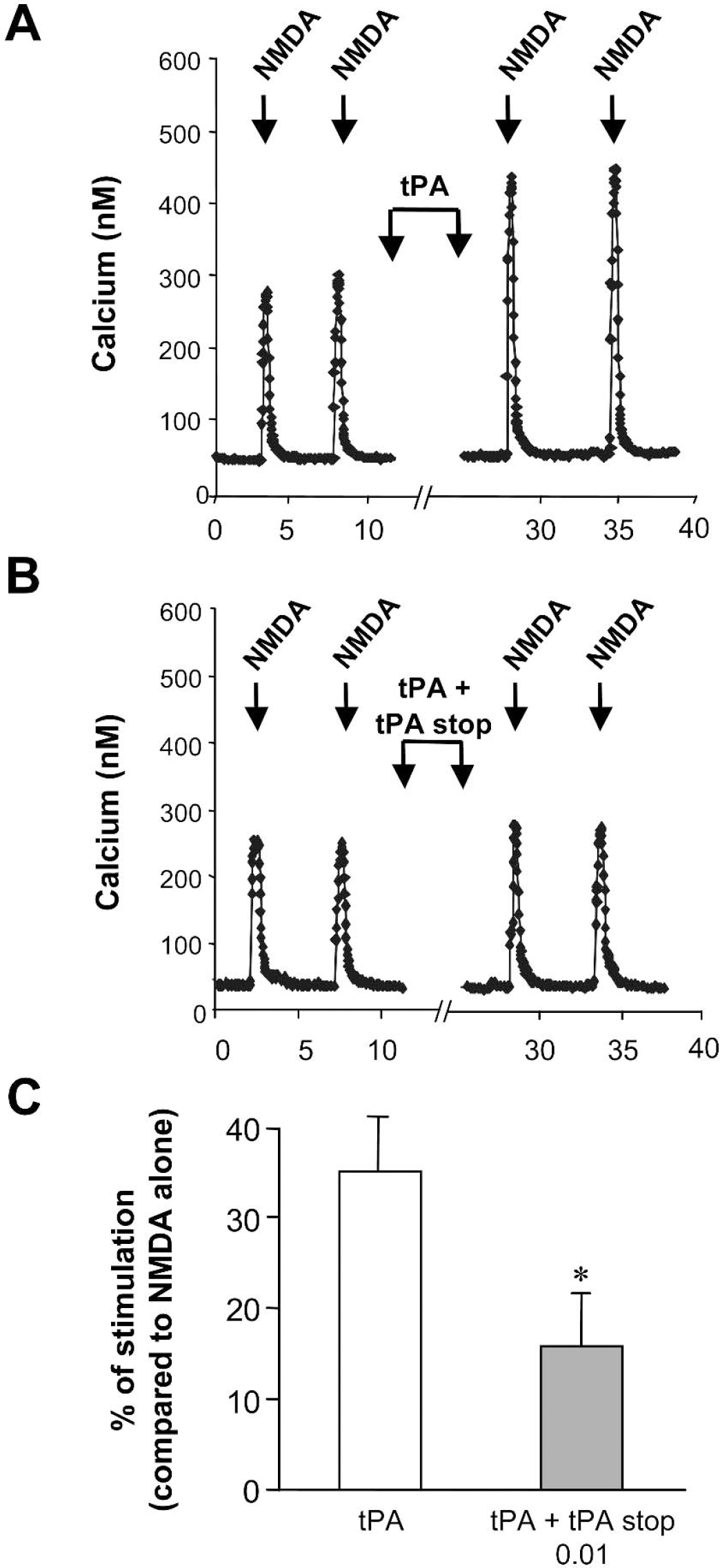
Tissue-type plasminogen activator (tPA) stop blocks tPA-dependent potentiation of N-methyl-
Inhibition of tPA by tPA stop protects neurons from NMDA-induced excitotoxicity in vivo
Finally, we have investigated whether the inhibition of excitotoxicity observed in vitro in the presence of tPA stop also occurred in vivo. We have tested the effect of the addition of tPA stop (1.5 nmol) on the lesion induced by the intrastriatal injection of NMDA (75 nmol) in rats. As shown in Fig. 7A, the volume of the intrastriatal lesion induced by the injection of NMDA was dramatically reduced by approximately 55% by the co-injection of tPA stop (46.5 mm3 + 12.39 for NMDA alone versus 20.7 mm3 + 11.06 for NMDA + tPA stop). In addition, the activity of tPA-stop was estimated in a model of AMPA-induced lesion after intrastriatal injection in rats. Experiments were performed in the presence of MK-801 to prevent secondary activation of NMDA receptors. Although AMPA alone (6 nmol) induced a brain lesion of around 25 mm3, co-injection of tPA stop (1.5 nmol) failed to influence the AMPA-induced lesion (Fig. 7B). These data are in agreement with those obtained in vitro (Fig. 4), suggesting that the deleterious effect of tPA on neuronal death is specific of the activation of NMDA receptors.
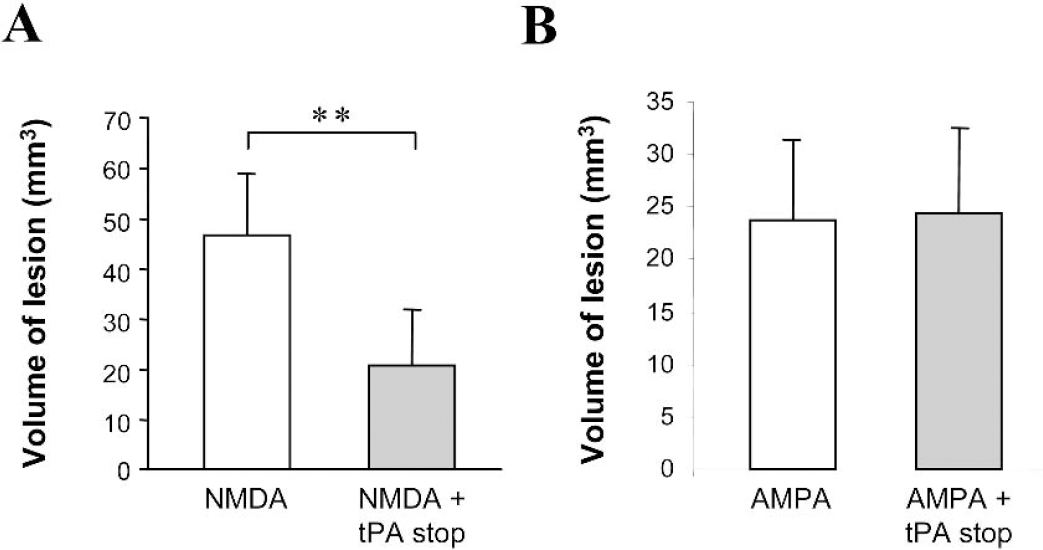
Tissue-type plasminogen activator (tPA) stop reduces N-methyl-
DISCUSSION
Once thrombotic occlusion occurs, the ischemic cascade leads to the activation of mechanisms involved in excitotoxicity. In fact, within minutes of ischemia onset, there is a marked increase in glutamate concentration in the affected portion of the brain, resulting in overactivation of NMDA receptors. In consequence, NMDA receptor antagonists represent an attractive target for neuroprotective therapy and have been assessed for clinical trials (Lee et al., 1999). Nevertheless, although a number of experiments performed by using neuroprotective agents have been proved to be effective in rodent models, unfortunately they were not in humans. Despite all these efforts, treatment of ischemic stroke remains one of the most challenging areas of medicine today. The only treatment approved by the Food and Drug Administration is early reperfusion by the thrombolytic agent, tPA. In 1995, a study performed by the National Institute of Neurological Disorders and Stroke, showed that tPA was both safe and beneficial in the treatment of stroke, with a 3-hour time window (NINDS, 1995). Further studies have extended this therapeutic window to 6 hours for intraarterial injection (del Zoppo et al., 1998). tPA has the ability to activate the zymogen plasminogen into plasmin in order to degrade fibrin clots (Henkin et al., 1991). In the central nervous system, tPA is expressed by neurons and glia (Sappino et al., 1993) and is known to participate in a large number of functions either in the developing brain or the adult brain. Most of its effects described so far have been related to its ability to degrade the extracellular matrix and potentially forming a path for extending processes in neurite outgrowth and neuronal development (Seeds et al., 1990). tPA was also characterized by its ability to potentiate the excitotoxic neurodegeneration (Tsirka et al., 1995). It was evidenced that tPA mediates excitotoxicity by cleaving plasminogen into plasmin and initiating the proteolytic cascade that leads neuronal death (Chen and Strickland, 1997). In a model of NMDA-induced excitotoxicity in cultured cortical neurons, we have evidenced that tPA mediated its deleterious effect on neuronal death by interacting with and cleaving the N-terminal end of the NR1 subunit of the NMDA receptor (Nicole et al., 2001). In addition to the direct neuronal effect that we have evidenced, it has been suggested that tPA could mediate its effect through the activation of plasminogen into plasmin (Matys and Strickland, 2003). Others have reported nonproteolytic activity of tPA in the central nervous system (Kim et al., 1999; Rogove and Tsirka, 1998). For example, tPA is also involved in microglial activation during excitotoxicity, but this function does not involve its catalytic activity (Rogove and Tsirka, 1998). Thus, based on its pleiotropic effects, the use of well-characterized in vitro models of neuronal death is a fundamental approach to further understand the real role of tPA in the central nervous system. Our findings in the present work support the idea that blocking the proteolytic activity of tPA could be an appropriate strategy to limit the neuronal death that occurs after excitotoxic brain damage. Indeed, we showed that an inhibitor of tPA, such as tPA stop, can prevent the overactivation of NMDA receptor. Although we cannot exclude that tPA stop could have other effects, our data clearly show that tPA stop is able to block the potentiating effect of exogenous tPA on NMDA signaling (Figs. 5 and 6). In addition, our experiments of NMDA-induced toxicity suggest that tPA stop is also able to significantly reduce the effect of endogenous tPA on NMDA-induced neuronal death (Fig. 2). In addition, although tPA stop at the doses used in our experiments could inhibit trypsin (if this one was present), our results with tPA stop are in agreement with our previous work using the type 1 plasminogen activator inhibitor (PAI-1), an endogenous inhibitor specific for tPA (Docagne et al., 2002). Overall, this demonstrates that inhibitors of the proteolytic activity of tPA are neuroprotective against NMDA-induced neuronal death. Finally, we evidenced that the use of a synthetic inhibitor of the proteolytic activity of tPA in a model of NMDA-induced excitotoxicity in rats leads to a dramatic neuroprotective activity. This observation is in agreement with the demonstration that neuronal damages that occur after either cerebral ischemia (Wang et al., 1998) or excitotoxin injection (Tsirka et al., 1995) are reduced in tPA-deficient mice as compared with wild-type animals.
Altogether, we can suggest that the use of a tPA inhibitor targeted to the cerebral parenchyma could be appropriate for the treatment of excitotoxic neuronal damages, which occur in a number of brain disorders such as stroke and head trauma. In this view, tPA stop may be a better candidate than peptidic inhibitors, such as PAI-1 and neuroserpin. Indeed, although they would all intravenously promote clot formation by inhibiting the proteolytic activity of t-PA in the blood, it may be easier to redesign a chemical compound and take advantage of its smaller size to target it to the brain and facilitate its passage through the blood–brain barrier. For this, the use of lipidic or chemical shuttles should be investigated.
Footnotes
Acknowledgments:
We thank O. Nicole for sharing data before publication and for critical comments on the manuscript. We also warmly thank P. Carmeliet for providing the antibody against tPA.