
Abstract
Various studies describe increased concentrations of transforming growth factor-β (TGF-β) in brain tissue after acute brain injury. However, the role of endogenously produced TGF-β after brain damage to the CNS remains to be clearly established. Here, the authors examine the influence of TGF-β produced after an episode of cerebral ischemia by injecting a soluble TGF-β type II receptor fused with the Fc region of a human immunoglobulin (TβRIIs-Fc). First, this molecular construct was characterized as a selective antagonist of TGF-β. Then, the authors tested its ability to reverse the effect of TGF-β 1 on excitotoxic cell death in murine cortical cell cultures. The addition of 1 μg/mL of TβRIIs-Fc to the exposure medium antagonized the neuroprotective activity of TGF-β 1 in N-methyl-D-aspartate (NMDA)-induced excitotoxic cell death. These results are consistent with the hypothesis that TGF-β 1 exerts a negative modulatory action on NMDA receptor-mediated excitotoxicity. To determine the role of TGF-β 1 produced in response to brain damage, the authors used a model of an excitotoxic lesion induced by the intrastriatal injection of 75 nmol of NMDA in the presence of 1.5 μg of TβRIIs-Fc. The intrastriatal injection of NMDA was demonstrated to induce an early upregulation of the expression of TGF-β 1 mRNA. Furthermore, when added to the excitotoxin, TβRIIs-Fc increased (by 2.2-fold, P < 0.05) the lesion size. These observations were strengthened by the fact that an intracortical injection of TβRIIs-Fc in rats subjected to a 30-minute reversible cerebral focal ischemia aggravated the volume of infarction. In the group injected with the TGF-β 1 antagonist, a 3.5-fold increase was measured in the infarction size (43.3 ± 9.5 versus 152.8 ± 46.3 mm3; P < 0.05). In conclusion, by antagonizing the influence of TGF-β in brain tissue subjected to excitotoxic or ischemic lesion, the authors markedly exacerbated the resulting extent of necrosis. These results suggest that, in response to such insults, brain tissue responds by the synthesis of a neuroprotective cytokine, TGF-β1, which is involved in the limitation of the extent of the injury. The pharmacologic potentiation of this endogenous defensive mechanism might represent an alternative and novel strategy for the therapy of hypoxic-ischemic cerebral injury.
It is well accepted that the toxic overstimulation of postsynaptic glutamate receptors may contribute to pathologic neuronal death resulting from certain acute insults to the CNS, including hypoglycemia, hypoxia-ischemia, trauma, and spinal cord injury (Choi et al., 1992). Such forms of excitotoxic neuronal death are mediated by the activation of ion channel-linked glutamate receptors, especially N-methyl-D-aspartate (NMDA) receptors (Choi et al., 1992), probably because of their high Ca2+ permeability (Nowak et al., 1984). Considerable interest exists for developing effective methods to attenuate excitotoxicity in various disease states. The most direct approach is to administer a pharmacologic compound able to block glutamate-induced responses. Several glutamate antagonists are under development and clinical trial. However, most of these drugs present relatively harmful side effects, which have limited their potential use in humans (Del Zoppo et al., 1997). Hence, there is an increasing interest for alternative strategies.
Whereas the consequences of the overstimulation of glutamate receptors in the mechanisms that lead to neuronal death have been studied extensively, little is known about the adaptive responses of brain tissue to limit the extent of such insults. Several studies describe an important increase in cytokine concentrations after cerebral ischemia, as well as various neurologic disorders (review Feuerstein et al., 1998). Among these cytokines, transforming growth factor-β (TGF-β) has elicited an increasing interest because of its patterns of expression in altered brain tissue. For example, one study shows an increased expression of TGF-β 1 in the brain after ischemic stroke in humans (Krupinski et al., 1996). Based on this information, TGF-β has been characterized as an injury-related cytokine (for review see Krieglstein and Krieglstein, 1998).
In vitro, several studies describe a neuroprotective role of TGF-β 1 against different metabolic and excitotoxic challenges (Unsicker et al., 1992; Prehn et al., 1993; Klempt et al., 1992). It also has been reported that, under certain conditions, TGF-β 1 is able to potentiate glutamate-induced neuronal injury (Prehn and Krieglstein, 1994; Chao et al., 1992; Prehn and Miller, 1996). These opposing results may reflect the use of different regimens of TGF-β treatment, which would induce multiple consequences, some protective and some deleterious.
In our laboratory, we demonstrated with a model of primary cortical murine cell cultures that TGF-β 1 fails to influence the survival of neurons when exposed to an apoptotic paradigm but exerts a neuroprotective effect mediated by astrocytes and restricted to NMDA-induced necrosis (Buisson et al., 1998).
In vivo, several studies describe a marked increase in TGF-β 1 mRNA after hypoxic-ischemic brain injury (Da Cunha et al., 1993; Prehn et al., 1993; Henrich-Noack et al., 1994; McNeill et al., 1994; Rimaniol et al., 1995; Vivien et al., 1998). Whereas the intracerebroventricular application of TGF-β 1 induces a slight reduction of the ischemic volume (Prehn and Krieglstein, 1994; McNeill et al., 1994), there is no clear evidence for the influence of the endogenously produced TGF-β, mainly because of the lack of a potent antagonist.
Transforming growth factor-β elicits its effects through cell surface receptors. Three major types of binding proteins are known to be ubiquitously distributed in TGF-β-responsive cells. They are referred as type I (TβR-I), type II (TβR-II), and type III (betaglycan; TβR-III) receptors. Both TβR-I and TβR-II are glycoproteins of 53 and 75 to 85 kDa, respectively, whereas TβR-III is a proteoglycan of 280 to 330 kDa (Massagué et al., 1994). In the murine brain, TGF-β type I and II receptors are expressed on both neurons and astrocytes (Vivien et al., 1998). Molecular cloning and functional analyses show that both TβR-I and TβR-II are serine/threonine kinase receptors necessary for the signal transduction of TGF-β (Massagué et al., 1994). The TGF-β type II receptor binds to its proper ligand, but the type I receptor requires the presence of bound TβR-II to interact with TGF-β (Wrana et al., 1992). After ligand-receptor interaction, TβR-I and TβR-II form a heteromeric complex (Wrana et al., 1992; Vivien et al., 1995), and the TβR-II so bound is able to transphosphorylate TβR-I (Wrana et al., 1992; Vivien and Wrana, 1995). Thereafter, the signaling of TGF-β occurs through the activation of Smad 2/3 and Smad 4 transcriptional factors (Kim et al., 1997; Dennler et al., 1998). Because the first step of TGF-β signaling requires an interaction between TGF-β and its type I and type II receptors, we used a molecular construct made of the extracellular domain of TGF-β type II receptor fused with the Fc region of human immunoglob-ulins (TβRIIs-Fc). This construct has been characterized as a powerful blocker of the interaction between TGF-β and its cell surface receptors (Komesli et al., 1998).
By injecting TβRIIs-Fc into brain tissue exposed to an excitotoxic insult or to a reversible cerebral ischemia, we were able to study, for the first time, the role of endogenously produced TGF-β.
MATERIALS AND METHODS
Materials
Polymerase chain reaction (PCR) kits were purchased from Life Technologies (Cergy Pontoise, France), and the reverse transcriptase system kit was obtained from Promega (Paris, France). The NMDA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA), kainate, and (+)5-methyl-10,11-dihydro-5H-dibenzo(a,d)cyclohepten-5,10-imine maleate were from Tocris (Bristol, U.K.). Eagle's minimal essential medium, horse serum, and fetal bovine serum were obtained from Sigma Chemical Co. (Isle D'Abeau, France); poly-d-lysine and laminin were from Life Technologies. The TGF-β was purchased from R & D System (Oxford, U.K.). All other chemicals used were obtained from Sigma Chemical Co..
Cell culture
Mixed cortical cell cultures containing both neuronal and glial elements were prepared from fetal mice at 15 to 16 days' gestation (Rose et al., 1993). Cerebral cortices were dissected and incubated for 20 to 30 minutes in 0.025% of trypsin in media stock (MS; modified Eagle's minimal essential medium with 2 mmol/L glutamine and 25 mmol/L glucose) and transferred to MS supplemented with 5% fetal bovine and 5% horse serum for trituration. Glial cell cultures from cortex were prepared similarly from 1- to 3-day-old postnatal pups (Rose et al., 1993) and plated in MS supplemented with 10% fetal bovine serum and 10% horse serum. Dissociated cortical fetal cells were plated at a density of about 3 × 105 cells per well on an established bed of glia in the same plating medium.
All cultures were kept at 37°C in a humidified 5% CO2-containing atmosphere. After 3 to 7 days in vitro, glial division was halted by exposure to 10 μmol/L cytosine arabinoside. Cells subsequently were shifted into a maintenance medium that was identical to that of the plating medium but lacking fetal bovine serum. The medium was changed twice weekly. Experiments were performed on cortical cultures after 14 to 15 days in vitro.
Slowly triggered excitotoxicity was carried out at 37°C and was induced by a 24-hour exposure to a low concentration of NMDA (12.5 μmol/L) in MS supplemented with glycine (10 μmol/L).
Neuronal cell death was estimated by examination of the cultures using phase-contrast microscopic study and measurement of lactate dehydrogenase (LDH) released by damaged cells into the bathing medium 1 day after experimentation (Koh and Choi, 1987). The LDH signal, corresponding to complete neuronal death, was determined by assaying sister cultures exposed to 100 μmol/L NMDA for 24 hours in MS supplemented with glycine. Background LDH levels were determined in sister cultures subjected to sham wash and subtracted from the levels in experimental conditions to yield the LDH signal specific to a given experimental injury.
Expression and purification of TbRIIs-Fc
The cDNA encoding the extracellular domain of human TbRII was amplified by PCR and cloned in the polylinker between EcoRI (position 2209 base pairs [bp]) and BamHI (position 2226 bp) sites of the vector plg-Tail (R & D System) containing a hygromycin-selectable marker. The cassette for hygromycin resistance was purified from Selecta Vecta-Hyg (R & D System) after digestion with EcoRV (sites at positions 14 and 1679 bp) and subcloned in pIg-Tail vector previously digested by Scal (site position 4585 bp). The restriction enzymes EcoRV and Scal both generated blunt ends, which could be joined using T4 DNA ligase (Fig. 1A). The pIg-Tail expression system enables the mammalian production of fusion proteins with a C-terminal Fc tail, and hygromycin-selectable marker enables selection of stable transfected cell lines. The chimeric protein TbRIIs-Fc was expressed into the medium of cultured Chinese hamster ovary cells (American Type Culture Collection CRL 9618) after stable transfection with modified recombinant pIg-Tail vector. The selected clone was cultured in AIMV medium (Gibco BRL Life Technologies) in which it loses its adherent properties. The TβRIIs-Fc was purified from the medium by one-step protein A affinity chromatography and lyophilized.
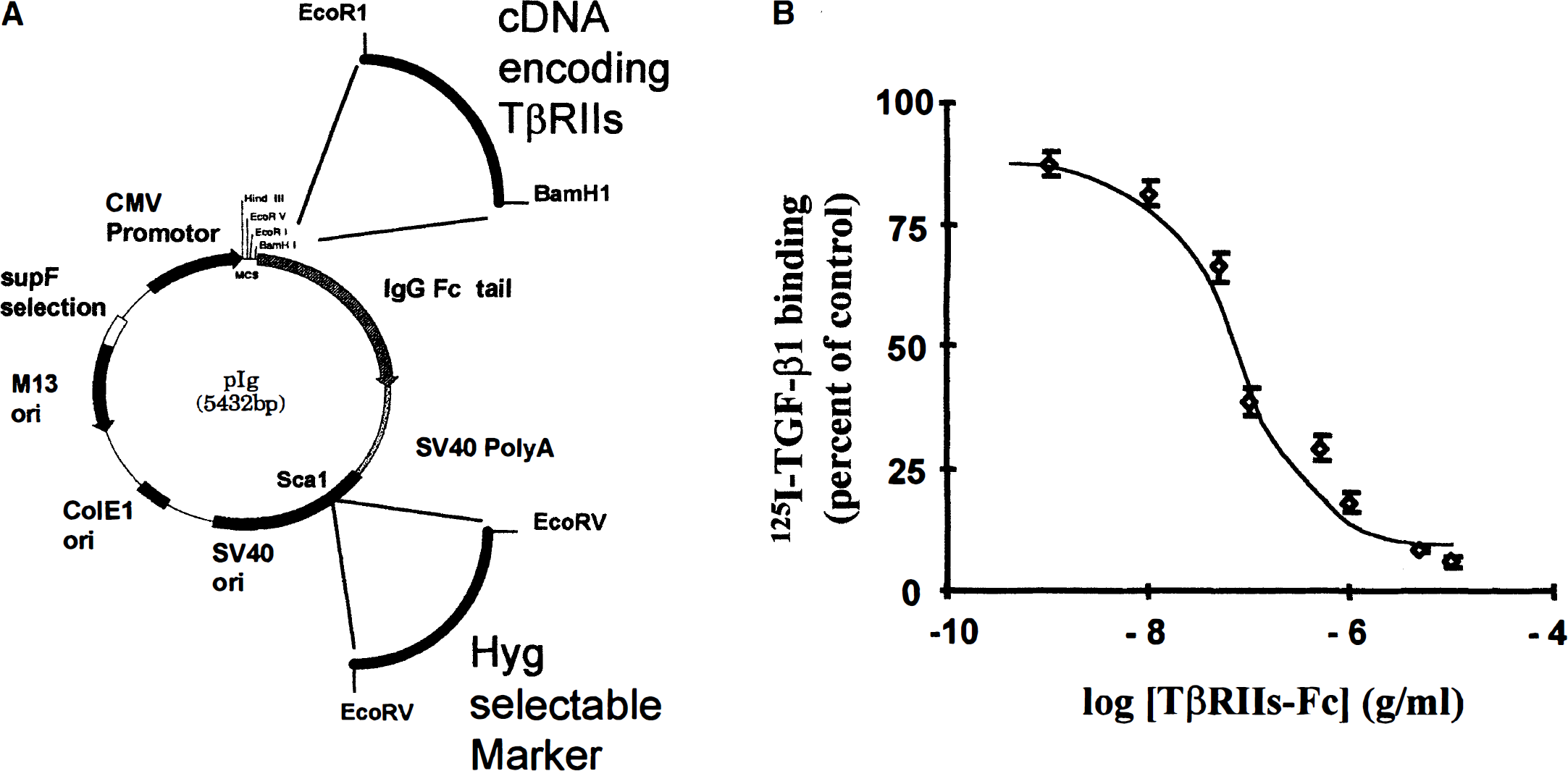
A soluble transforming growth factor-β (TGF-β) type II receptor used as a TGF-β1 antagonist.
Competition of 125I-TGF-β1 binding on Mv1Lu cells by TβRIIs-Fc
The Mv1Lu cell line (American Type culture collection CCL-64) was maintained in Eagle's minimal essential medium supplemented with 10% fetal calf serum (Life Technologies).
The 125I-TGFβ1 (NEN, Life Sciences Products, Les Ulis, France) binding to monolayer cells was performed according to published procedures (Massagué J, 1987). For the competition binding assay, the final concentration of radiolabeled ligand was 40 pmol/L (specific activity 3000 to 4500 Ci/mmol), with TbRIIs-Fc final concentration ranging from 0.001 to 10 mg/mL.
Semiquantitative reverse transcriptase polymerase chain reaction
Total RNA samples were prepared from cerebral cortex by a phenol/chloroform extraction method using the RNA Ble extraction kit (Eurobio, Paris, France). Samples (1 μg) of total RNA were transcribed into cDNA using poly-dT oligonucleotides. For the semiquantitative experiments, an aliquot of cDNA libraries (1μL from 20 μL) was amplified by PCR with specific oligonucleotides for β-actin (sense 5′ GTG GGC CGC TCT AGG CAC AA 3′ [25 to 45 bp] and antisense 5′ CTC TTT GAT GTC ACG CAC GAT TTC 3′ [564 to 540 bp]; 30 cycles of PCR were chosen corresponding to the 50% of the saturation curve of the PCR product). Then, an aliquot (1 μL from 20 μL) of the same cDNA libraries was amplified with sets of specific oligonucleotides for TGF-β1 (sense 5′ CCG AAG CTT GCC CTG GAC ACC AAC TAT TGC 3′ [808 to 838 bp] for antisense 5′ CCG TCT AGA GCT GCA CTT GCA GGA GCG 3′ [1201 to 1174 bp]). Amplification was performed in a Thermolyne thermocycler with the Gibco BRL PCR kit. Conditions of amplification were 30 seconds at 95°C, 30 seconds at 55°C, and 1 minute at 72°C. Amplified products were separated by agarose gel electrophoresis and visualized by ethidium bromide staining.
In vivo procedures
All experiments were performed on male Sprague-Dawley rats weighing 280 to 320 g provided from CERJ (France). Anesthesia was induced by halothane at 4% and maintained at 1% to 1.5% administrated through a vaporizer in a gas mixture of 30% O2 to 70% N2O.
Excitotoxic lesions
Rats received a unilateral injection into the left striatum: coordinates 0.2 mm posterior, 3 mm lateral, 5.5 mm ventral to the bregma (Paxinos et al., 1985) with either vehicle (phosphate-buffered saline, pH 7.4, n = 4), NMDA (75 nmol in 3 (μL phosphate-buffered saline, pH 7.4, n = 6), 75 nmol NMDA + 3 ng TGF-β1 (n = 6), 75 nmol NMDA + 1.5 μg TβRIIs-Fc (n = 6), or 1.5 βg TβRIIs-Fc alone (n = 4). Five minutes after the insertion of the needle, the solution was injected over the next 6 minutes using a Hamilton syringe pump at a rate of 0.5 μL/min. The needle was removed 5 minutes later. After 24 hours, rats were killed, and the brains were removed and frozen in isopentane for histologic analysis.
Middle cerebral artery occlusion
All of the rats used for the ischemia procedure were intubated, and a femoral artery was cannulated for continuous arterial pressure monitoring and gas analysis. Body temperature was maintained at 37.5° ± 0.2°C with a heating pad. Thirty minutes before the middle cerebral artery occlusion (MCAO), the rats received a 3-μL intracortical injection (coordinates: 0.2 mm posterior, 5.5 mm lateral, 5.5 mm ventral to the bregma) of either vehicle (phosphate-buffered saline, n = 6) or 1.5 μg of TβRIIs-Fc (n = 6). A control experiment was performed by the intracortical injection of TβRIIs-Fc in the absence of MCAO (n = 3).
Focal cerebral ischemia was induced by the intraluminal MCAO method (Longa et al., 1989). Briefly, a midline neck incision was made, and the right common carotid artery was exposed. After coagulation of its branches, the external carotid artery was distally severed. A nylon thread (0.18 mm in diameter), which had a distal cylinder (3 mm long and 0.38 mm diameter) of thermofusible glue (3M:3764, Radiospares Composantes, France), was inserted in the lumen of the external carotid artery and pushed into the internal carotid artery up to the origin of the middle cerebral artery. To restore the middle cerebral artery blood flow, the nylon thread was removed and cut 30 minutes later.
Histologic analysis
Twenty-four hours after the beginning of surgery, euthanasia was performed by vertebral dislocation; the brains were rapidly removed, frozen in isopentane at −50°C, and stored at −80°C. Cryostat-cut coronal brain sections (20 μm) were stained with thionine and analyzed using an image analyzer. The lesioned areas were delimited by the paleness of histologic staining in altered tissue compared with the color of healthy tissue. Regions of interest were determined through the use of a stereotaxic atlas for the rat (Paxinos and Watson, 1982), and an image analysis system (BIOCOM RAG 200, Paris, France) was used to measure the lesioned area.
RESULTS
The soluble TGF-β type II receptor prevents the interaction of TGF-β with its endogenous receptors.
Transforming growth factor-β type I and type II receptors form a ligand-dependent heteromeric signaling complex, in which the TGF-β type II receptor tends to act as the primary and necessary receptor. In the current study, we used the extracellular domain of the TGF-β type II receptor fused with the Fc region of a human immunoglobulin (TβRIIs-Fc) as a soluble TGF-β antagonist. The TβRIIs-Fc is a product with a molecular mass around 45 to 65 kDa, which corresponds to the purified recombinant protein. Electrophoresis analysis performed without reducing agent showed a product of molecular mass around 90 to 130 kDa, corresponding to the homodimeric form associated through its Fc domains (Komesli et al., 1998). To produce this molecular construct on a large scale, CHO cells were stably transfected with the modified expression vector, pIg-Tail, containing the cDNA encoding the truncated TGF-β type II receptor (amino acid residues 1 to 159) and a cassette for hygromycin resistance, as described in Fig. 1a. After one-step purification by affinity chromatography on protein-A sepharose, a pure soluble chimeric protein was recovered from the culture media of CHO cells. Western blot analysis showed that the purified recombinant protein was recognized both by a polyclonal antibody raised against the extracellular domain of TβRII and a polyclonal antibody raised against the human Fc fragment (data not shown). To characterize the antagonistic activity of recombinant chimeric TβRIIs-Fc, we performed a binding experiment between membrane-bound 125l-labeled TGF-β1 and serial dilutions of TβRIIs-Fc (0.001 to 10 mg) in mink lung epithelial cells, a cell line that expresses both TGF-β receptors on its cell surface (Massagué et al., 1994) (Fig. 1B). As shown in the Fig. 1B, the addition of increasing concentrations of TβRIIs-Fc was able to counteract the binding of 125 l-labeled TGF-β to its receptors (CI-50 of 0.1 μg/mL).
In vitro, the neuroprotective effect of TGF-β1 against NMDA-induced necrosis is prevented by a soluble TGF-β type II receptor.
First, we tested the influence of TGF-β1 on various types of excitoxic stress thought to mimic the overstimulation of the postsynaptic glutamate receptors observed in hypoxic-ischemic insults. We used the following glutamatergic agonists: NMDA, AMPA, and kainate (Choi et al., 1992). The exposure of mixed cortical neuron-glia cultures (days in vitro 14) to these agonists produced an acute swelling of neuronal bodies followed 24 hours later by widespread neuronal degeneration, whereas the glia remained intact. The TGF-β1 (1 ng/mL) selectively reduced the NMDA-induced necrosis, whereas it had no effect on either AMPA- or kainate-mediated toxicity (Table 1).
TGF-β1 selectively protects neurons against NMDA-induced necrois in mixed neuron/glia cultures
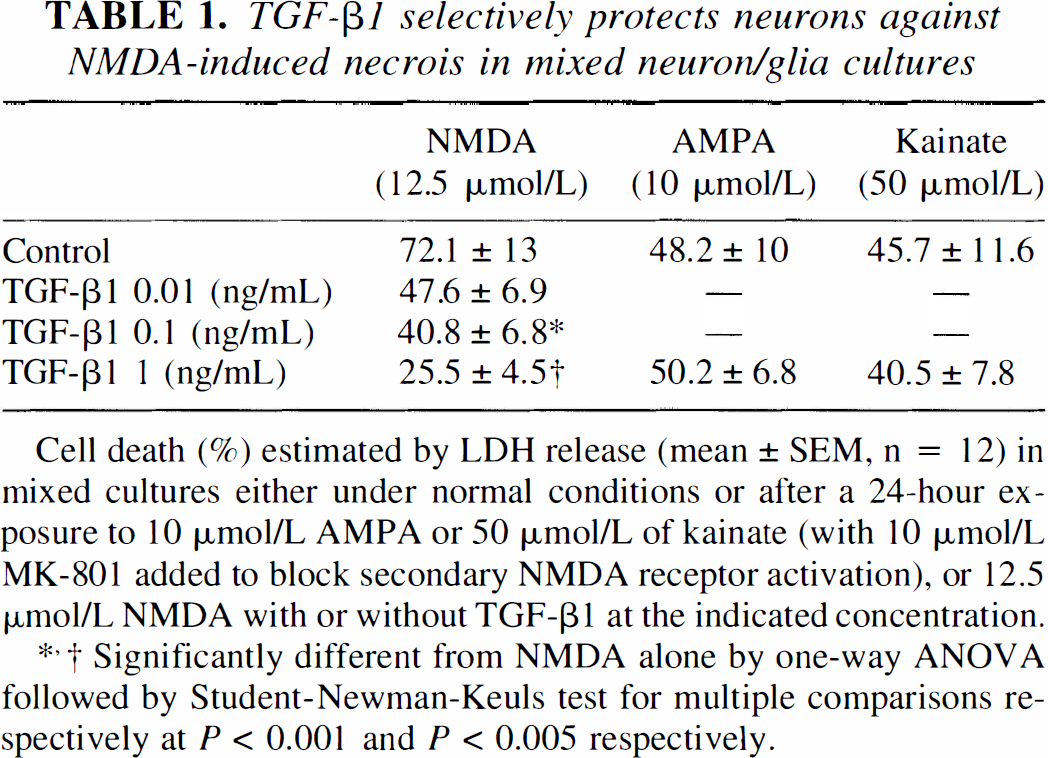
Cell death (%) estimated by LDH release (mean ± SEM, n = 12) in mixed cultures either under normal conditions or after a 24-hour exposure to 10 μmol/L AMPA or 50 μmol/L of kainate (with 10 μmol/L MK-801 added to block secondary NMDA receptor activation), or 12.5 μmol/L NMDA with or without TGF-β1 at the indicated concentration.
Significantly different from NMDA alone by one-way ANOVA followed by Student-Newman-Keuls test for multiple comparisons respectively at P < 0.001 and P < 0.005 respectively.
We then tested the antagonistic activity of TβRIIs-Fc on TGF-β1-induced neuroprotection. When neurons were exposed to NMDA (12.5 μmol/L) for 24 hours, 80% of the cells were killed. The addition of TGF-β1 induced a dose-dependent decrease in neuronal cell death (Table 1). This neuroprotective effect induced by TGF-β1 was abolished by TβRIIs-Fc (1 μg/mL). The TbRIIsFc alone failed to exert any deleterious effect (Fig. 2).
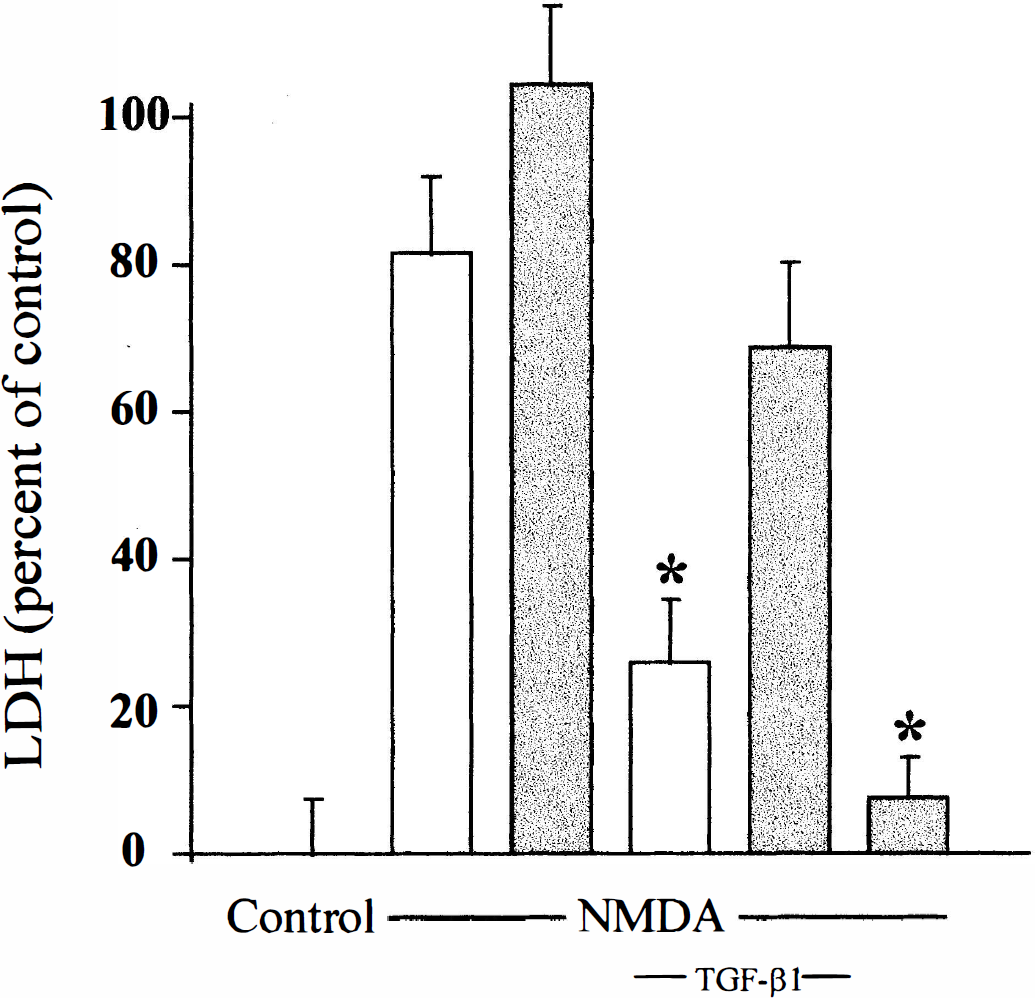
The TβRIIs-Fc receptor antagonizes the TGF-β neuroprotective effect against N-methyl-d-aspartate (NMDA)-induced neuronal death in vitro. Cell death (percent) is estimated by lactate dehydrogenase (LDH) release (mean ± SD, n = 12) in mixed cultures either under control conditions or after a 24-hour exposure to 12.5 μmol/L of NMDA with or without 1 ng/mL of TGF-β1, which was or was not coincubated with the soluble TGF-β type II receptor (1 μg/mL) (shaded columns). Asterisk indicates values significantly different from NMDA alone or TGF-β1 alone, respectively, by one-way analysis of variance followed by Student-Newman-Keuls test for multiple comparisons (P < 0.05).
Overexpression of TGF-β1 mRNA after intrastriatal injection of NMDA
Total RNA samples (1 μg) from rat striatum were harvested and submitted to reverse transcription using poly-dT oligonucleotides. Then, 1 μL (one twentieth) of each cDNA sample was amplified by PCR using oligonucleotides for TGF-β1 and β-actin studied under the same conditions of amplification. Each set of oligonucleotides gave the product of the expected size (318 bp for TGF-β1 and 539 bp for β-actin). To further clarify the specificity of the PCR products, digest controls through the use of the internal restriction sites of our expected PCR products were performed (data not shown). All of our PCR reactions showed the same level of expression of the chosen housekeeping gene (β-actin) (Fig. 3). Furthermore, a PCR control using β-actin-specific primers was performed with a representative RNA sample harvested from cerebral cortices of mice to rule out any genomic DNA (data not shown).
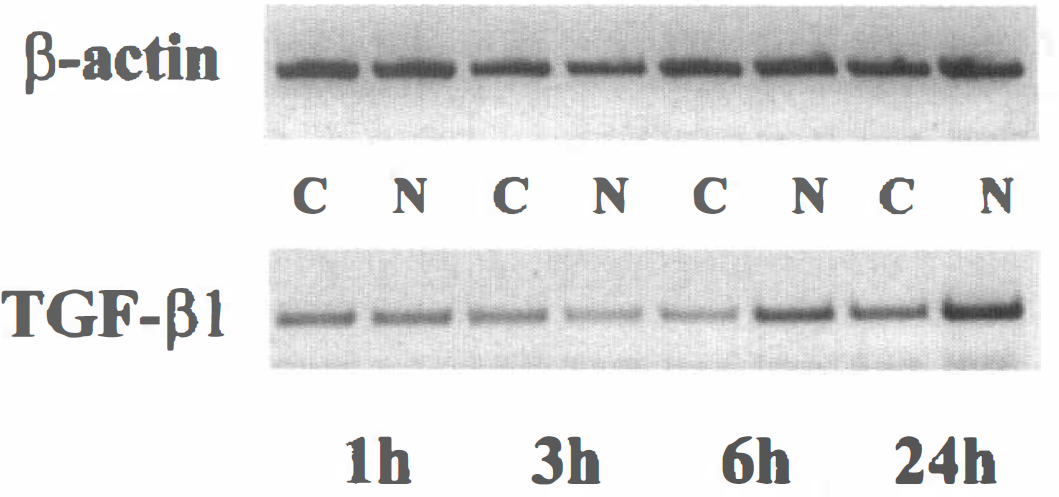
Intrastriatal injection of NMDA induces an overexpression of TGF-β1 mRNA. Total RNA samples from injected (N) and contralateral (C) striata were harvested at the indicated times and analyzed by reverse transcriptase PCR for TGF-β1 and β-actin, as described in the methods section.
Total mRNA samples were harvested from both ipsilateral and contralateral striata 24 hours after the injection of NMDA. As observed in Fig. 3, the expression of TGF-β1 mRNA was clearly enhanced as early as 6 hours and maintained until 24 hours in the striatum subjected to the NMDA injection.
TGF-β antagonist potentiates the NMDA-induced necrosis in vivo
To investigate the influence of TGF-β1 produced in response to an NMDA-induced necrosis, we performed a coinjection of NMDA with either recombinant TGF-β1 or TβRIIs-Fc in the rat striatum. The NMDA alone (75 nmol in 3 μL of phosphate-buffered saline) induced a highly reproducible lesion, which was measured 24 hours later (27 ± 7 mm3, mean ± SD, n = 8). Coadministration of the recombinant human TGF-β1 (3 ng) with NMDA reduced the lesion volume by 42%, thus confirming the beneficial influence observed in vitro for TGF-β1 against NMDA-induced necrosis (15.7 ± 4.5 mm3, mean ± SD, n = 8, P < 0.005).
Moreover, when we coinjected NMDA and TβRIIs-Fc (1.5 μg in 3 μL), the excitotoxic lesion volume was increased by 90% (51.4 ± 11.3 mm3, mean ± SD, n = 8, P < 0.001) (Fig. 4).
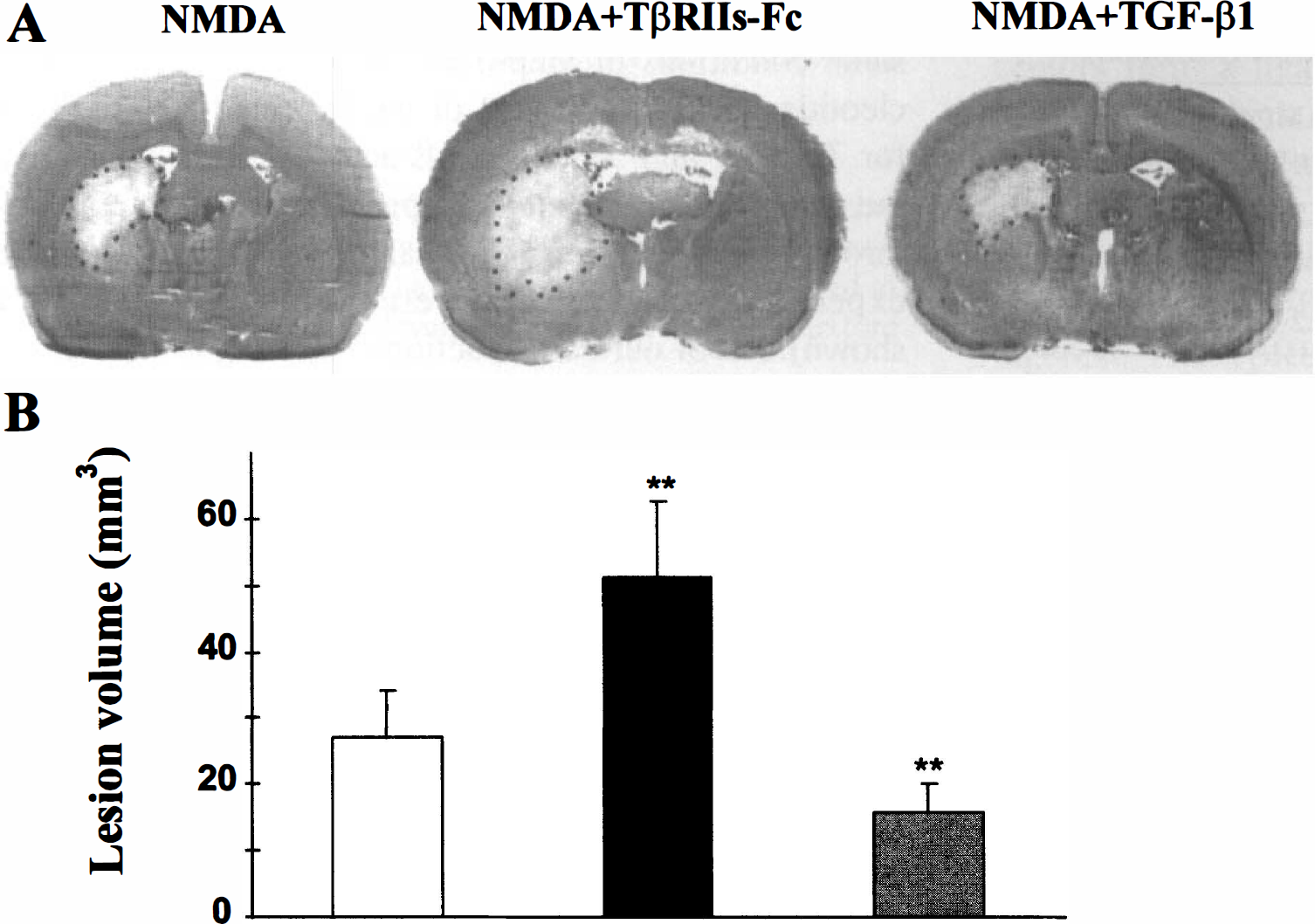
The TβRIIs-Fc receptor potentiates the extent of NMDA-induced lesion in vivo.
To rule out the possibility that the effect observed in the presence of the soluble TβRII receptor could result from the Fc fragment of human immunoglobulin, we injected NMDA in the presence of 1.5 μg of the whole human immunoglobulin (IgG). The IgG did not modify the extent of the lesion. Similarly, the soluble TβRII receptor alone produced only a discrete lesion (4.1 ± 0.5 mm3, mean ± SD, n = 4) (data not shown).
Overexpression of TGF-β1 mRNA after transient middle cerebral artery occlusion
The interruption of cerebral perfusion that provokes stroke initiates a cascade of changes in the extracellular compartment of the brain. Here, we demonstrate modifications in the level of expression of TGF-β1 mRNA after a 30-minute transient focal ischemia induced by MCAO in the rat (Fig. 5). Total RNA samples from ipsilateral and contralateral cortices and striatum were harvested. The RNA samples (1 μg) were reverse transcribed with poly-dT oligonucleotides. An aliquot of cDNA libraries (1 μL from 20 μL) was amplified by PCR with specific oligonucleotides for β-actin. Then, 1 μL of the same cDNA libraries was amplified with sets of specific oligonucleotides. As shown in Fig. 5, all of our PCR reactions showed the same level of expression of the chosen housekeeping gene (β-actin). The TGF-β1 mRNA in the injured striatum was clearly enhanced 3 hours after the transient MCAO and maintained 3 days after ischemia. Similarly, an enhanced expression of the TGF-βl mRNA was observed in the cortex ipsilateral to MCAO. The cortical expression of the TGF-β1 mRNA was clearly enhanced 6 hours after the transient MCAO and maintained 3 days after the insult.
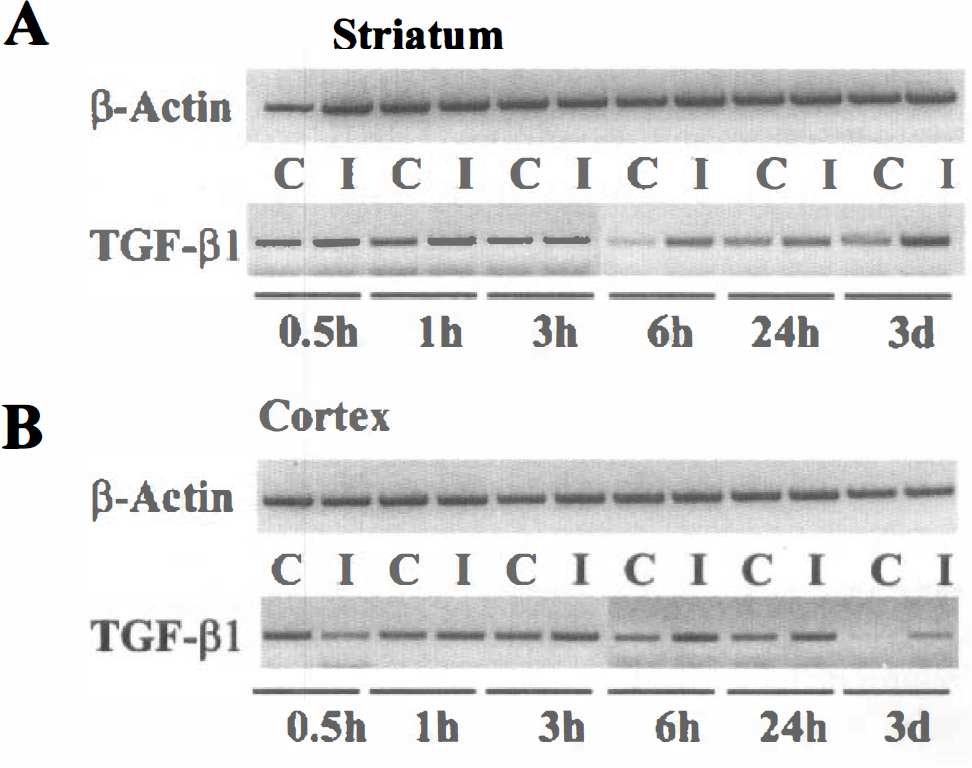
Transient middle cerebral artery occlusion induces overexpression of TGF-β1 mRNA both in injured striatum and cortex. Total RNA from ischemic ipsilateral (I) and contralateral (C) striata
Effect of an intracortical injection of the soluble TβRII receptor on the extent of infarction induced by a reversible focal ischemia
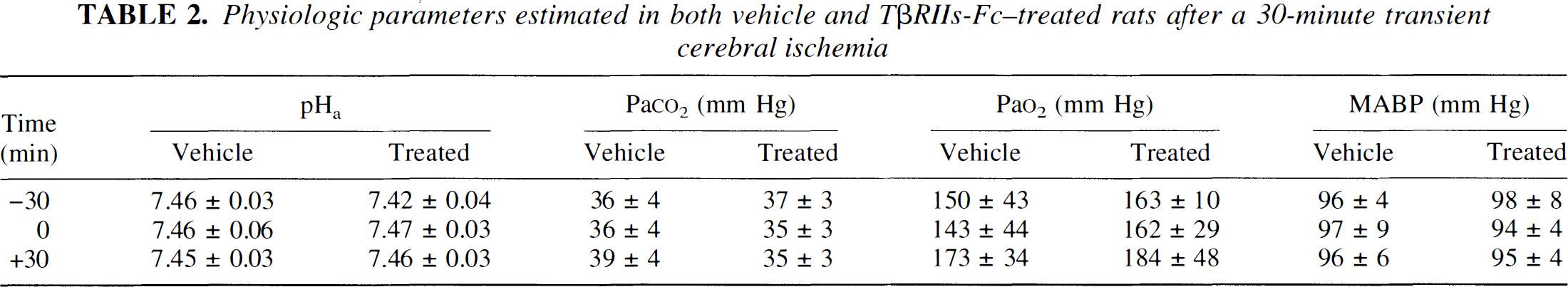
A 30-minute occlusion of the middle cerebral artery induces a highly reproducible infarction affecting both the cortex and the striatum. When we injected into the cortex 3μL of vehicle (phosphate-buffered solution) 30 minutes before transient occlusion of the middle cerebral artery, we measured a total infarcted volume of 43.3 ± 9.5 mm3 (n = 6). The lesion was primarily striatal (30.2 ± 10.5 mm3, mean ± SD), whereas the cortical lesion was more limited (7.7 ± 29.4 mm3, mean ± SD). The intracortical injection of TβRIIs-Fc markedly potentiated the total volume of the ischemic lesion (152.8 ± 46.3 mm3, mean ± SD, n = 6, P < 0.0001), whereas the injection of the soluble TβRII receptor in the absence of the ischemic insult induced only a small lesion (2.0 ± 0.5 mm3, mean ± SD, n = 3). This exacerbation induced by the soluble type II receptor for TGF-β was exclusively localized in the cortex (109.9 ± 32.3 mm3, mean ± SD, P < 0.0001), since there was no difference in the striatal lesion (Fig. 6). During these experiments, arterial pressure, pH, Po2 and Pco2 pressure were monitored. There was no differences in the physiologic parameters between the two experimental groups (Table 2).
Physiologic parameters estimated in both vehicle and TβRIIs-Fc-treated rats after a 30-minute transient cerebral ischemia
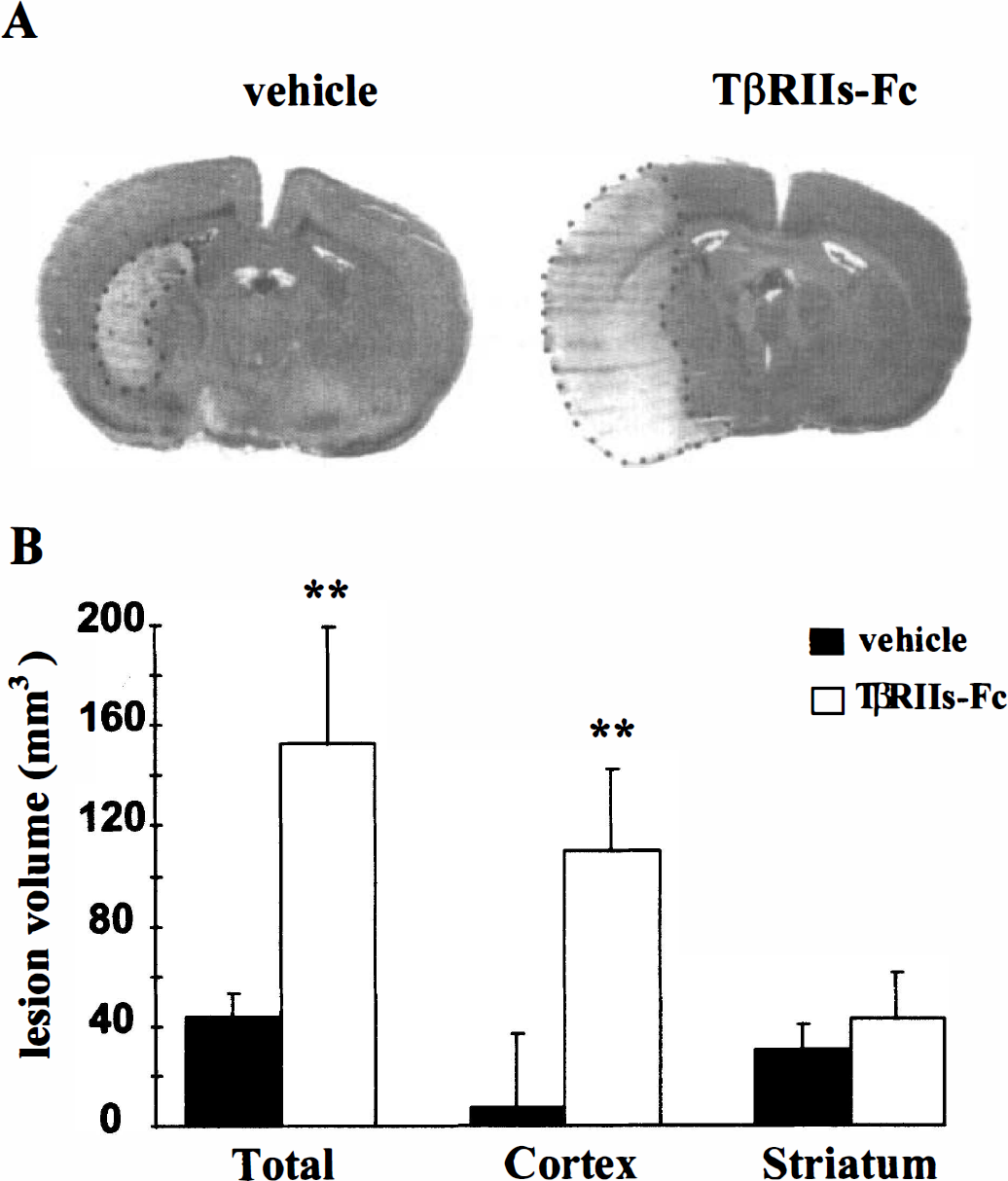
The TβRIIs-Fc receptor potentiates the extent of the infarct after transient cerebral ischemia in rats.
DISCUSSION
In the current study, we demonstrate that the expression of the mRNA for TGF-β1 increases after an excitotoxicity-induced or an ischemia-induced brain lesion. The observation that the expression of the mRNA for TGF-β1 is dramatically enhanced after brain injury is consistent with previous studies (Prehn et al., 1993; Vivien et al., 1998). However, to exert its biological activity, the protein TGF-β1 needs to be activated. In fact, TGF-β1 is secreted in a latent form made of a TGF-β1 active dimer associated with a set of proteins (Harpel et al., 1992; Souchelnitsky et al., 1995). The activation of this complex is an essential step for this cytokine to exert its activity (Wrana et al., 1994; Vivien and Wrana, 1995). Thus, the presence of high levels of mRNA does not predict the implication of this cytokine in the outcome of the injury. To investigate the role of active TGF-β1, it was necessary to antagonize its biological action. Since there is no synthetic and selective antagonist available, we developed a soluble TGF-β type II receptor that was characterized for its ability to prevent the interaction of TGF-β with its native receptors. First, we studied the influence of this soluble receptor in a model of excitotoxicity in cortical murine cultures. We confirm previous observations that TGF-β1 reduces the neuronal death induced by NMDA (Prehn et al., 1993; Henrich-Noack et al., 1996). Moreover, the neuroprotective effect of TGF-β1 is reversed by the soluble TβRII receptor, thus validating the functional antagonist property of this construct on TGF-β1 signaling.
The core investigation of this study was to determine the effect of TβRIIs-Fc on an excitotoxic lesion induced by the intrastriatal injection of NMDA in rats. Recombinant TGF-β1 reduces the NMDA-induced lesion size by 40% and would thus support the neuroprotective effect observed in vitro. Soluble TβRII provokes an increase of the NMDA-induced lesion by 92%. Even if injection of the soluble receptor TbRIIs-Fc alone induces a small lesion, the increase in the lesion size observed cannot be explained by simple additive effects. Thus, these results demonstrate that a cerebral tissue exposed to an excitotoxic stress is able to produce a cytokine that limits the extent of the injury.
Excitotoxicity may contribute in an important manner to several forms of acute injury to the nervous system, including stroke. Accordingly, it was thought essential to evaluate the influence of TGF-β1 in an experimental model of cerebral ischemia. By injecting the TβRIIs-Fc into the cortex 30 minutes before the induction of a reversible focal cerebral ischemia, we were able to selectively block the influence of endogenously produced TGF-β1 in this structure. This strategy resulted in a dramatic enhancement of the volume of damaged cortical tissue. These findings indicate that the activation of TGF-β receptors modulates NMDA-induced neuronal cell death both in vitro and in vivo.
The role of TGF-β in brain diseases has not been adequately resolved. This cytokine has been proposed to exert antiapoptotic effects in vitro (McNeill et al., 1994; Henrich-Noack et al., 1996) and to modulate astrocytic reactivity (Lindholm et al., 1992), both of which might suggest a possible direct or indirect effect of TGF-β on the survival of neurons. In addition, this factor is involved in the deposition of the extracellular matrix (Westerhausen et al., 1991) and thus increases wound healing. Also, TGF-β may be involved in antiinflammatory processes (Kulkarni et al., 1993, McNeill et al., 1994). Although Prehn et al. (1993) describe that the intracerebroventricular injection of recombinant TGF-β1 induces a slight reduction in infarct size after cerebral focal ischemia in mice, no clear evidence of a role for endogenous TGF-β has been proposed. The major finding of the current experiments is that an intrastriatal injection of a recombinant antagonist for TGF-β, TβRIIs-Fc, dramatically enhances an NMDA-induced lesion. This observation is the first demonstration that a cytokine, produced in response to an excitotoxic stimulus, exerts a neuroprotective activity. This result is strengthened by the beneficial activity of exogenously applied TGF-β, observed in vivo, against NMDA-induced lesion. These data strongly indicate that the activation of the TGF-β signaling pathway initiates a cascade of events beneficial for brain tissue exposed to an excitotoxic stress. The early activation of this pathway or its amplification might represent a novel therapeutic approach for the treatment of acute brain injury. However, the molecular mechanisms underlie the neuroprotective effect of TGF-β1 in vivo remain to be elucidated. Previously, we demonstrated in vitro that TGF-β1 may exert its neuroprotective effect by upregulating a protein synthesized by astrocytes: the plasminogen activator inhibitor-1, a protein characterized by its ability to inhibit tissue-type plasminogen activator activity (Inagaki et al., 1994). How might tissue-type plasminogen activator be involved in excitotoxic injury? The question remains unresolved, but experimental data obtained from tissue-type plasminogen activator knock-out mice reveal that such animals are relatively resistant to an excitotoxic stress (Tsirka et al., 1997) and to ischemia (Wang et al., 1998). Finally, the current study validates the use of recombinant soluble receptors as a powerful tool to investigate the role of cytokines in vivo. A better knowledge of the influence of various cytokines, including TGF-β produced by injured tissue after cerebral ischemia, may promote several innovative therapeutic approaches.