
Abstract
Cardiovascular and neurologic surgeries often involve a temporary reduction in cerebral blood flow. In these conditions, as well as during cerebral ischemia and traumatic brain injury, the temporary loss of oxygen and glucose initiates a cascade of cellular events that culminate in neuronal death and damage. Understanding the mechanisms that contribute to neuronal death after hypoxia/ischemia is critically important for treatment of such brain injury. Here, we use a model of combined cerebral hypoxia/ischemia (H/I) to examine the role of protease-activated receptor-1 (PAR-1) in hypoxic/ischemic neuronal damage. Our data show that PAR-1-deficient mice have smaller lesion volumes than wild-type controls after 45 minutes of H/I. The results of the genetic block of PAR-1 were corroborated using a PAR-1 antagonist, which decreased infarct volume in wild-type C57Bl6 mice. Examination of cellular responses to H/I reveals that PAR-1 -/- animals have less cellular death and diminished glial fibrillary acidic protein expression. Additionally, PAR-1 -/- mice exhibit less motor behavior impairment in rotorod and inverted wire-hang tests. These data suggest that PAR-1 contributes to hypoxic/ischemic brain injury and are consistent with other studies that implicate serine proteases and their receptors in neuropathology after cerebral insults.
Understanding neuronal disease after cerebral hypoxia/ischemia (H/I) is critically important for treatment of conditions in which cerebral blood flow is reduced. Temporary occlusion of cerebral arteries often accompanies neurovascular surgeries (Samson et al., 1994) and cerebral hypoxia is a common complication of both neurologic and cardiovascular surgeries (Kessler et al., 1997). Pathologic situations such as stroke and traumatic brain injury also deprive the brain of oxygen and glucose. Such conditions initiate a variety of cellular events within the brain that culminate in neuronal death and damage (Choi and Rothman, 1990).
Although they are best known for their role in coagulation and wound healing, serine proteases, such as thrombin and plasmin, have been implicated in the pathophysiology of CNS injury (Jiang et al., 2002; Xi et al., 2003). They are generated by proteolytic cleavage of their precursors prothrombin and plasminogen, respectively (Gingrich and Traynelis, 2000). Prothrombin is activated by tissue factor to form thrombin while tissue plasminogen activator (tPA) cleaves plasminogen to form plasmin, all of which are expressed in the CNS (Dihanich et al., 1991; Macfarlane et al., 2001; Tsirka et al., 1995; Weinstein et al., 1995). tPA gene expression is upregulated after focal ischemia (Kim et al., 2002), and tPA enters the brain after ischemia (Wang et al., 1998). Likewise, mRNA encoding prothrombin increases in the brain after cerebral ischemia (Riek-Burchardt et al., 2002). Thrombin and other serine proteases activate a family of receptors known as protease-activated receptors (PARs). Four PARs have been identified (PAR-1, −2, −3, and −4) (Macfarlane et al., 2001). PAR-1, the most abundant PAR in the CNS, is expressed in neurons, astrocytes, microglia, and the endothelial cells that line the blood–brain barrier (BBB) (Dihanich et al., 1991; Weinstein et al., 1995).
Activation of PAR-1 by thrombin or plasmin initiates a variety of cellular events in the brain including neurite outgrowth inhibition, astrocytic proliferation, microglia activation, and apoptosis induction (Choi et al., 2003; Gingrich and Traynelis, 2000; Wang et al., 2002). Additionally, PAR-1 activation by thrombin and plasmin potentiates N-methyl
MATERIALS AND METHODS
Animals
Young adult male PAR-1 -/- C57Bl/6 mice (n = 22) and wild-type control mice (n = 17) between 90 and 120 days old and from the same genetic background were used in these studies. PAR-1 -/- and wild-type controls were generated by breeding male PAR-1 +/- mice, a gift from Dr. Shaun Coughlin, University of California San Francisco (Connolly et al., 1996), with female C57Bl/6 wild-type mice from Jackson Laboratories (Bar Harbor, ME). Breeding heterozygous littermates generated homozygous null mutants and genetically matched wild-type controls that were subsequently used to establish PAR-1 -/- or wild-type control breeding pairs. Animals used for these studies were within four generations of the initial homozygous null mutant or wild-type breeding pairs. C57Bl/6 mice (Jackson Labs) were used for the PAR-1 antagonist studies. Animal procedures were reviewed and approved by Emory University Institutional Animal Care and Use Committee.
Hypoxia/ischemia model
Hypoxia/ischemia was produced by a modification of the method described originally in juvenile rats (Rice et al., 1981; Vannucci et al., 1996) and in adult mice (O'Donnell et al., 2002; Vannucci et al., 2001). Animals were anesthetized with halothane anesthetic until loss of righting and maintained with 1% to 2% halothane for the duration of the surgery. The right carotid artery was isolated, ligated in two locations with 6–0 surgical silk, and transected between the sutures. Animals were returned to their cages and were allowed to recover for a minimum of 2 hours with access to food and water. Mice were then placed into a plastic, screw-top container with inlet and outlet holes on the lid. The container was placed in a 35.5°C water bath and perfused with 8% oxygen at a rate of 1 L/min for 45 minutes. Oxygen (Specialty Gas, Suwanee, GA) tanks were certified by the company at 8% O2 with nitrogen balance. Furthermore, the oxygen level was verified using an oxygen sensor (Biosystems, Middletown, CT) before the animals were placed into the container. Previous studies have demonstrated that submersion of the container in a 35.5°C water bath is sufficient to maintain body temperature of 37.5 to 37.7°C (O'Donnell et al., 2002). After hypoxia, animals were removed from the container and allowed to recover in their cages with access to food and water.
In a previous experiment, we found that neuronal damage was dependent on duration of hypoxia. Forty-five minutes of hypoxia consistently produced large lesions that averaged 32.6% of the ipsilateral hemisphere in wild-type C57Bl/6 mice (data not shown). Therefore, we chose to examine neurologic damage after 45 minutes of H/I for these studies. Neuronal damage after exposure to 45 minutes of H/I was examined by 2,3,5-triphenyltetrazolium chloride (TTC) staining, cresyl violet, staining and glial fibrillary acidic protein (GFAP) immunohistochemistry.
Histology
After a survival period of 2 days, all mice were euthanized and infarct volume was assessed via staining with 2% TTC (Sigma-Aldrich, St. Louis, MO) staining in 2-mm-thick coronal brain sections (Khan et al., 2000). TTC is reduced by succinate dehydrogenase to form a red formazan product in viable tissue; healthy tissue is stained red and damaged tissue is unstained and remains white. TTC stained coronal sections were digitized on a flatbed scanner. Infarct volume was calculated using NIH Image software by measuring the area of the lesion in each coronal section.
For analysis with cresyl violet and GFAP, TTC-stained sections were postfixed in 4% paraformaldehyde and then submerged in 30% sucrose. After freezing, the 2-mm sections were cut into 10-μm slices on a cryostat and mounted on gelatin-coated slides. Some slides were stained with cresyl violet (EM Science, Gibbstown, NJ). GFAP expression was examined via fluorescence immunohistochemistry. Cryostat sections were incubated overnight with rabbit polyclonal GFAP antibody, 1:500 (Accurate Chemical and Scientific Corp., Westbury, NY). The next day sections were incubated in biotinylated goat anti-rabbit secondary antibody, 1:100 (Vector Laboratories, Burlingame, CA) followed by incubation with Streptavidin Texas Red, 1:100 (Amersham Biosciences, Piscataway, NJ) and viewed with a Leitz Laborlux microscope (Leica, Wexler, Germany) equipped with fluorescent optics.
Images were collected and staining density was quantified using Open Lab software (Improvision, Lexington, MA). The density slicing option was used to set a minimum intensity value, from a 0 to 250 binary scale, which served as the threshold intensity level. This value was consistent for all images analyzed. Each image was converted to a binary image according to the threshold intensity value. The binary image contains only pixels with intensity above the threshold value and reflects the positively stained area within the image. Pixels were calibrated to micrometer area, and the stained area was calculated in three separate sections per animal. The average of the three sections was divided by the total area of the image to obtain staining density.
PAR-1 antagonist
A peptide antagonist of PAR-1, trans-cinnamoyl-p-fluoro Phe-p-D13, L87-guanidinoPhe-LRR-amide (BMS 200261) (Bernatowicz et al., 1996), was synthesized by the Emory University Microchemical facility and administered immediately after carotid artery transection and before exposure to 8% hypoxia. One microliter of a 6-mmol/L solution of BMS 200261 in HEPES (10 mmol/L) buffered saline (150 mmol/L) was stereotaxically injected into the lateral ventricle (0.6 mm lateral, 1.5 mm posterior and 2.5 mm ventral to bregma) under isoflurane anesthesia over a 3-minute period. Vehicle control mice received 1 μL of HEPES buffered saline.
Behavioral tests
Rotorod: Mice were trained for 2 days before the H/I. Each trial consisted of 60 seconds at 10 rpm, 60 seconds rest, and acceleration from 10 rpm at 0.1 rpm/s until the animal fell from the rod. Each test day contained two trials with an intertrial interval of 5 minutes. The latency to fall was recorded for both acceleration segments, and the two scores were averaged. Inverted wire hang: Mice were placed on wire bars and inverted to 180°. The latency to fall was recorded up to 3 minutes.
Statistical analysis
Quantitative data are presented as mean ± standard deviation. Comparisons were made using Mann Whitney test and Kruskal-Wallis test with Dunn's post hoc test. The pre- and posthypoxia behavior results were compared using paired t-tests. P < 0.05 was regarded as statistically significant.
RESULTS
Hypoxia/ischemia-induced cell death
In this study, we used a relatively new model of H/I to examine the role of PAR-1 in H/I. Recently, it has been shown that PAR-1-deficient mice have significantly smaller lesions after focal cerebral ischemia induced by occlusion of the middle cerebral artery (Junge et al., 2003). Here, we use a different model of stroke and global hypoxia/hypoperfusion to give further evidence that PAR-1 contributes to neuronal damage after H/I. This model involves unilateral transection of the common carotid artery followed by a 45-minute exposure to 8% oxygen. The combined reduction of blood flow and oxygen confers damage to the ipsilateral cortex and hippocampus (Fig. 1A).
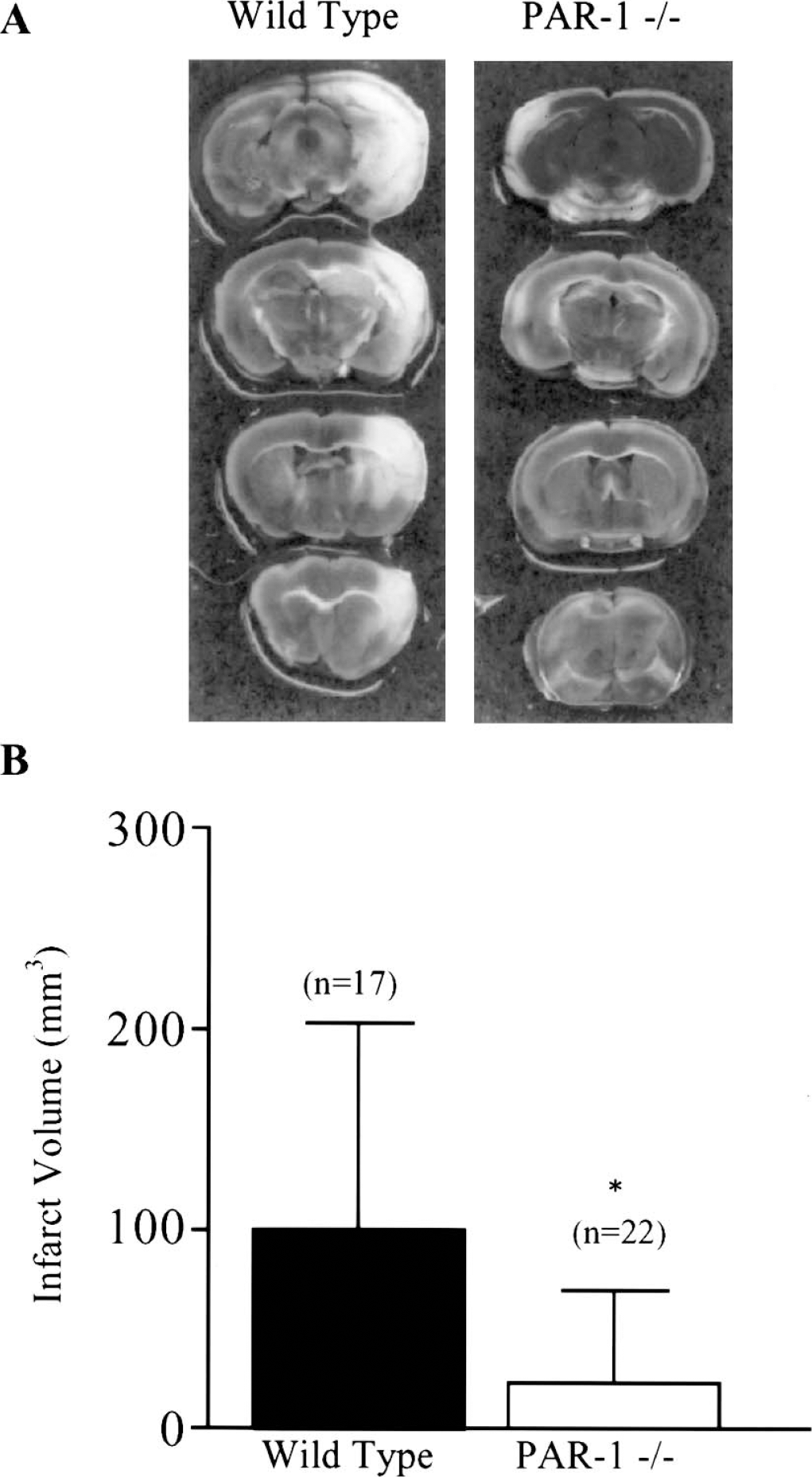
Wild-type and PAR-1 -/- infarct volume 48 hours after 45 minutes hypoxia/ischemia. (
Role of PAR-1 in hypoxia/ischemia-induced cell death
Exposure to 45 minutes H/I resulted in significantly smaller infarct volumes in PAR-1 -/- mice than in wild-type controls 48 hours after onset of H/I (Fig. 1). PAR-1 deficiency conferred a 75% reduction in infarct volume. To corroborate our genetic results pharmacologically, we examined the effect of a PAR-1 antagonist on infarct volume after H/I in wild-type C57Bl/6 mice. The PAR-1 antagonist, BMS 200261, blocks activation of the receptor and significantly reduces phosphoinositide (PI) hydrolysis in hippocampal neurons (Bernatowicz et al., 1996; Junge et al., 2003). Pretreatment with the PAR-1 antagonist reduced infarct volume dramatically compared to vehicle-treated controls (Fig. 2). This result further substantiates our hypothesis that blocking PAR-1 activation protects the brain from damage after hypoxic/ischemic injury.
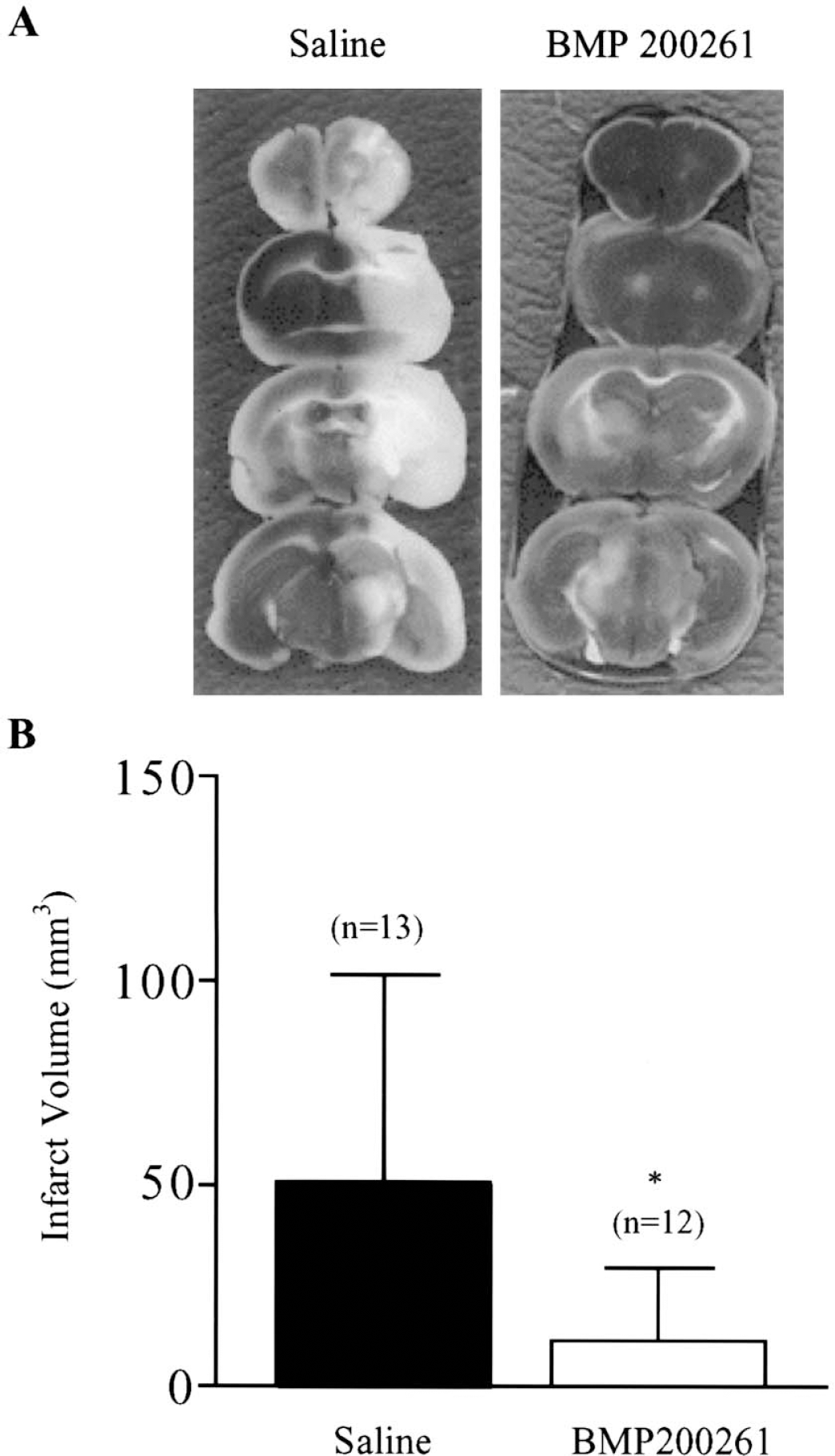
Effect of a PAR-1 antagonist on lesion volume. (
To further characterize the cellular response in wild-type and PAR-1 -/- animals, we examined cresyl violet staining and GFAP immunostaining. Lesions were associated with a loss of cresyl violet staining in the ipsilateral cortex and CA1 region of the hippocampus. Widespread cell loss was observed in the ipsilateral cortex (Fig. 3) and hippocampus (Fig. 4) of wild-type, but not PAR-1 -/- mice. The core of the lesion was observed as a clearly delineated zone of damage within the cortex (Fig 3A). This area is sparsely stained, reflecting a decreased density of cellular profiles (Fig 3B). Neurodegeneration is also evident in the CA1 pyramidal cells of the ipsilateral, versus contralateral, hippocampus and the dentate gyrus in wild-type mice (Fig. 4A and 4C). The density of CA1 pyramidal neurons on the ipsilateral side (Fig. 4B) was noticeably decreased compared to either the contralateral side (Fig. 4D) or to the staining density in the hippocampus and dentate gyrus on both the ipsilateral and contralateral sides in PAR-1 -/- mice (Figs. 4 E–H). Quantification of the damage in the core of lesion revealed a significant loss of staining in the wild-type ipsilateral cortex compared to the contralateral side (Fig. 5). Conversely, there was no significant difference in staining density between the hemispheres in the PAR-1 -/- mice, suggesting a protective effect of the PAR-1 deficiency.
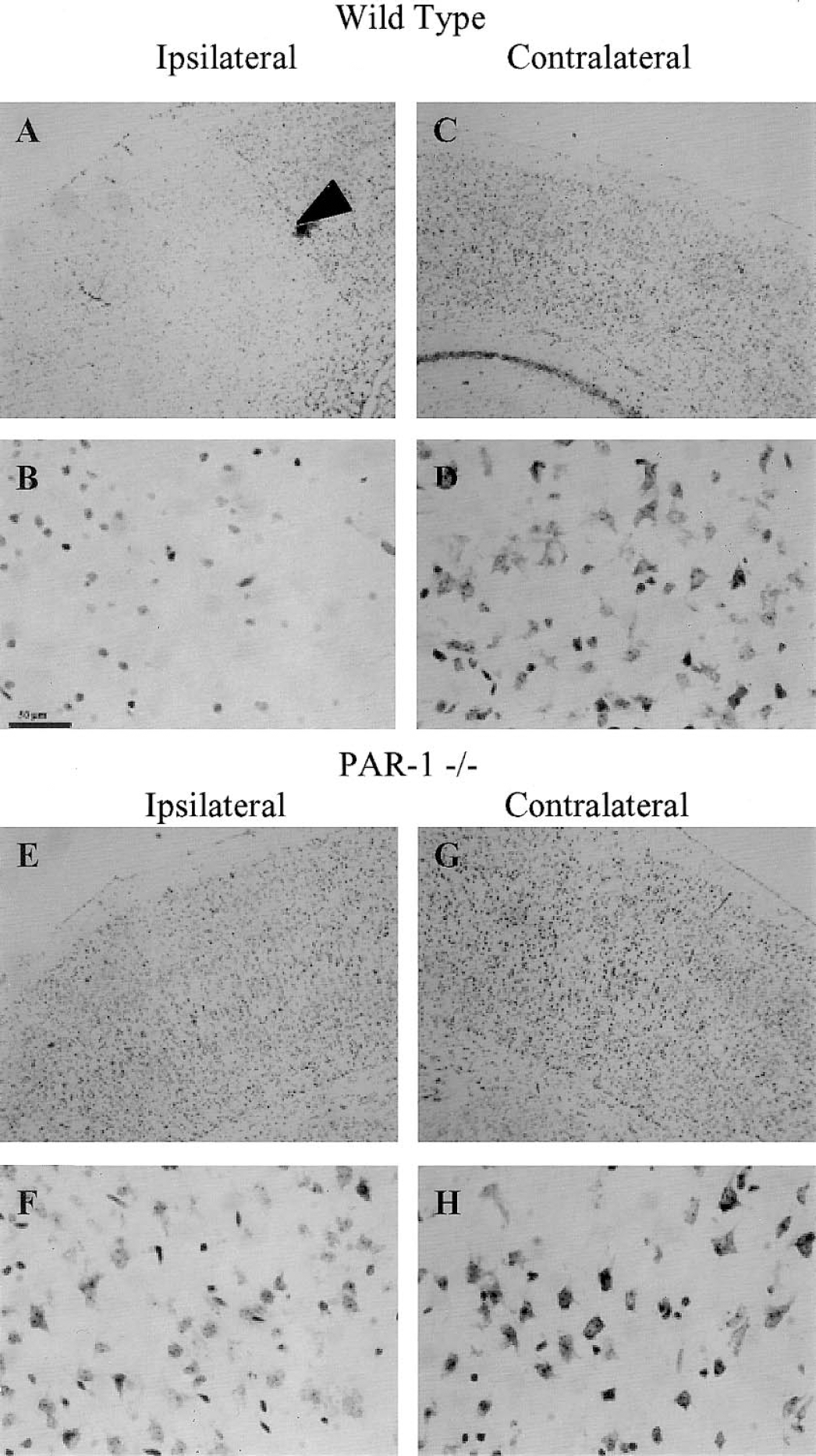
Neurodegeneration in the ipsilateral cortex of wild-type, but not PAR-1 -/- mice after hypoxia/ischemia. Representative photomicrographs at low and high magnification of (
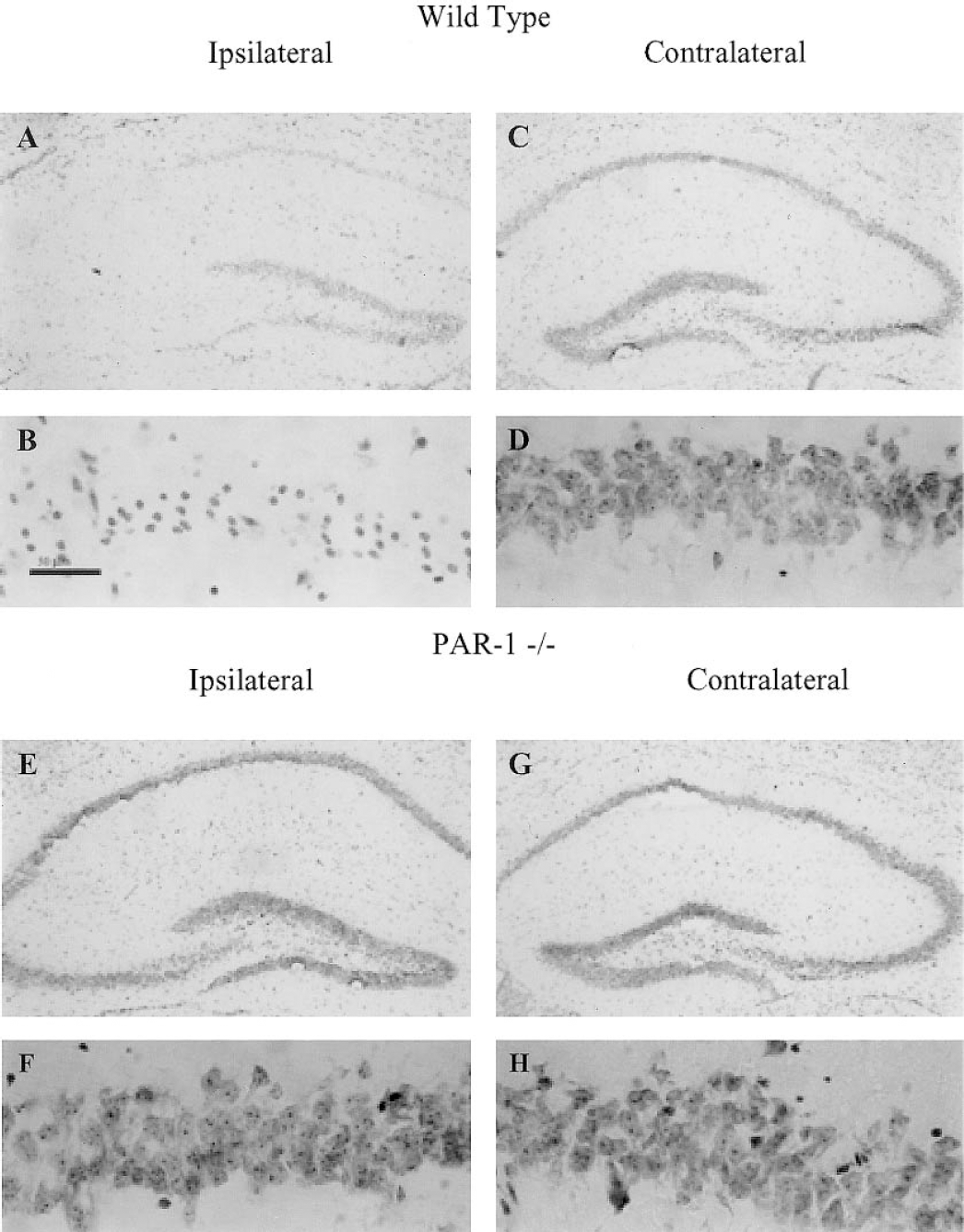
Neurodegeneration in the ipsilateral, but not contralateral hippocampus of wild-type but not PAR-1 -/- mice after hypoxia/ischemia. Representative photomicrographs of (A) wild-type ipsilateral hippocampus and (B) CA1 region, (C) wild-type contralateral hippocampus, and (D) CA1 region; (E) PAR-1 -/- ipsilateral hippocampus and (F) CA1 region, and (G) PAR-1 -/- contralateral hippocampus and (F) CA1 region. Wild type (n = 4). PAR-1 -/- (n = 4). Scale bar = 50 μm.
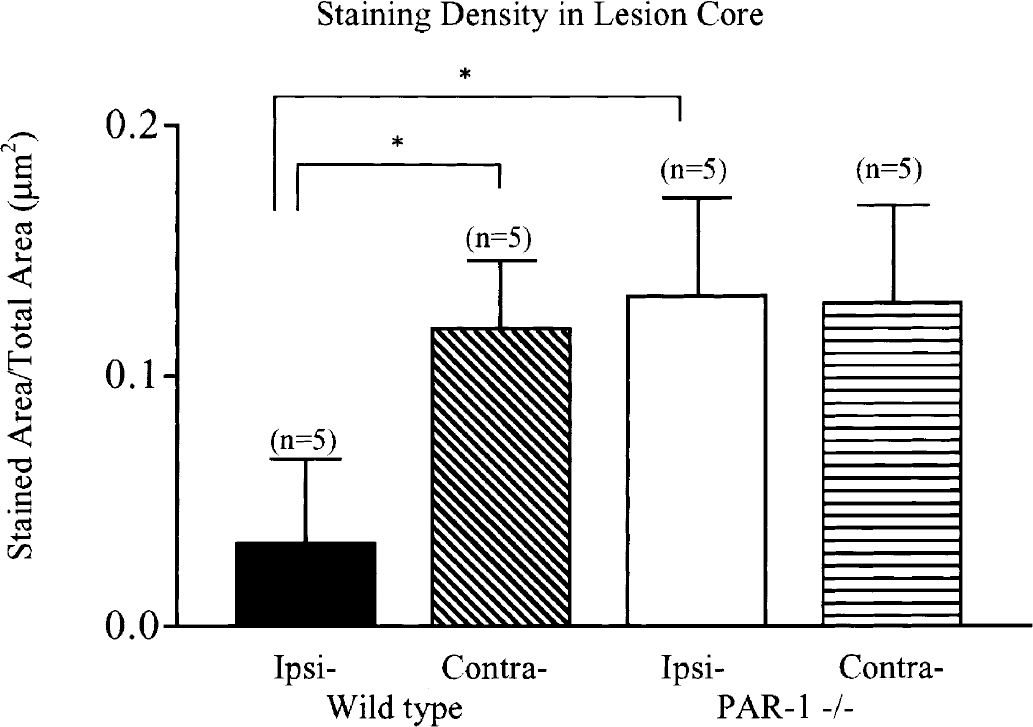
Quantification of staining density in the cortical lesion core. The staining density in the ipsilateral cortex of wild-type mice was lower than the contralateral hemisphere, 0.03 ± 0.03 μm2 versus 0.12 ± 0.03 μm2. Conversely, there was no difference in staining densities between ipsilateral and contralateral cortices in PAR-1 -/- mice, 0.13 ± 0.04 μm2 versus 0.13 ± 0.04 μm2. Ipsilateral, cortical staining density was lower in wild-type mice compared to PAR-1 -/- mice. *P < 0.05 Wild type (n = 5); PAR-1 -/- (n = 5).
Reactive astrogliosis in damaged areas
One hallmark of brain injury is the activation of astrocytes and their consequent expression of GFAP. PAR-1 activation has been shown to induce astrocyte proliferation (Wang et al., 2002). In this study, wild-type lesions are associated with strong GFAP immunoreactivity (Fig. 6). The GFAP staining density is significantly greater in the ipsilateral penumbral region of the lesion in wild-type mice compared to the contralateral side, whereas there is very faint GFAP staining in both the ipsilateral and contralateral hemispheres in PAR-1 -/- mice (Fig. 6E). The lack of GFAP in PAR-1 -/- mice is consistent with the small lesion volumes and limited neuronal damage in these animals.
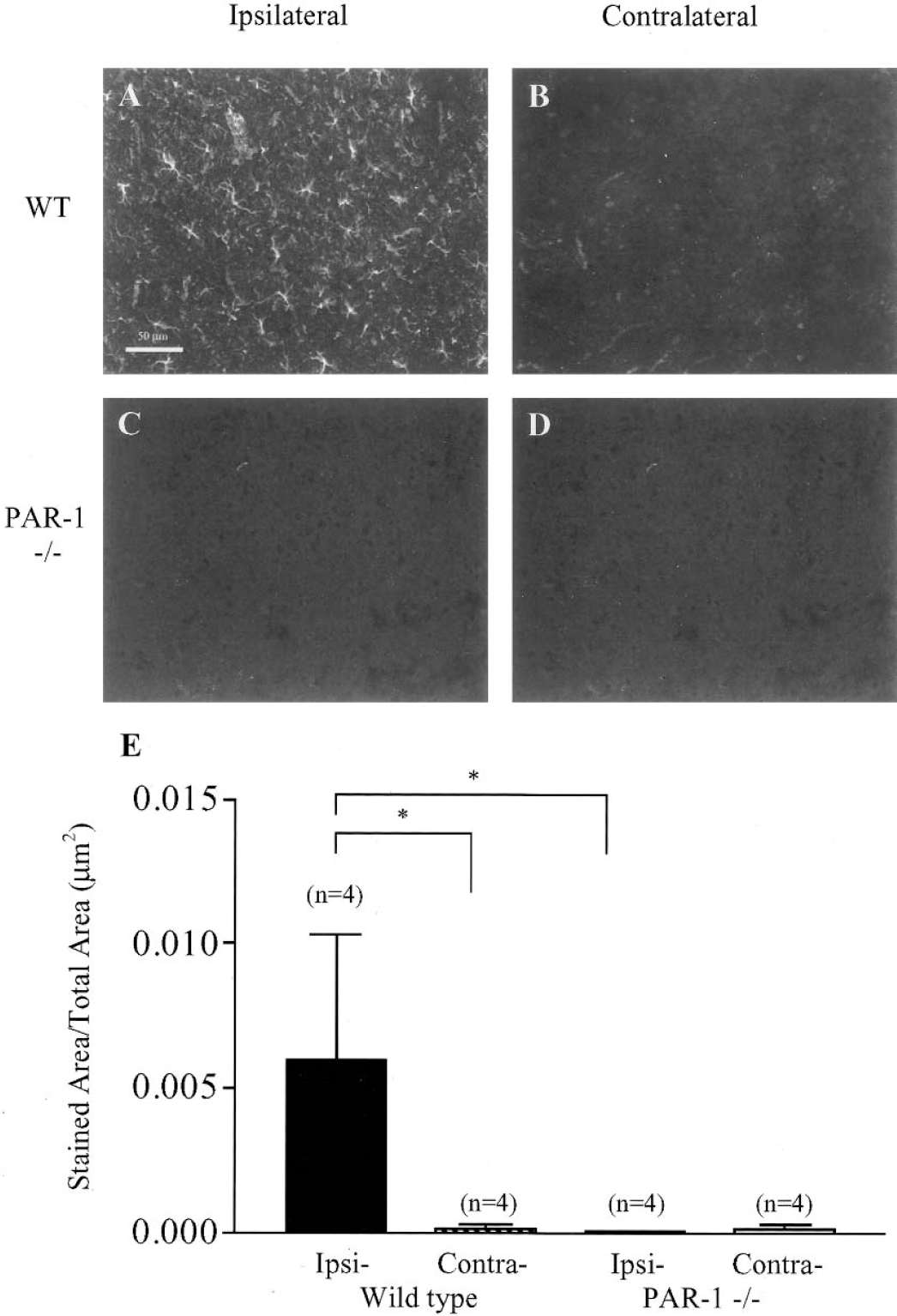
Glial fibrillary acidic protein (GFAP) immunoreactivity in wild-type and PAR-1 -/- mice after hypoxia/ischemia. Representative photomicrographs of GFAP immunostaining in (
Neurologic deficits are less severe in PAR-1 -/-
PAR-1 deficiency appears to protect against neuronal cell damage after H/I as assessed by histologic techniques. It has been suggested that disparity between basic animal studies and clinical studies of stroke may be due to the lack of functional and behavioral data in animal studies (STAIR, 1999). Thus, we performed tests aimed at examining motor behavior to address the questions of functional neuronal preservation in PAR-1 -/- mice. To examine whether PAR-1 knockout can provide functional protection after H/I, we examined neurologic impairment using two well-characterized motor behavior tests. Motor function was assessed using rotorod and wire hang tests 48 hours after onset of H/I. Wild-type mice subjected to H/I were significantly impaired in the ability to remain on the rotorod compared to baseline (pre-H/I) performance (Fig. 7A). The PAR-1 -/- group, however, maintained the ability to remain on the rotorod for approximately as long as their pre-H/I test. The percent change from baseline performance showed that the wild-type mice were significantly more impaired than PAR-1 -/- mice after H/I (Fig. 7B). The wild-type group showed a 40% reduction in latency to fall, whereas the PAR-1 -/- group's ability to perform the test was unchanged.
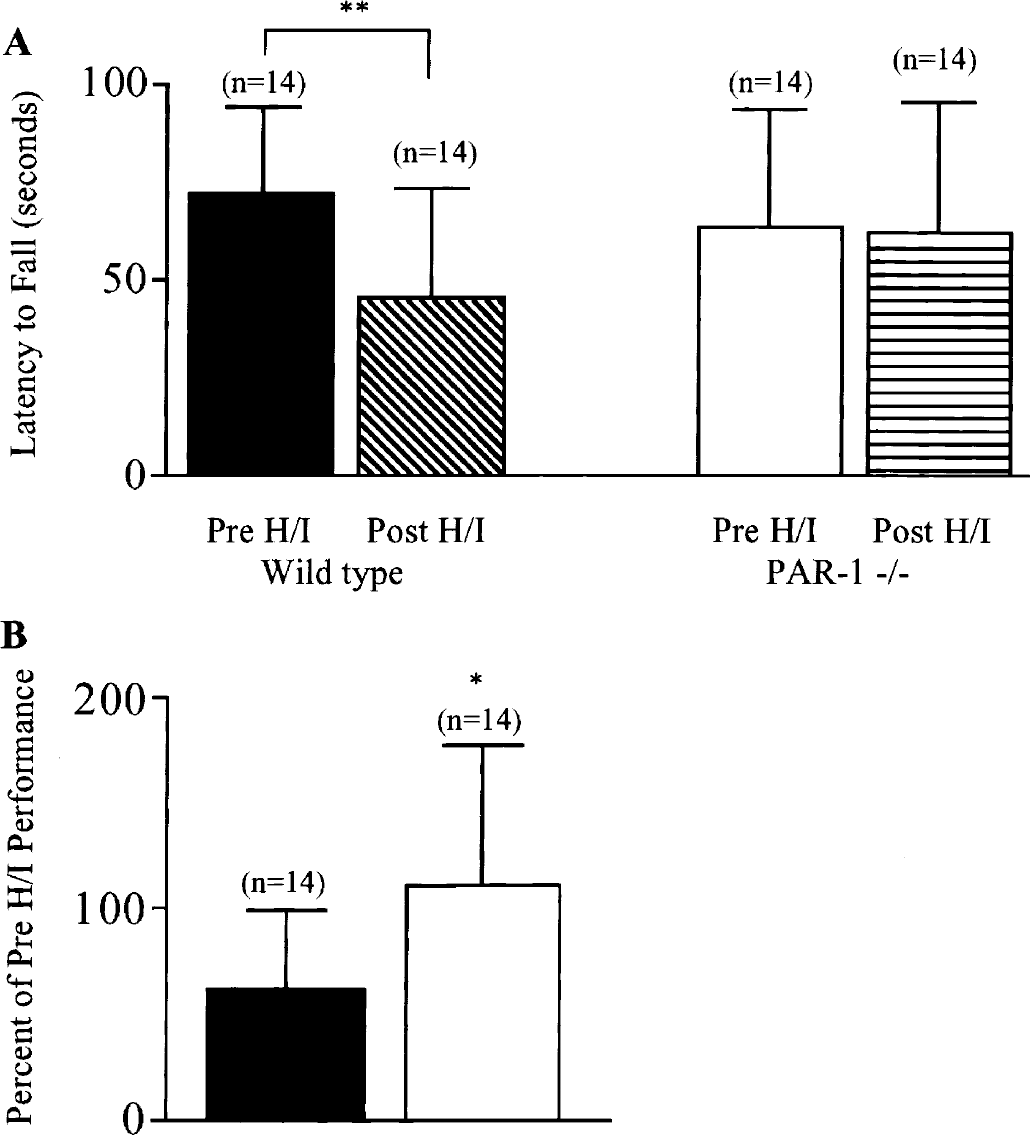
Rotorod testing of sensorimotor deficits after hypoxia/ischemia (H/I). PAR-1 -/- and wild-type mice were tested 1 day before (Pre H/I) and 2 days after (Post H/I) 45 minutes of H/I. Latency to fall (
Both wild-type and PAR-1 -/- mice demonstrated impaired performance on the wire hang test (Fig. 8A). However, the deficit in wild-type mice was significantly more pronounced than in PAR-1 -/- mice (Fig. 8B). Wild-type animals exhibited a nearly 65% reduction in latency to fall; however, PAR-1 -/- animals showed a 30% reduction. The percent change was significantly more pronounced in wild-type animals than PAR-1 -/- animals, suggesting that PAR-1 -/- offers a degree of neurologic protection. Together these data demonstrate that the absence of PAR-1 is neuroprotective and preserves motor function after H/I.
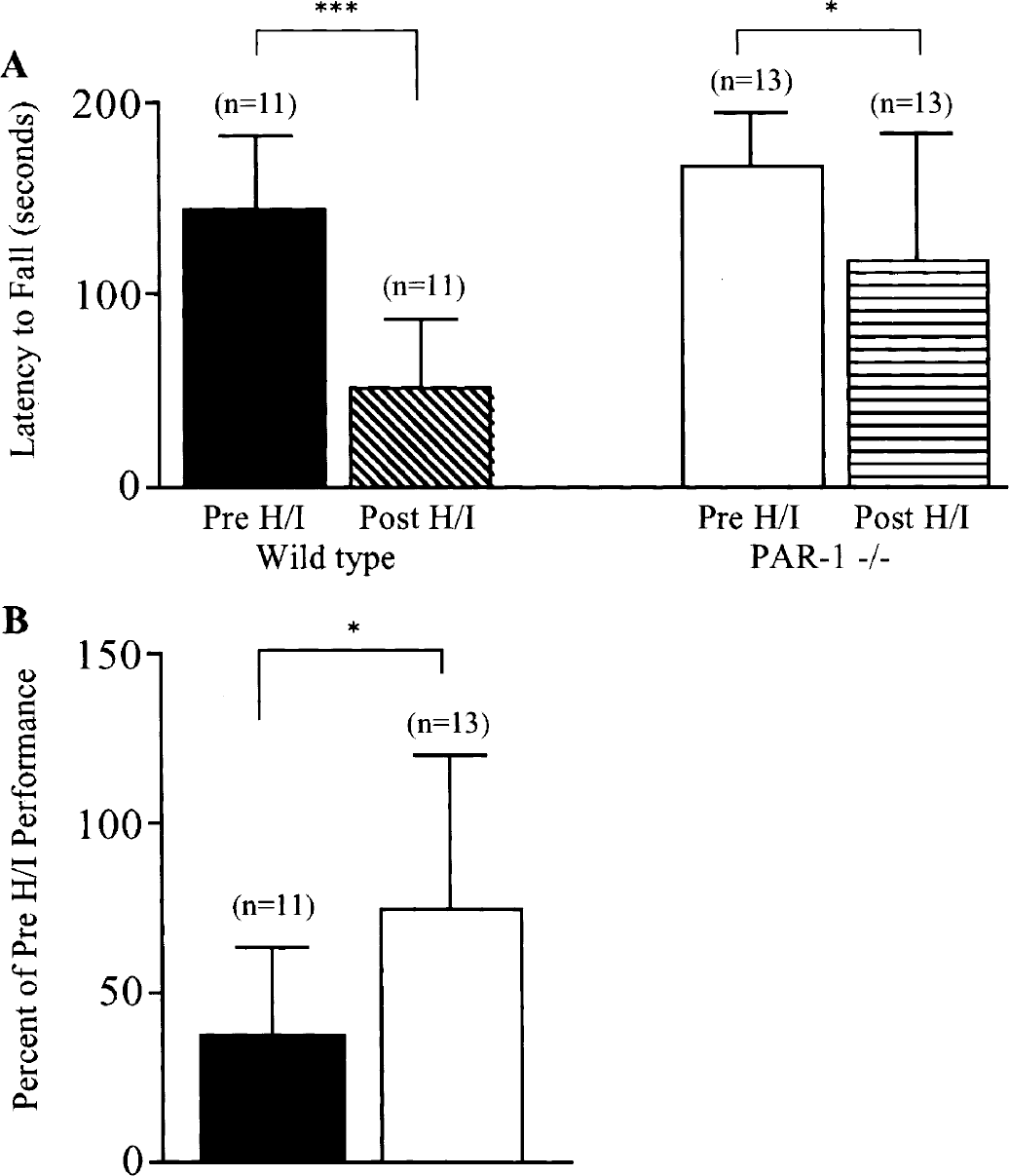
Inverted wire hang testing of motor deficits after hypoxia/ischemia (H/I). PAR-1 -/- and wild-type mice were tested 1 day before (Pre H/I) and 2 days after (Post H/I) 45 minutes of H/I. Latency to fall (
DISCUSSION
PAR-1 deficiency protects against hypoxic/ischemic injury
The most important finding of this study is that PAR-1 activation contributes to neuronal death after hypoxic/ischemic injury. Two lines of evidence support this conclusion. First, PAR-1 null mutant mice have nearly 75% smaller infarct sizes than genetically matched wild-type mice. Second, in wild-type C57Bl/6 mice, a PAR1 antagonist significantly reduced infarct volume. This result is consistent with the results in knockout animals, supporting the working hypothesis that PAR-1 activation contributes to neuronal damage. Furthermore, cresyl violet staining revealed that the reduction of lesion volume in PAR-1 -/- mice involves the sparing of neurons in the cortex and the CA1 region of the hippocampus. As expected, the glial response was more robust in wild type, consistent with their larger lesion volumes.
The Stroke Therapy Academic Industry Roundtable recommends that studies of cerebral injury in animals demonstrate consistency between histological and functional data (STAIR, 1999). Both histopathologic and behavioral assessments are essential, considering that slight cellular changes may not be detected using standard histologic methods. In the rotorod and wire hang tests, PAR-1 deficiency attenuated the motor behavior impairment seen in wild-type controls subjected to hypoxia/ischemia, suggesting that preserving neuronal survival is important for behavioral recovery in this model. Taken together, these data suggest that PAR-1 is an important contributor to the neuropathology and behavioral deficits after hypoxia/ischemia and are consistent with other studies that implicate PAR-1 in neurotoxicity (Debeir et al., 1996; Derian et al., 2003; Festoff et al., 2000; Junge et al., 2003).
Serine proteases and protease receptors in brain injury
Protease-activated receptor-1 is a G-protein-coupled receptor that is activated by serine proteases such as thrombin and plasmin. Plasmin and thrombin are produced via activation of their precursors, prothrombin and plasminogen, respectively. Prothrombin and plasminogen are abundant in circulating blood, but are also present in the brain on neurons and glia (Gingrich and Traynelis, 2000). Proteolytic cleavage of PAR-1 by thrombin or plasmin results in a new amino acid sequence at the N-terminus, which operates as a tethered ligand to initiate signaling. PAR-1 has been shown to couple to Gαi, Gαq, and Gα12/13 families of Gα-proteins in neurons, astrocytes, and peripheral cells to induce a variety of signaling events including adenylyl cyclase inhibition, PI hydrolysis, mitogen-activated protein kinase activation, and calcium mobilization (Grand et al., 1996; Macfarlane et al., 2001; Wang et al., 2002).
Although we did not measure the levels of different serine proteases in this model, both prothrombin and tPA gene expression are upregulated in the ischemic brain (Kim et al., 2002; Riek-Burchardt et al., 2002), and tPA has been shown to enter the brain after ischemia (Wang et al., 1998). In pathologic situations that induce BBB breakdown, circulating prothrombin, plasminogen, and tPA may enter the brain parenchyma and produce active thrombin and plasmin, both of which can activate PAR-1 (Xi et al., 2003). These studies demonstrate that the components that generate activated thrombin or plasmin are present in the brain after ischemia. Additionally, administration of tPA has been shown to impair BBB integrity and allow extravasation of circulating proteins into the brain (Busch et al., 1997). Extravasated proteins will not only affect vulnerable neurons but also astrocytes that help maintain BBB integrity or respond to cellular damage. For example, thrombin-mediated stimulation of PAR-1 induces astrocyte proliferation and downstream activation of ERK 1/2 and calcium mobilization (Motohashi et al., 1997; Nishino et al., 1993; Sorenson et al., 2003; Wang et al., 2002). Thrombin has also been shown to stimulate GFAP expression (Sorenson et al., 2003), consistent with the expression of PAR-1 on astrocytes (Weinstein et al., 1995). These data suggest that PAR-1 activation may contribute to the astrocytic response to injury. In the present study, GFAP levels were reduced in the injured PAR-1 -/- mice compared to wild-type animals. This may be because of the limited amount of damage in these animals after H/I, but may also reflect changes in either astrocytic proliferation or GFAP expression in these animals. This latter possibility is consistent with additional data from our laboratories demonstrating a decreased expression of GFAP after cortical stab injury in PAR-1 -/- mice (not shown).
Within the CNS, serine protease activity and PAR-1 stimulation generate an array of cellular responses, many of which appear to be harmful. Thrombin potentiates NMDA receptor responses in a PAR-1-dependent manner (Gingrich et al., 2000). Given the implication of NMDA-mediated glutamate toxicity in ischemic cell death, these results provide a potential mechanism by which serine proteases may be deleterious to neuronal survival during stroke and H/I. PAR-1 activation via thrombin treatment activates microglia and mediates neuronal damage in the substantia nigra (Carreno-Muller et al., 2003; Choi et al., 2003). Thrombin and PAR-1 activation cause cell death in spinal motoneurons in vitro and in vivo (Turgeon et al., 1998, 1999). Thrombin has also been shown to induce cell death in hippocampal neurons through apoptotic mechanisms and have neurotoxic actions on cultured septal neurons (Debeir et al., 1996; Donovan et al., 1997). Thus, serine proteases appear to play a number of potentially harmful roles after neural injury. Consistent with this idea as well as the data presented here, both removal of PAR-1 or blockade of PAR-1 receptors can reduce infarct volume in a transient model of ischemia (Junge et al., 2003).
Because PAR-1 is expressed on astrocytes, neurons, and endothelial cells, its actions in vivo will likely be complex. As an example of this complexity, inhibition of PAR-1 with neutralizing antibodies blocks the neuroprotective effect of activated protein C (Cheng et al., 2003). Furthermore, several reports confirm that large doses of thrombin are toxic to neurons, whereas low doses stimulate survival and can protect against future insults (Masada et al., 2000; Smith-Swintosky et al., 1995; Striggow et al., 2000). These data suggest that the ability of thrombin pretreatment at low concentrations to protect against later damage, referred to as thrombin preconditioning, was found to depend on activation of p44/42 MAP kinase, the same pathway utilized in astrocyte proliferation (Jiang et al., 2002). This complexity is compounded by the potential interactions between tPA, plasmin, thrombin, and PAR1. Junge et al. (2003) showed that tPA administration can lead to PAR-1 activation in hippocampal slices after cleavage of brain-derived plasminogen, suggesting that some of the neuroprotective effects observed in tPA -/- animals may reflect reduced PAR-1 activation, in addition to other tPA-specific mechanisms (Tsirka et al., 1995; Wang et al., 1998).
Clearly, a better understanding of the mechanisms of protease activation of PAR-1 is important considering the broad actions of PAR-1 and evidence suggesting that PAR-1 activation can have harmful effects. The data presented here support the idea that PAR-1 activation can contribute to neuronal damage and that blockade of PAR-1 in nervous tissue might be a novel therapeutic target for the treatment of a range of pathologic CNS insults.
Footnotes
Abbreviations used
Acknowledgments:
The authors thank Dr. Donald Stein for providing the rotorod apparatus.