
Abstract
Experimental studies have shown the role of excitotoxicity in the pathogenesis of ischemic brain lesions, and glutamate levels have been found to be elevated in CSF and plasma from patients, early after stroke. In this study, we investigated whether platelets could be involved in the mechanism of altered plasma glutamate levels after stroke. Forty four patients, from 6 hours to 9 months after ischemic stroke, 15 age-related healthy controls and 15 controls with stroke risk factors or previous transient ischemic attack were enrolled. Glutamate plasma levels, platelet glutamate release after aggregation and platelet glutamate uptake were assessed. Plasma glutamate levels were increased up to 15 days after the ischemic event in stroke patients, and the levels at day 3 were inversely correlated with the neurologic improvement between day 3 and 15. Ex vivo platelet glutamate release was decreased by 70% in stroke patients, suggesting previous in vivo platelet activation. Moreover, platelet glutamate uptake in these patients was decreased by 75% up to 15 days and was still reduced 90 days after stroke. Our data show a prolonged increase of glutamate in plasma after stroke, which might presumably be linked to altered platelet functions, such as excessive release of the amino acid or impaired uptake.
Introduction
Glutamate has been shown to play a key role in the pathogenesis of brain ischemia (Dirnagl et al, 1999), and elevated concentrations of the amino acid have been reported in brain tissue surrounding the necrotic core both in animal models of ischemia (Benveniste et al, 1984; Hillered et al, 1989) and in stroke patients (Bullock et al, 1995; Kanthan et al, 1995), shortly after symptom onset. This elevation could be linked to increased release from neurons, resulting from energy failure, or to reduced clearance of glutamate by glial transporters. In fact, in animal models of hypoxia–ischemia, an altered expression of glutamate transporter subtypes has been described (Raghavendra Rao et al, 2000; Torp et al, 1995). High levels of glutamate were also found in CSF (Davalos et al, 1997) and in blood (Castillo et al, 1996) of patients, up to 48 hours after stroke, and apparently correlated with poor clinical outcome. Increased glutamate levels in blood have been assumed to result from leakage of the amino acid from the necrotic core through the damaged blood–brain barrier, but so far no clear evidence supporting this view has been provided, and blood glutamate may derive from peripheral sources as well. Platelets might be one possible alternative source of glutamate release. As a matter of fact, they have been shown to display high-affinity glutamate transport (Mangano and Schwarcz, 1981) and to express the three major glutamate transporters (Zoia et al, 2004), and may contribute significantly to the clearance of this amino acid from the circulation (Cananzi et al, 1995). Interestingly, a putative role for glutamate in modulating the functions of blood cells has already been proposed. In fact, platelets (Franconi et al, 1996), white blood cells (Kuo et al, 2001), and endothelial cells (Sharp et al, 2003) express functional NMDA receptors, suggesting that they are possible peripheral targets for glutamate released into the circulation.
Of note, experimental evidence in animal models of ischemia suggests that platelets, besides determining vessel occlusion and thrombus formation, may contribute to the pathogenesis of stroke by releasing soluble factors, which may alter the permeability of the blood–brain barrier and possibly damage neurons directly (Fujimoto et al, 1985; Ballabh et al, 2004). Moreover, several groups showed that, compared with age-matched healthy controls, platelets of stroke patients show morphologic and metabolic alterations (Joseph et al, 1988, 1989; O'Malley et al, 1995) likely linked to energy exhaustion.
Although never so far investigated, glutamate might be released during platelet activation or aggregation, and this source may contribute to the elevation of the amino-acid levels reported after ischemic stroke. Therefore, in the present study, we tested the hypothesis that the glutamatergic system in platelets might be dysfunctional in stroke patients, possibly leading to the elevation of glutamate blood levels. Hence, we investigated glutamate in blood after brain ischemia from different perspectives, by assessing glutamate concentrations in plasma, and its possible release and uptake from platelets of ischemic stroke patients, with respect to controls. To establish if the alterations in platelet function of stroke patients were secondary to the recent activation linked to the stroke or were related to possible stroke risk factors, we also assessed the same parameters in subjects with known stroke risk factors or previous transient ischemic attack (TIA), which occurred at least one year before enrolment in the present study.
Methods
Fifty-five patients with acute ischemic stroke (within three days from the onset) were enrolled at the Department of Neurology of the University of Milano-Bicocca, San Gerardo Hospital, Monza, Italy. Patients with history of recent (within three months) infection, current major cardiac, renal, hepatic, autoimmune and cancerous disease, history of TIA, stroke or head trauma, as well as those who received immunosuppressive drugs or acquired major infection during the study, were excluded. Other risk factors for stroke were recorded, along with demographic and clinical data of patients (Table 1).
Clinical and demographic data of stroke patients, controls, and subjects with stroke risk factors
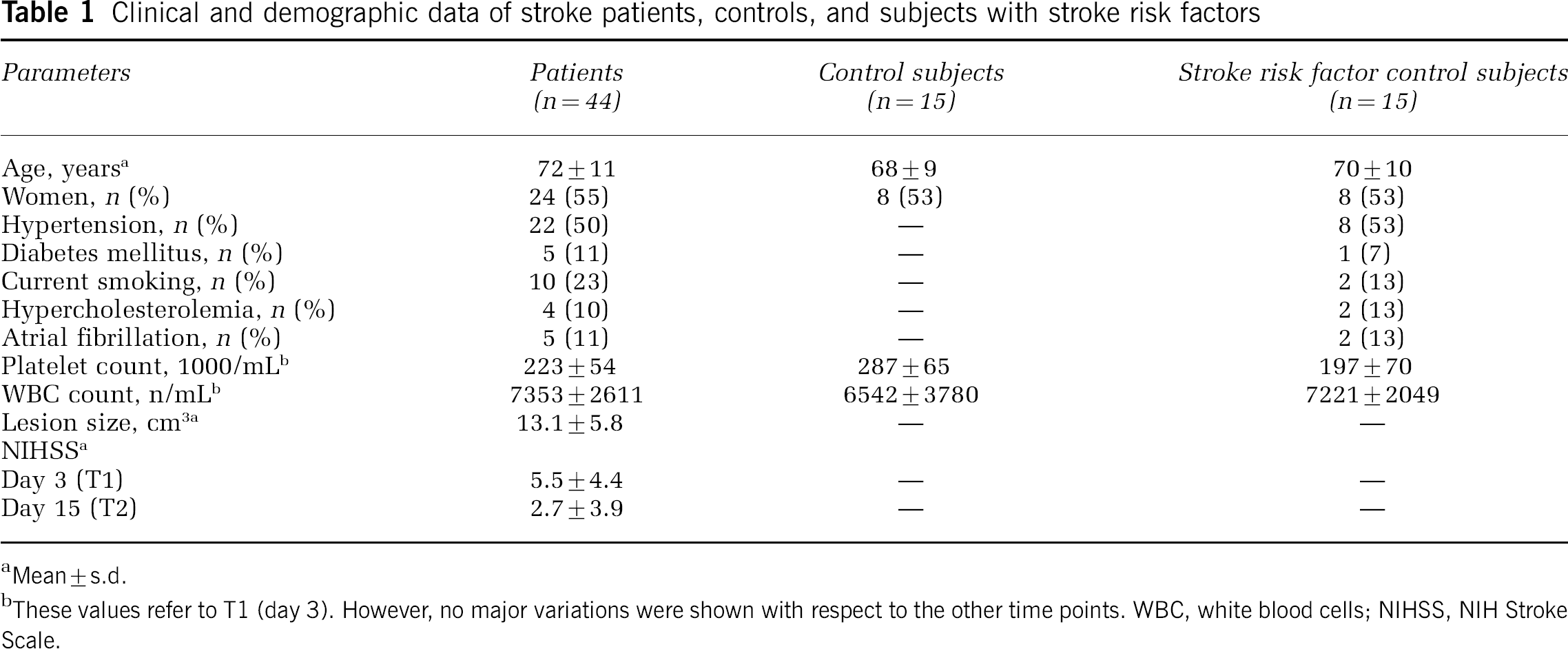
Mean±s.d.
These values refer to T1 (day 3). However, no major variations were shown with respect to the other time points. WBC, white blood cells; NIHSS, NIH Stroke Scale.
Because seven patients were lost due to infections and four died, 44 out of 55 completed the study. All patients underwent a complete neurological examination at entry, and stroke severity was determined by the National Institute of Health Stroke Scale (NIHSS) (Brott et al, 1989) at the onset of the event and then on days 3 (T1), 15 (T2), 90 (T3), and at 9 months (270 days, T4) after stroke. At the same time points, a venipuncture of the forearm was performed for the measurement of plasma glutamate levels and platelet glutamate uptake. A computerized tomography was performed in each patient within 24 hours and on the 5th day after stroke, and the volume of the lesion was measured using Cavalieri's Direct Estimator (Clatterbuck and Sipos, 1997). Five patients developed hemorrhagic transformation, as documented by the brain CT scan.
A subgroup of 15 patients was recruited within 6 hours of symptom onset (T0), before administration of any antiaggregating treatment. A blood sample was obtained to analyze platelet aggregation and glutamate release.
Antiaggregating (acetylsalicylic acid, ticlopidine, triflusal) or anticoagulant (heparin, warfarin, acenocumarol) treatment was started as soon as the patient entered the department, in accordance with clinical indication.
In all, 15 age- and sex-matched control subjects, seven men and eight women, apparently healthy and without known stroke risk factors, were recruited among blood donors of the San Gerardo Hospital.
Finally, the same analysis was performed in 15 age-related control subjects with known stroke risk factors or previous TIA (at least one year before study enrolment), treated with antiaggregating drugs (see Table 1).
The study was approved by the Ethical Committee of San Gerardo Hospital and the subjects enrolled gave informed consent to all the described procedures.
Platelet Preparation
A whole blood aliquot of 30 mL was collected with informed consent, after overnight fasting from the antecubital vein, in plastic tubes containing 0.17 mol/L K2-EDTA, and immediately processed to obtain platelets and plasma. To avoid spurious platelet activation, the blood withdrawal was performed by releasing the tourniquet and using a 19-gauge needle (Colciaghi et al, 2004). No specific inhibitors of platelet activation, such as prostaglandins, were used since they could impair glutamate release, as previously reported in vitro (Bezzi et al, 1998). Platelet-rich plasma (PRP) was collected after centrifugation at 210g for 20 mins. One aliquot of PRP was removed and centrifuged at 3600g for 15 mins to obtain a pellet of platelets, which was resuspended in 1 mL 0.32 mol/L sucrose containing 5% dimethylsulfoxide (Sigma-Aldrich, St Louis, MO, USA) to maintain membrane integrity, frozen in stratacooler (Nalgene, Rochester, NY, USA), and then stored at −80°C. After thawing, cytological studies with Trypan blue and May-Grunwald Giemsa stains showed 80% vital platelets with intact organelles.
Glutamate HPLC Assay
An aliquot of 900 μL of plasma was immediately deproteinized, to avoid enzymatic glutamate degradation, with 100 μL of 4 mol/L H2ClO4, rapidly neutralized with 50 μL of 7 mol/L K2CO3, and subsequently stored at −80°C for the following assessment of glutamate levels, performed with reverse-phase HPLC. After precolumn derivatization with o-phthaldialdehyde, samples were run through a C18 reverse-phase column (Beckman Instruments, Fullerton, CA, USA; 25 cm × 4.6 mm) by a multistep gradient of two solvents (A: 0.1 mol/L sodium acetate buffer, pH 7.2; B: methanol/tetrahydrofuran, 97:3 vol/vol) at a flow rate of 1.5 ml/min. Amino acids were detected by fluorimetry, as previously reported (Ferrarese et al, 1993).
Platelet Glutamate Uptake
The sodium-dependent glutamate uptake in platelets was performed after the original method of Mangano and Schwarcz (1981) with minor modifications, as previously described (Ferrarese et al, 2001). Platelets were resuspended and washed in 0.32 mol/L sucrose; aliquots of 50 μL were added to 425 μL Tris-citrate buffer pH 7.0, and samples were preincubated for 10 mins at 37°C. To investigate the sodium-dependent high-affinity uptake, blank samples were preincubated into a similar Tris buffer that had sodium chloride substituted by equimolar choline chloride and sodium citrate by equimolar potassium citrate. Uptake was started by the addition of 25 μL [3H]glutamate (specific activity=44 Ci/mmol, NEN Life Science Products, Milan, Italy) 60 μmol/L final concentration, and stopped after 30 mins by the addition of 3-mL ice-cold Tris buffer containing 1 mmol/L cold glutamate. Kinetic analysis, with [3H]glutamate concentrations ranging from 1 to 120 μmol/L, was also performed in 10 subjects selected from patients and controls. Platelets were separated by centrifugation, and washed pellets were dissolved in Formula-989 (Du Pont) and counted in a beta-counter with 60% efficiency. All assays were routinely performed in triplicate. Net high-affinity uptake was determined by subtracting no-sodium blanks from the uptake in the presence of sodium and expressed as pmol glutamate/mg proteins per 30 mins. Sodium-independent low-affinity uptake was also calculated as the difference between uptake at 37°C and at 4°C, in either the presence or absence of sodium. Protein concentrations were determined by a spectrophotometer with the Bradford reagent.
Platelet Glutamate Release
Platelet glutamate release was investigated after platelet in vitro aggregation, performed in the subgroup (n=15) of stroke patients recruited within 6 hours from stroke onset (before antiaggregating therapy) and in controls. A 9-mL whole blood sample was collected in tubes containing 1 mL, 3.8% sodium citrate and centrifuged to obtain PRP as described previously. The PRP was divided into 2 aliquots of 500 μL; platelet count in the PRP was standardized between 150,000 and 200,000 per μL by dilution with plasma from the same patient. One aliquot was incubated for 30 secs at 37°C with 3.5 μL of collagen 0.4 μmol/L and the rate of aggregation was evaluated with an Aggrego-Meter (Crono-Log), while the sec aliquot was kept untreated at 37°C. The two aliquots of PRP were then spun to obtain platelet poor plasma, which was then inactivated for glutamate HPLC assay, as described above.
Statistical Analysis
Since normal distribution of the parameters could not be assumed and group size was relatively small, we employed the Kruskal–Wallis and Mann–Whitney U-test to compare different groups. Analysis of variance (ANOVA) followed by Bonferroni post hoc comparison test was used to analyze the influence of various risk factors on a parameter. The correlation among glutamate plasma levels, platelet uptake, and clinical, neuroradiological, and laboratory parameters were assessed by linear regression analysis.
Results
Plasma glutamate levels were significantly increased by 40% at 6 hours (P<0.01), by 48% at 3 days (P<0.005), and by 59% at 15 days (P<0.0001) after stroke, while they returned to the same range as controls after 90 days from symptom onset (Figure 1). Plasma glutamate level at day 3 after stroke was inversely correlated (r=−0.36, P<0.05) (Figure 2) with improvement of the NIHSS score between days 3 and 15. In the group of patients with stroke risk factors or previous TIA at least 1 year earlier, glutamate levels were not significantly different from controls (Figure 1). To investigate whether platelets may release glutamate and if increased glutamate levels in plasma could be linked to its release from platelets, we studied plasma glutamate levels after ex vivo platelet exposure to proaggregating stimuli, such as collagen, in 15 patients enrolled before antiaggregating therapy. A 2-fold increase of extracellular glutamate levels was shown in the PRP of healthy controls (P<0.0001) after aggregation (Figure 3). On the contrary, a slight and nonsignificant increase in extracellular glutamate levels after platelet aggregation was observed in PRP from stroke patients, where basal plasma levels were significantly increased before aggregation, compared with control subjects. Thus, ex vivo platelet glutamate release after collagen-induced aggregation was decreased by 70% in stroke patients compared with controls (P<0.01) (Figure 3). Finally, we investigated the high-affinity platelet glutamate uptake in stroke patients, which showed 75% decrease at 3 and 15 days from stroke onset compared with controls (P<0.0001), and a minor (60%) but significant reduction (P<0.005) even at 3 months (90 days) after stroke, while no significant difference was found after 9 months (270 days) (Figure 4). Glutamate uptake in subjects with stroke risk factors did not show any difference compared with healthy controls (Figure 4). No differences were shown for all these parameters between the small subgroup of patients with hemorrhagic infarction and the rest of the recruited patients.
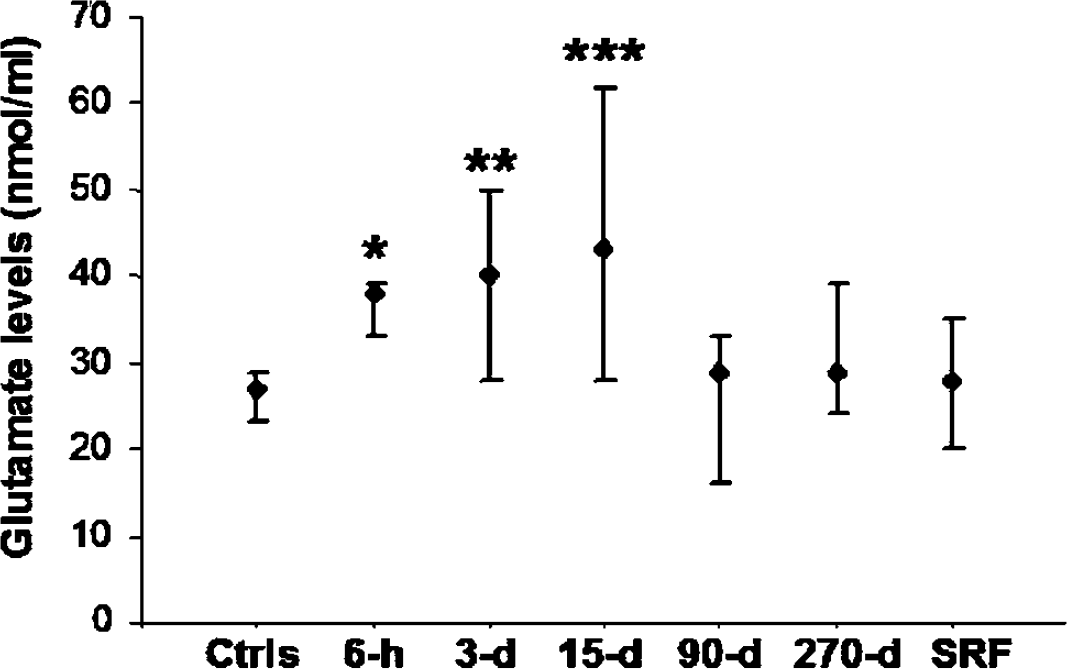
Glutamate plasma levels at various time points after stroke. Glutamate plasma levels increase early after stroke, and remain elevated up to 15 days. At 90 days after stroke, glutamate plasma levels are not different compared with both healthy subjects (Ctrls) and subjects with stroke risk factors (SRF). *P<0.01, **P<0.005, ***P<0.0001 versus Ctrls and SRF. Values are expressed as median and quartiles (25% and 75%). h=hours; d=days.
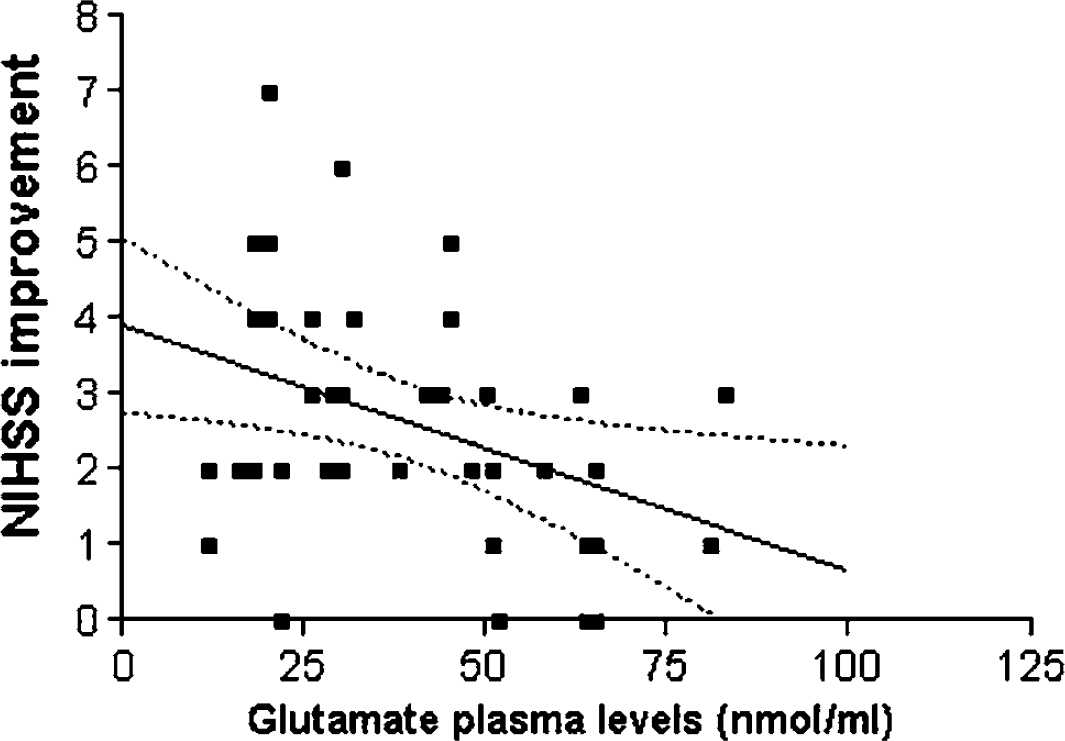
Correlation between glutamate plasma levels (day 3) and NIHSS improvement between days 3 and 15. r=−0.36, P<0.05. 95% confidence interval is shown.
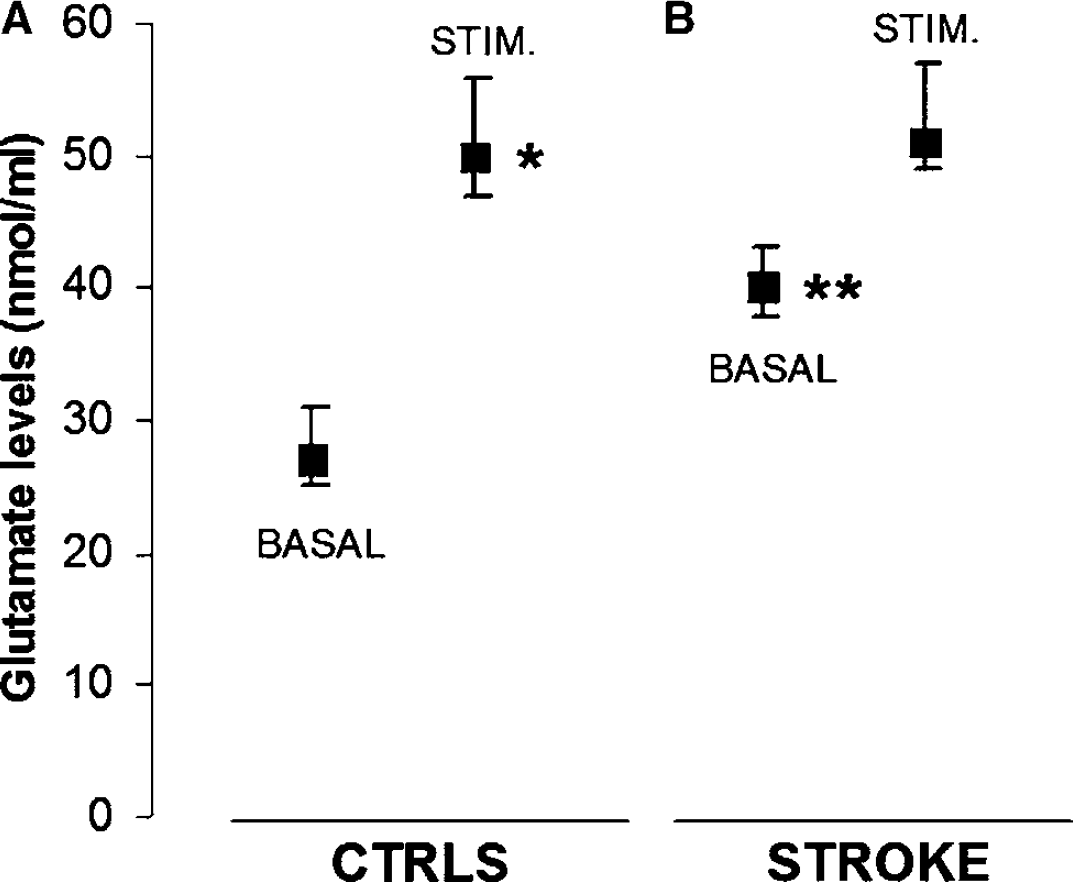
Glutamate release from platelets of stroke patients and healthy controls. Ex vivo platelets from healthy subjects (
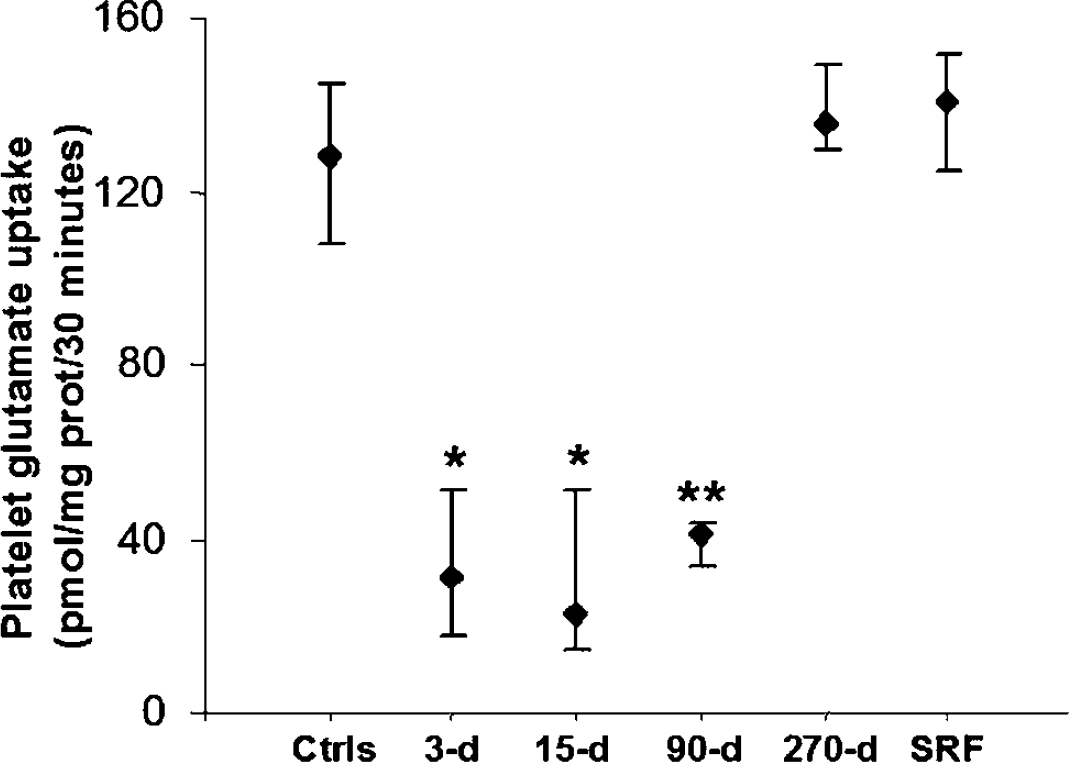
Platelet glutamate uptake at various time points after stroke. Platelet glutamate uptake is decreased approximately 75% up to 15 days after stroke. At 90 days after stroke, platelet glutamate uptake is still reduced (∼65%), although after 9 months (270 days), no difference is shown between stroke patients and both healthy subjects (Ctrls), and subjects with stroke risk factors (SRF). *P<0.0001, **P<0.0005 versus Ctrls and SRF. Values are expressed as median and quartiles (25% and 75%). d=days.
Moreover, no correlation was demonstrated among glutamate plasma levels, glutamate uptake, and brain lesion size measured by Cavalieri's Direct Estimator.
Furthermore, no significant correlation was found among these biological markers and laboratory data (erythrocyte sedimentation rate, reactive C protein, fibrinogen and iron levels, leukocyte and platelet count) or risk factor exposure (hypertension, diabetes mellitus, current smoking, hypercholesterolemia, atrial fibrillation).
Discussion
In this report, we have shown that plasma glutamate levels are increased in stroke patients, from the first up to the 15th day after symptom onset. Moreover, we have reported clear indications that glutamate is normally released from platelets after ex vivo aggregation, and that this process is impaired in acute ischemic stroke patients, suggesting previous in vivo activation and subsequent exhaustion. Finally, our results showed that platelet glutamate uptake is reduced in patients up to 3 months after stroke, whereas it returns to the same range of controls within 9 months.
Plasma glutamate levels reported in this study are consistent with previous papers, which have shown elevated blood glutamate levels in ischemic stroke patients (Castillo et al, 1996). Furthermore, we found that glutamate levels in the early phase of stroke are inversely correlated with subsequent clinical improvement, measured by the difference in NIHSS score between days 3 and 15. Thus, in agreement with previous observations (Castillo et al, 1996), high plasma glutamate levels 3 days after stroke may predict poor outcome, although because of the wide dispersion of our data, this correlation would hardly be useful for clinical purposes.
Moreover, the longer follow-up of our study allowed us to show that plasma glutamate increase can still be observed up to 15 days after symptom onset.
In this work, we also reported that platelets release glutamate ex vivo, when challenged with aggregating stimuli. Although the biological meaning of this process is still unclear, it could be linked to the regulation of platelet activation and aggregation itself. As a matter of fact, platelets have been shown to possess NMDA receptors, which may play an antiaggregating role on activation (Franconi et al, 1996). Additionally, glutamate might be involved in modulating the functions of white blood cells (Kuo et al, 2001), possibly playing a role in the interaction between platelets and leukocytes at the level of the tissue injury. Moreover, human cerebral endothelial cells have been shown to express functional NMDA receptors, the stimulation of which, theoretically after glutamate release, may lead to breakdown of the blood–brain barrier (Sharp et al, 2003).
Interestingly, ex vivo glutamate release was reduced in stroke patients before antiaggregating therapy, whereas basal extracellular plasma glutamate levels were increased. Since other studies have shown signs of platelet metabolic exhaustion in stroke patients, such as a reduced number of mitochondria (Joseph et al, 1989), a possible explanation of this finding might be related to a downregulation of platelet function after the previous recent in vivo overactivation. Thus, in vivo glutamate release from platelets during thrombus formation and vessel occlusion may also contribute to the elevation of plasma levels of the amino acid in the early phase of stroke. Although it is tempting to speculate that platelet glutamate release may also contribute to the pathogenesis of brain ischemia, further research is needed to establish a clear cause–effect relationship.
However, plasma glutamate levels are increased up to 15 days after stroke, in patients treated with antiaggregating drugs, suggesting that mechanisms different from release from platelets might be involved. One possibility could be that glutamate is increased in blood as a result of impaired clearance of the amino acid from the bloodstream, because our data showed impaired platelet uptake in patients until three months after stroke. These data are in agreement with recent findings by Marquardt et al (2002), showing increased markers of platelet activation up to 3 months after stroke. Since platelet life span is limited to 8 to 10 days, these authors conclude that ongoing platelet stimulation must be present in stroke patients, and propose that inflammatory mediators such as cytokines might be involved in this process. Indeed, platelets have been shown to possess cytokine receptors (Soslau et al, 1997; Limb et al, 1999; Loppnow et al, 1998), which seem to be able to regulate platelet function (Soslau et al, 1997). Furthermore, cytokines have been demostrated to impair glutamate uptake in glial cultures (Liao and Chen, 2001), and this kind of modulatory action might be working also in platelets, as also suggested by our preliminary in vitro results (unpublished observations). Therefore, the reduction of platelet glutamate uptake observed in this study, as well as platelet prolonged activation, might well be consequent to the action of pro-inflammatory soluble mediators, such as IL-6 or TNF-α, which were found increased in the blood of patients up to three months after stroke (Fassbender et al, 1994; Ferrarese et al, 1999).
This interpretation is further supported by the fact that in our study the group of subjects with stroke risk factors or previous TIA displayed the same levels of plasma glutamate and platelet uptake as healthy controls. In our opinion, these data, as well as the time course of the alterations we observed in stroke patients, clearly indicate that the modifications of platelet function in stroke are subsequent to the cerebrovascular event itself and do not depend on chronic factors, such as the presence of atheroma, or vessel inflammation/over-reactivity. Also, the finding that stroke patients with or without vascular risk factors had similarly decreased glutamate uptake and increased plasma levels is in line with this view.
In conclusion, the pathological and clinical meaning of this study is more evident when considering that glutamate transport may be regulated in a similar way in the CNS as in platelets, which may be considered as putative peripheral markers of central glutamatergic dysfunction. Present observations also suggest that excitotoxic mechanisms may continue after the acute phase of stroke, when neuronal damage directly linked to ischemia and energy failure has already subsided. Further studies on larger patient populations should be performed to validate peripheral glutamate and inflammatory alterations as possible prognostic markers and therapeutic targets in stroke patients.
Footnotes
Acknowledgements
This work was partially supported by Scharper SPA, Italy, to which we address our sincere gratefulness.