
Abstract
Different brain regions exhibit differing sensitivities to ischemia/excitotoxicity. Whether these differences are due to perfusion or intrinsic factors has not been established. Herein, we found no apparent association between sensitivity to ischemia/excitotoxicity and the level of expression or basal phosphorylation of calcium/calmodulin-stimulated protein kinase II (αCaMKII) or glutamate receptors. However, we demonstrated significant differences in CaMKII-mediated responses after ischemia/excitotoxic stimulation in striatum and cortex.
INTRODUCTION
Calcium/calmodulin-stimulated protein kinase II (CaMKII) is a multifunctional serine (Ser)/threonine (Thr) kinase, and is the most abundant calcium-activated kinase in brain. One or more members of this family are found in virtually every tissue, and mediate a diverse range of physiological responses.1,2 The biological properties of CaMKII are controlled by multisite phosphorylation and targeting to subcellular microdomains through interactions with specific target proteins.2,3 The roles of two phosphorylation sites, Thr286 and Thr305/6, have been well characterized. Phosphorylation at these sites has a profound effect on the activity of CaMKII, as well as targeting to various subcellular locations.
2
Recently, the consequences of phosphorylation at Thr253 have begun to be elucidated. Thr253 phosphorylation occurs
Among its many roles, CaMKII colocalizes with and regulates glutamate receptors (especially AMPA and NMDA receptors (AMPA-R and NMDA-R). 6 These receptors are not only essential to normal neuronal function, but are central to the process of excitotoxic cell death, which occurs in stroke, seizures, and other conditions. Recently, a CaMKII inhibitor was shown to be neuroprotective in experimental stroke, even when administered 1 hour after ischemia. 7
Ischemic stroke is the most common form of stroke, and occurs after occlusion of a cerebral blood vessel. The resultant ischemia causes widespread membrane depolarization, release of excitatory neurotransmitters, opening of ion channels, and sustained elevation of intracellular calcium, leading to eventual cell death. 8 Different brain regions show varying sensitivity to ischemia in humans and animals and the reasons for this differential sensitivity remain uncertain. For example, after stroke, central (basal ganglia and deep white matter) injury progresses more rapidly than peripheral (cortical) injury in rodents and humans. 9 Possible contributors to this differential sensitivity include variations in collateral circulation, 10 proportion of white matter, 11 and endogenous tissue differences. 12 Additionally, different animal strains exhibit differing sensitivities to ischemia. For example, Spontaneously Hypertensive Rats (SHRs) and their parent Wistar Kyoto (WKY) strain exhibit dramatically different sensitivities to ischemia/excitotoxicity, unrelated to the presence of hypertension. 13 These differences have been proposed to be due to poorer collateral circulation causing decreased perfusion to the ischemic cortex. 14 However, Lecrux et al (2007) have shown increased expression of αCaMKII and increased basal phosphorylation of the GluA1 subunit of the AMPA-R at Ser831 in SHR striatum, and proposed that these changes may at least partly explain the enhanced vulnerability of this strain to AMPA-mediated excitotoxicity.
We have examined the expression and phosphorylation of αCaMKII and glutamate receptors in brain regions with differing sensitivities to ischemia/excitotoxicity to investigate whether this proposed model could explain the regional sensitivity to ischemia/excitotoxicity in striatum and cortex. We have confirmed Lecrux's findings in the striatum of SHRs and WKY, and extended the molecular analysis to different brain regions. Our results indicate that there are tissue-specific responses to injury, independent of perfusion factors. However, neither the expression levels nor basal phosphorylation of αCaMKII or glutamate receptors are associated with regional sensitivity to ischemia/excitotoxicity. Our data indicate that the enhanced susceptibility of brain regions to ischemia may be due to altered phosphorylation of αCaMKII at Thr253 after an excitotoxic stimulus.
MATERIALS AND METHODS
MATERIALS
Preparation of Brain Tissue
All procedures were performed approval from the University of Newcastle Animal Care and Ethics Committee, as well as in accordance with the relevant guidelines and regulations, including the NSW Animal Research Act, the NSW Animal Research Regulation, and the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes.
Middle Cerebral Artery Occlusion
All procedures were performed in accordance with national guidelines, and with the approval of institutional Animal Ethics Committees. Male SD rats (270 to 500 g;
Western Blotting
Tissue homogenates (10 to 20
Data Analysis
All statistical analyses were conducted using GraphPad Prism software V4.0 (GraphPad, San Diego, CA, USA). Comparisons between ischemic and contralateral hemisphere values, brain regions, and stimulated and nonstimulated brain slices were made using a paired Student's two-tailed
RESULTS
Differences in Level of Expression or Basal Phosphorylation of αCaMKII and Glutamate Receptors Do Not Account for Regional Sensitivity to Excitotoxicity or Ischemia
Different brain regions within the one animal exhibit differing sensitivities to excitotoxicity/ischemia. However, the mechanisms governing this enhanced vulnerability have not been identified.
To test whether increased regional sensitivity to ischemia/excitotoxicity is due to variations in αCaMKII and glutamate receptor expression and/or basal phosphorylation, we examined expression in two brain regions (striatum and cortex) in three rat strains (SHRs, SDs, and WKYs). In each strain, striatum progresses to infarction more rapidly than cortex. Therefore, if the mechanism proposed by Lecrux et al (2007) to account for the species difference in sensitivity to ischemia/excitotoxicity is also responsible for the regional difference in sensitivity we predicted that there would be higher levels of αCaMKII expression and basal phosphorylation of Ser831-GluA1 in the striatum when compared with cortex. However, the expression of αCaMKII (Figure 1E), GluA1 (Figure 1A), and basal phosphorylation of Ser831-GluA1 (Figure 1B) did not differ between striatum and cortex in any strain. Additionally, no difference was observed between brain regions for basal Thr286-αCaMKII (Figure 1F) or Thr253-αCaMKII (Figure 1G) phosphorylation.
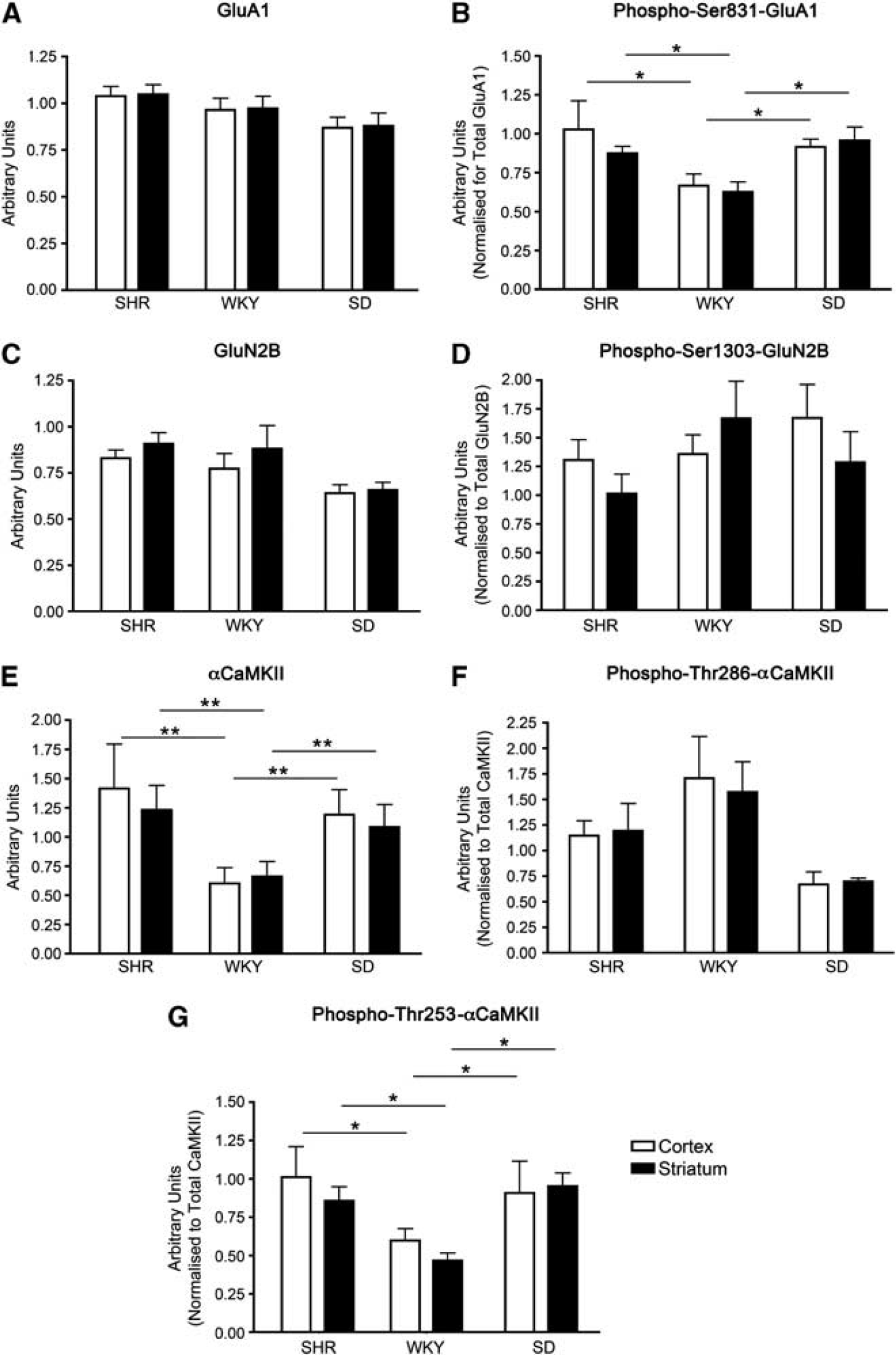
Levels of protein expression and basal protein phosphorylation of αCaMKII and glutamate receptor expression in the cortex and striatum of Spontaneously Hypertensive (SHR), Wistar Kyoto (WKY), and Sprague-Dawley (SD) rats. (
We next investigated other variables that may contribute to differences in susceptibility to excitotoxicity/ischemia, including the expression and basal phosphorylation of the GluN2B subunit of the NMDA-R. We found no difference in the level of expression of total GluN2B (Figure 1C) or basal phosphorylation of Ser1303-GluN2B (Figure 1D) between these brain regions. Furthermore, we found no difference in the expression of the GluA2 subunit of the AMPA-R between SD cortex and striatum (2.72 ± 0.45 and 2.48 ± 0.39 arbitrary units/mg protein, respectively). In addition, as previously described, 18 a similar level of PSD-95 (a postsynaptic protein used as a measure of excitatory synapse numbers 19 ) was observed in both the cortex and striatum of SDs (1.437 ± 0.083 and 1.222 ± 0.065 arbitrary units/mg protein, respectively). This indicates that a difference in the density of synapses is also an unlikely explanation for the difference in regional susceptibility to excitotoxicity/ischemia.
As these three rat strains exhibit differing sensitivities to excitotoxicity/ischemia (after a given period of focal ischemia, SHRs have the largest infarcts, followed by SDs and then WKYs 20 ), we hypothesized that SHRs would exhibit the highest αCaMKII and glutamate receptor expression and basal phosphorylation, WKYs would possess the lowest, and SDs expression/basal phosphorylation would be of an intermediate level. We found that αCaMKII expression (Figure 1E) and basal phosphorylation of Ser831-GluA1 (Figure 1B) were significantly higher in SHRs than in WKYs, without any difference in the level of expression of GluA1 (Figure 1A), confirming the findings of Lecrux et al (2007). However, αCaMKII expression (Figure 1E) and basal phosphorylation of Ser831-GluA1 (Figure 1B) in SDs were not significantly different from SHRs though they were significantly higher than in WKY; the same pattern was observed for basal Thr253-αCaMKII phosphorylation (Figure 1G). Additionally, no significant difference was observed between rat strains for basal Thr286-αCaMKII phosphorylation (Figure 1F). Therefore, while differences in expression and basal phosphorylation of αCaMKII or glutamate receptors may be a major contributor to the differences in sensitivity to ischemia/excitotoxicity between SHR and WKY, additional factors are likely to be important in other rat strains.
Striatum and Cortex Respond Differently to Excitotoxic Stimulation In Vitro
Since variations in the level of expression and basal phosphorylation of αCaMKII and glutamate receptors did not appear to be associated with susceptibility of different brain regions to ischemia/excitotoxicity, we determined whether the phosphorylation of αCaMKII or glutamate receptors in the striatum and cortex would differ in response to an excitotoxic stimulus. Brain microslices derived from the striatum and cortex of SDs were stimulated with AMPA, NMDA + glycine, or glutamate + glycine. These three stimuli were chosen as they allowed the selective activation of the two main excitotoxic pathways (AMPA and NMDA-R mediated), as well as a stimulus that activated multiple excitotoxic pathways (glutamate + glycine stimulation activates all ionotropic and metabotropic glutamate receptors). SDs were used for these experiments as they are an outbred strain, and they are of intermediate susceptibility to stroke/ischemia when compared with SHRs and WKYs. 20 Phosphorylation of αCaMKII, sites on glutamate receptors known to be phosphorylated by CaMKII, and a control site that is phosphorylated by another protein kinase were examined.
To establish time courses for each phosphorylation site, preliminary experiments were performed over 5 minutes for each stimulus. A series of 5 time points, ranging from 30 seconds to 5 minutes were examined. There was considerable variation among the phosphorylation sites in the rates of phosphorylation (Supplementary Figures 1 to 3). The optimum poststimulus time at which to compare the responses was found to be 90 seconds as this was on the linear portion of the time course for the phosphorylation sites examined (except Thr305-αCaMKII) and the differences in phosphorylation at this time best reflected the pattern of overall differences between cortex and striatum. Therefore, the results in Figures 2 to 4 are all quantitative comparisons at 90 seconds after stimulus.
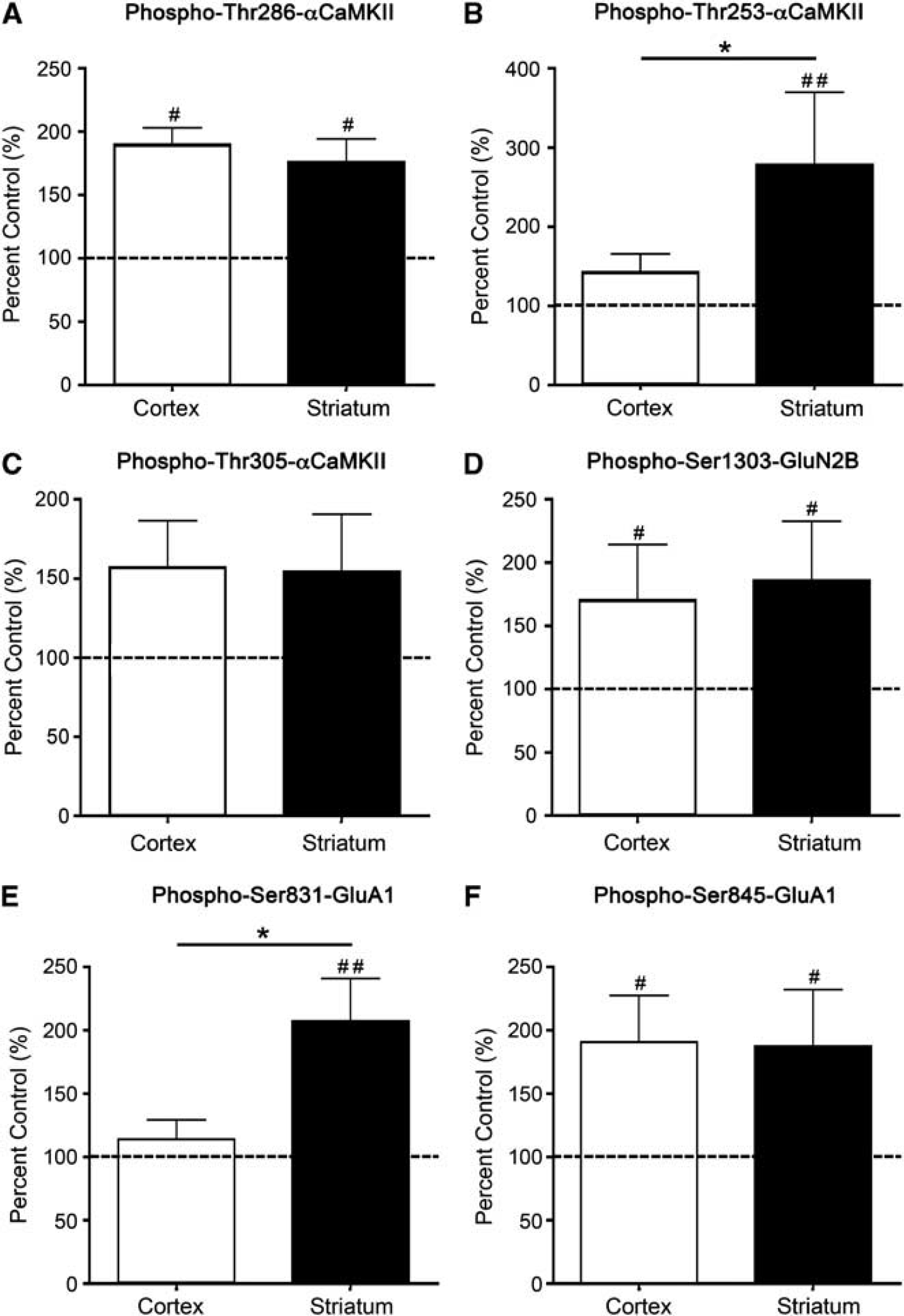
αCaMKII, GluA1, and GluN2B phosphorylation after stimulation with 100
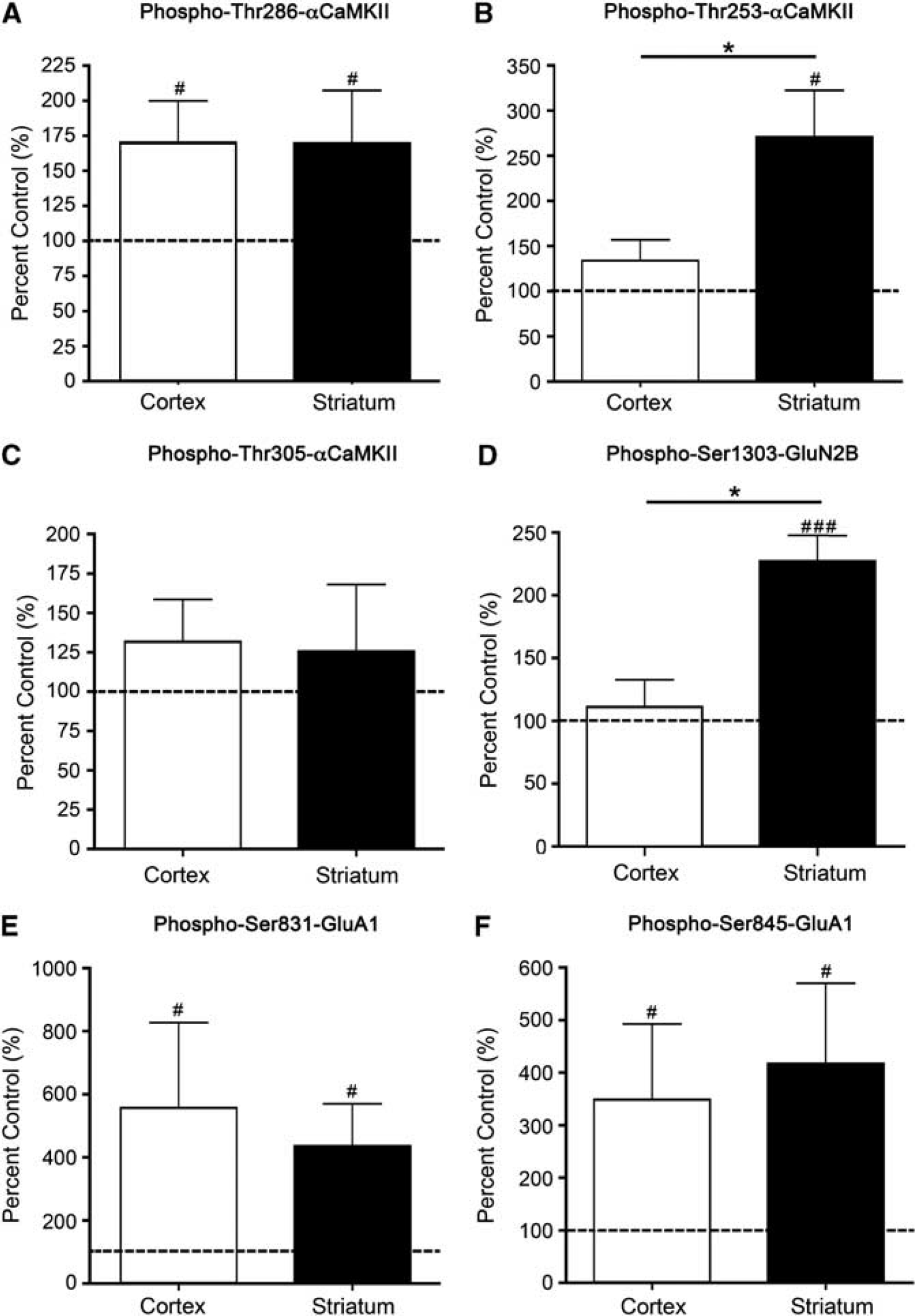
αCaMKII, GluA1, and GluN2B phosphorylation after stimulation with 100
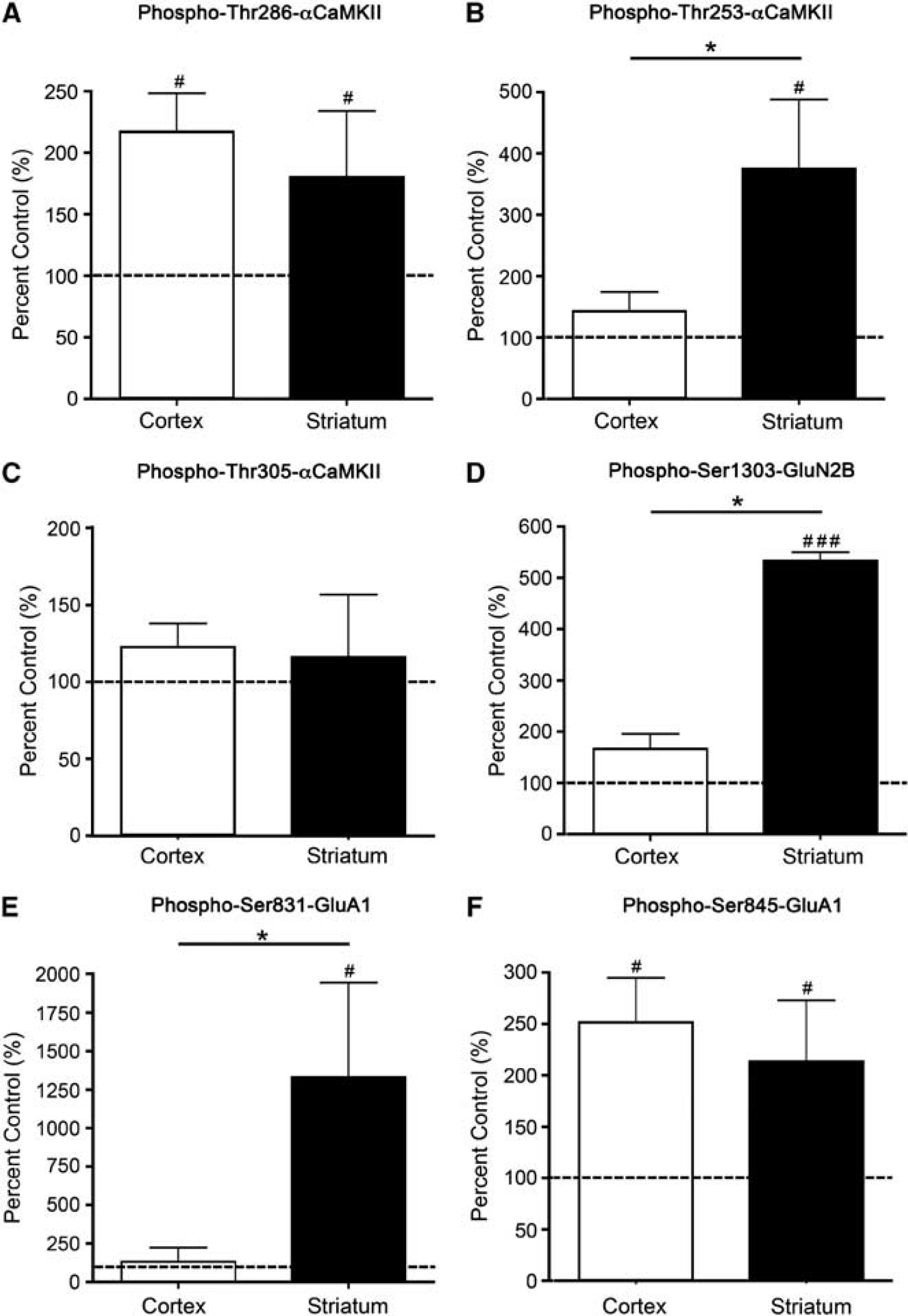
αCaMKII, GluA1, and GluN2B phosphorylation after stimulation with 2.5 mmol/L glutamate + 50
In both cortex and striatum, stimulation of microslices with AMPA produced a significant elevation in the phosphorylation of Thr286-αCaMKII (Figure 2A; Supplementary Figure 4). However, the AMPA-induced phosphorylation of Thr253-αCaMKII was very different in the two tissues: striatum showed an almost threefold increase in phosphorylation compared with nonstimulated control tissue after 90 seconds, whereas the response in cortex was not significantly different from control at this time (Figure 2B). At 5 minutes after stimulation, Thr253-αCaMKII phosphorylation was increased in both the cortex (203% control;
To determine whether certain glutamate receptor sites are phosphorylated to a greater extent in the striatum compared with the cortex after an excitotoxic stimulus, we examined Ser1303-GluN2B phosphorylation (phosphorylated by CaMKII or protein kinase C (PKC)21,22), and GluA1 phosphorylation at Ser831 (phosphorylated by CaMKII or PKC23,24) and Ser845 (phosphorylated by PKA but not CaMKII or PKC
25
) in these brain regions at various times poststimulus. AMPA-induced stimulation produced a significant increase in Ser1303-GluN2B (Figure 2D) and Ser845-GluA1 (Figure 2F) phosphorylation in both regions. By contrast, the AMPA-induced stimulation in Ser831-GluA1 phosphorylation was different in the two tissues: after 90 seconds the phosphorylation had doubled in striatum (Figure 2E) whereas no significant increase above control was detectable in cortex, although by 5 minutes there was a significant increase (146% control,
Stimulation of microslices by NMDA + glycine (an obligatory coagonist for NMDA-R activation) produced a significant elevation in the phosphorylation of Thr286-αCaMKII (Figure 3A; Supplementary Figure 4), Ser831-GluA1 (Figure 3E), and Ser845-GluA1 (Figure 3F) at 90 seconds that was similar in cortex and striatum. The phosphorylation of Thr305-αCaMKII was not significantly increased in either tissue after 90 seconds (Figure 3C), but was significantly elevated after 5 minutes (cortex: 595% control, striatum: 486% control;
To provide a more physiological stimulus, glutamate + glycine was used. The overall pattern of phosphorylation resembled a combination of those observed in response to AMPA and NMDA + glycine (Figures 2 and 3). Adding glutamate + glycine produced an increase in Thr286-αCaMKII (Figure 4A; Supplementary Figure 4) and Ser845-GluA1 (Figure 4F) phosphorylation that did not significantly differ between cortex and striatum. The glutamate + glycine-induced phosphorylation of Thr305-αCaMKII was not significantly increased compared with the control at 90 seconds (Figure 4B), but was significantly elevated at 5 minutes after stimulation (cortex: 198% control, striatum: 182% control,
CaMKII is Responsible for the Observed Glutamate Receptor Phosphorylation
The data presented in Figures 2 to 4 suggest a potential relationship between Thr253-αCaMKII phosphorylation and glutamate receptor phosphorylation at Ser831-GluA1 and Ser1303-GluN2B. However, both PKC and CaMKII can phosphorylate these sites. To ascertain whether CaMKII or PKC is responsible for the enhanced phosphorylation of Ser831-GluA1 or Ser1303-GluN2B in the striatum after an excitotoxic stimulus, brain microslices were treated with inhibitors of either CaMKII (myr-AIP, KN-93, and the inactive analog, KN-92) or PKC (chelerythrine chloride). The two CaMKII inhibitors were used at concentrations that are relatively specific for CaMKII, 1 and have no effect on PKC activity.
Our results show that preincubation of microslices with myr-AIP and KN-93, but not KN-92 or chelerythrine, blocked the AMPA- and NMDA-induced increases in Thr286-αCaMKII (Figures 5A and 5B) and Thr253-αCaMKII (Figures 5C and 5D) phosphorylation.
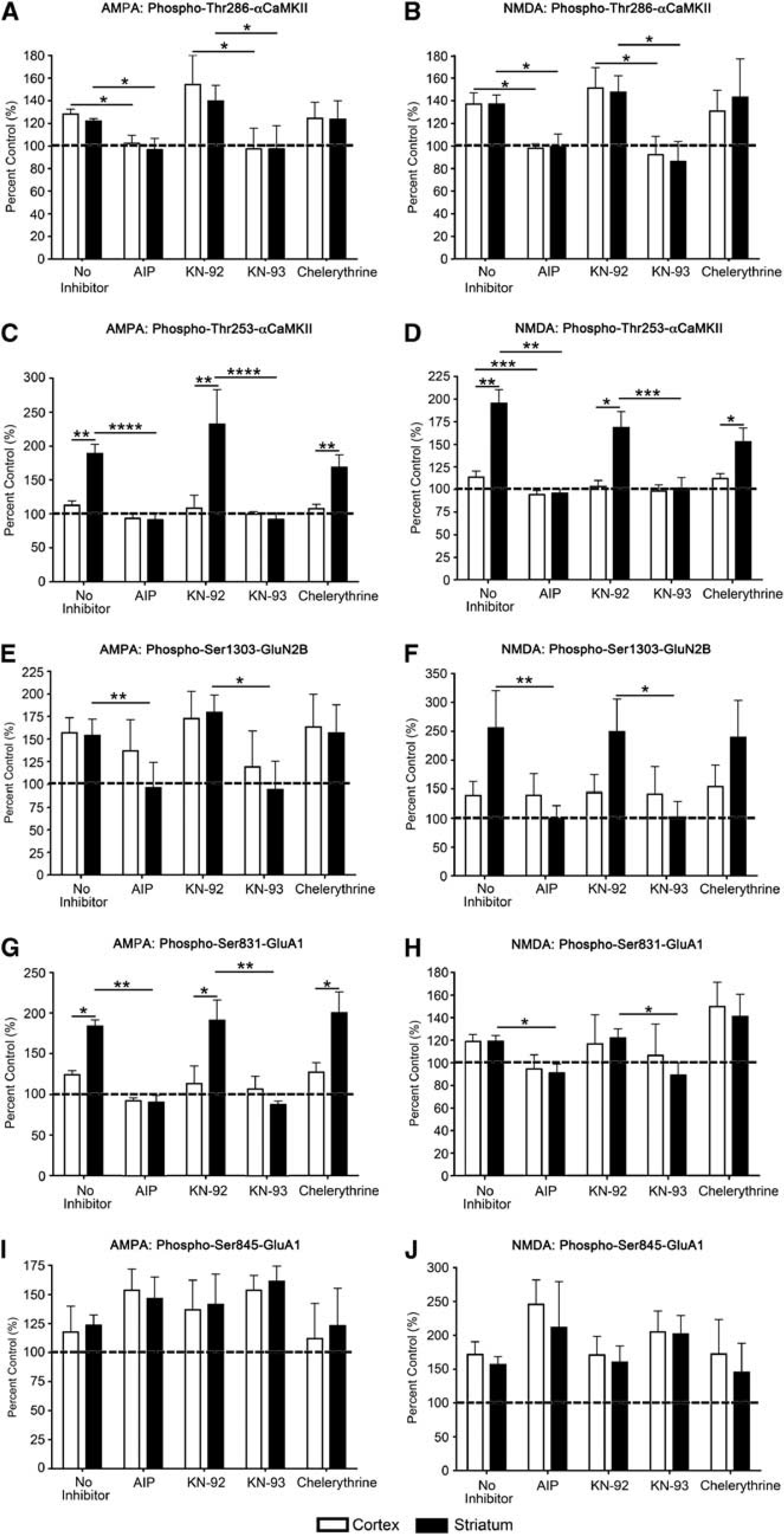
αCaMKII and glutamate receptor phosphorylation after inhibition of CaMKII and protein kinase C (PKC) activation, and stimulation with 100
No change in total αCaMKII expression after AMPA or NMDA stimulation was observed (data not shown). Both CaMKII inhibitors (myr-AIP and KN-93) significantly blocked the stimulus-induced increase in the phosphorylation of Ser831-GluA1 and Ser1303-GluN2B after AMPA or NMDA stimulation in the striatum (Figures 5E, 5F, 5I and 5J). The PKC inhibitor chelerythrine had no significant effect on the stimulus-induced phosphorylation of any site (Figure 5). No change was observed in total GluN2B or GluA1 expression after AMPA or NMDA stimulation (data not shown). These results show that, although PKC is potentially capable of phosphorylating both Ser831-GluA1 and Ser1303-GluN2B, in these microslices under the conditions used, CaMKII activity appears to be responsible for the observed phosphorylation of these receptors at these sites. However, a constitutively active form of death-associated protein kinase 1 can phosphorylate Ser1303-GluN2B, 26 so a role for death-associated protein kinase 1 in regulating this site after an excitotoxic stimulus cannot be ruled out.
The Time Course of αCaMKII Phosphorylation is Different in Striatum and Cortexafter Experimental Stroke In Vivo
The results using microslices show that there are inherent differences in the response of CaMKII in striatum and cortex to activation by an excitotoxic stimulus. We hypothesized that this would also be true after a stroke
After a short occlusion (90 seconds), the relative differences in αCaMKII phosphorylation
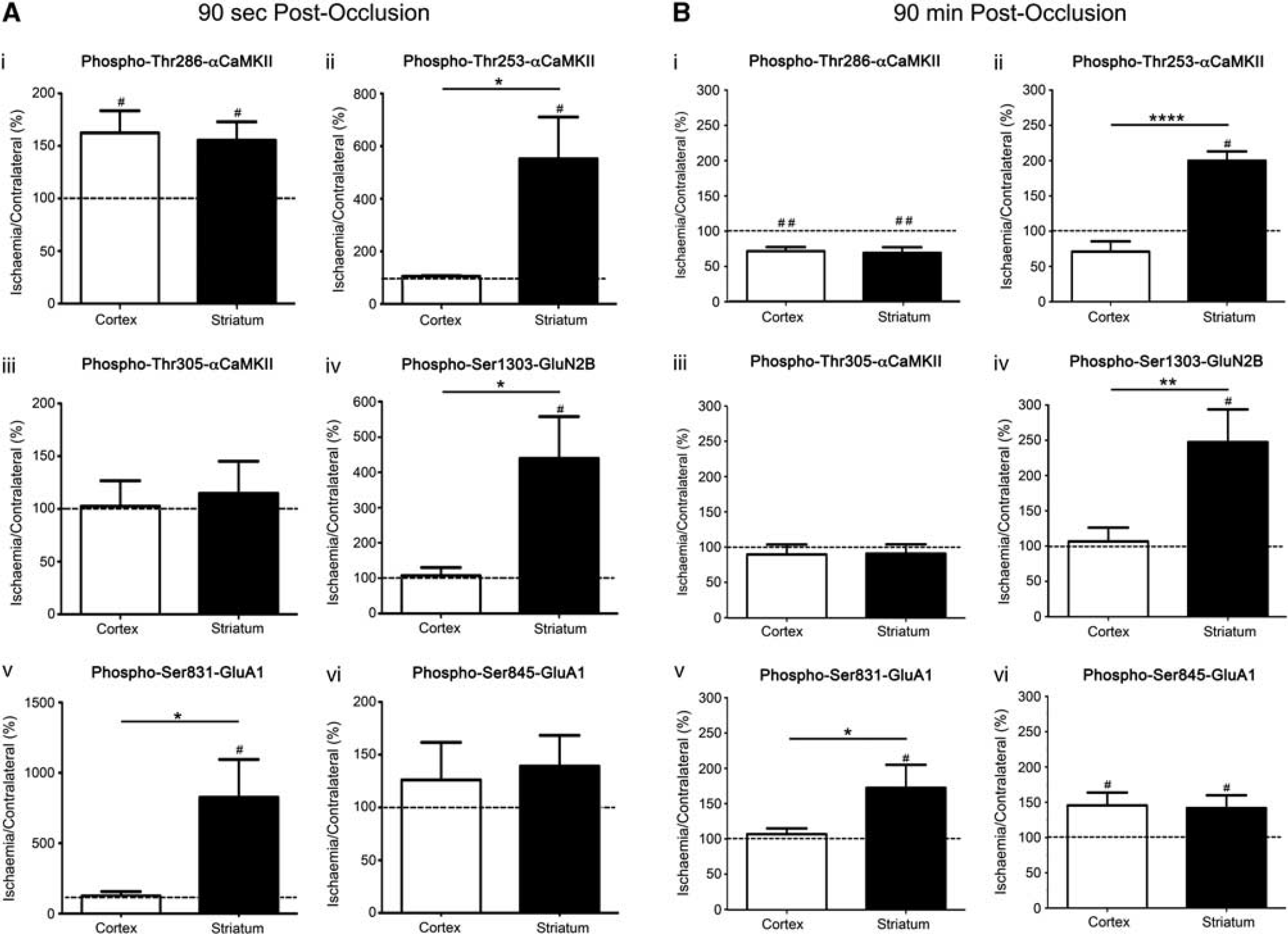
αCaMKII, GluA1, and GluN2B phosphorylation after occlusion of the middle cerebral artery for 90 seconds or 90 minutes. (i) Thr286-αCaMKII, (ii) Thr253-αCaMKII, (iii) Thr305-αCaMKII, (iv) Ser1303-GluN2B, (v) Ser831-GluA1, and (vi) Ser845-GluA1 phosphorylation in the striatum and cortex of Sprague-Dawley (SD) rats after (
At the end of the occlusion period (90 minutes with no reperfusion), Thr286-αCaMKII (Figure 6B(i)) and Ser845-GluA1 (Figure 6B(vi)) phosphorylation was significantly decreased in both the ischemic cortex and striatum, whereas the levels of phosphorylation at Thr253-αCaMKII (Figure 6B(ii)), Ser1303-GluN2B (Figure 6B(iv)), and Ser831-GluA1 (Figure 6B(v)) were significantly elevated in the ischemic striatum, but not in the cortex. Thr305-αCaMKII phosphorylation was not increased in the ischemic tissue compared with the contralateral side for either brain region (Figure 6B(iii)).
Next, we examined the changes in phosphorylation that occurred at various reperfusion times after 90 minutes MCAo. After 10 minutes of reperfusion, the level of phosphorylation of Thr286- and Thr253-αCaMKII was significantly increased in ischemic striatum, whereas the level of phosphorylation in ischemic cortex was not significantly different from contralateral (Figures 7A and 7C). However, after 60 minutes of reperfusion, Thr286-αCaMKII phosphorylation was elevated in the ischemic cortex but not in the striatum (Figure 7C), whereas Thr253-αCaMKII phosphorylation was elevated in both the ischemic cortex and striatum (Figure 7A). By 6 hours after reperfusion, Thr253-αCaMKII phosphorylation had returned to control levels in both regions (Figure 7A), whereas Thr286-αCaMKII phosphorylation levels had decreased in the cortex, but had risen in the striatum (Figure 7C). Reperfusion produced a significant transient increase in the phosphorylation of Ser1303-GluN2B in both striatum and cortex at 10 minutes after reperfusion, but was not different at 60 minutes or 6 hours after reperfusion (Figure 7D). Additionally, the phosphorylation of Ser831-GluA1 in cortex and striatum did not show a statistically significant change after reperfusion (Figure 7B).
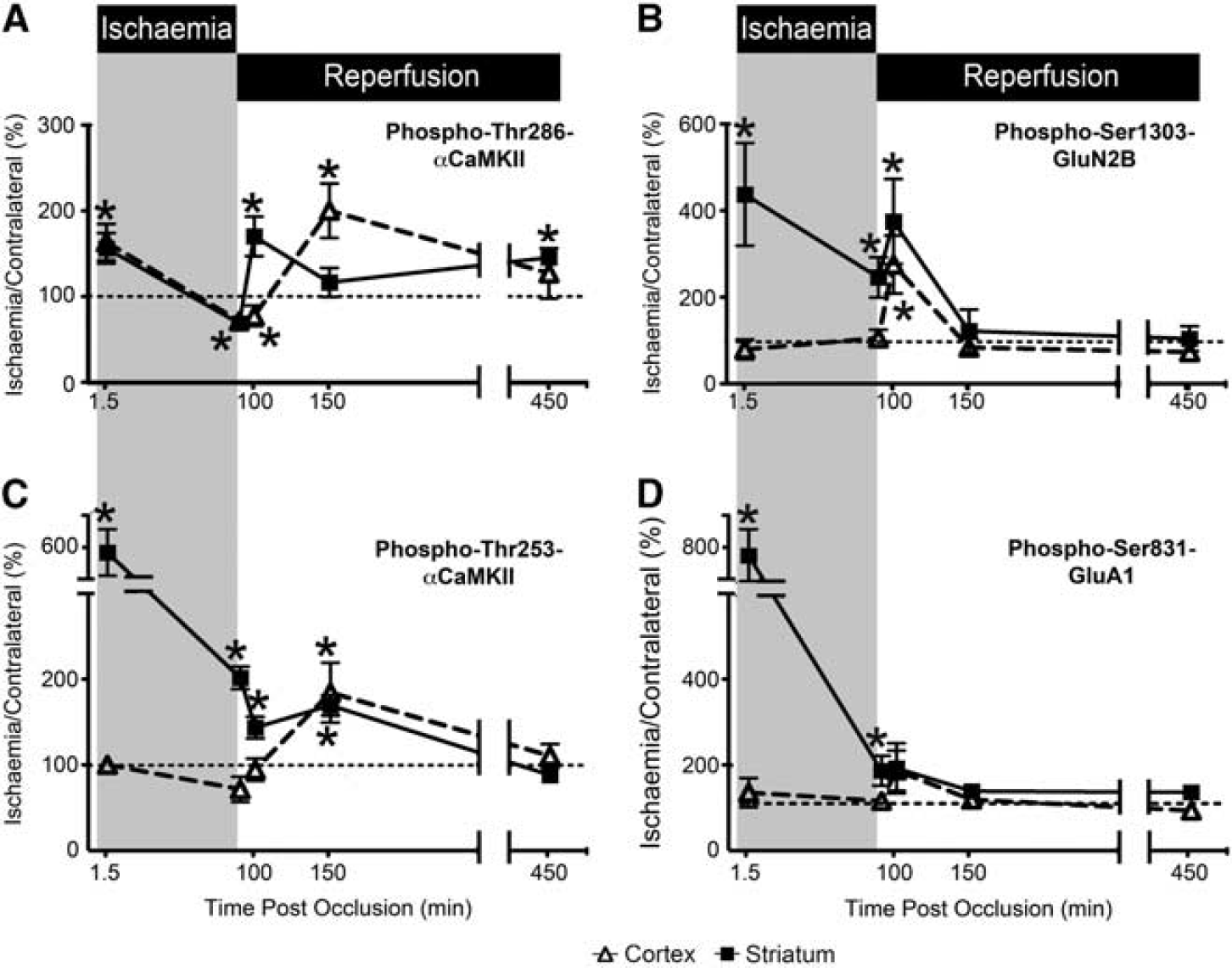
αCaMKII, GluA1, and GluN2B phosphorylation after varying periods of ischemia and reperfusion. (
These results show that striatum and cortex show different αCaMKII phosphorylation responses
DISCUSSION
Different brain regions exhibit differing sensitivities to ischemia; for example, the striatum is more vulnerable to ischemic damage than the overlying cortex after MCAo.
27
While factors such as collateral blood supply could account for this variation, there is an unresolved controversy about whether endogenous differences in such brain regions could be responsible for this differential susceptibility. Herein, we report that rapid changes in protein phosphorylation involving CaMKII, an enzyme involved in ischemia-induced cell death, are strikingly different in striatum and cortex after MCAo
Autophosphorylation of αCaMKII at Thr253 induced by either
We have shown that the differences in CaMKII activation and control between the striatum and cortex after ischemia/excitotoxicity are not accounted for by differences in the levels of expression of αCaMKII, AMPA-R, or NDMA-R, or the density of synapses in these regions. One explanation for the different CaMKII responses in striatum and cortex is the presence of different CaMKII interacting proteins that alter the molecular microenvironment for CaMKII by complex formation leading to allosteric modification and/or localization to different cellular microdomains. As Thr253 phosphorylation has no direct effect on CaMKII activity, but alters CaMKII targeting through interactions with particular binding proteins, and the pattern of expression of CaMKII binding proteins varies between brain regions and cell types, 3 tissue differences in phospho-Thr253-mediated CaMKII targeting may be responsible for the differences in sensitivity to ischemia/excitotoxicity between the striatum and cortex.
CaMKII has been implicated in neuronal death and survival pathways after excitotoxic insults, with reports of CaMKII inhibition either being neuroprotective29–31 or exacerbating cell death.32–34 While there is some controversy surrounding this issue, the balance of available data favors the hypothesis that short-term CaMKII inhibition postischemia, or after an excitotoxic stimulus, is neuroprotective in mature brain. Recent studies have shown that inhibitors of autonomously active CaMKII (calcium/calmodulin-independent activity) are neuroprotective when applied after an ischemic/excitotoxic insult, both
Since autophosphorylation at Thr286 makes CaMKII autonomously active,
2
it is assumed that the long-lasting excitotoxicity/ischemia-induced autonomous CaMKII activity is due to long-lasting autophosphorylation at Thr286. The finding that overexpression of a wild-type, but not a Thr286 phospho-null (T286A) form of αCaMKII increased glutamate induced cell death in primary hippocampal cultures
7
supports this assumption. In this light, it is interesting to consider our results with Thr286 phosphorylation after MCA occlusion and reperfusion. We observed an oscillatory pattern of phosphorylation–dephosphorylation of Thr286-αCaMKII in both the striatum and cortex (Figure 7A) that is consistent with events known to elevate intracellular calcium during ischemia and reperfusion: the onset of ischemia after occlusion, the early consequences of reperfusion and the waves of spreading depression associated with infarct evolution.
35
This suggests that repeated CaMKII activation by calcium/calmodulin maintains or boosts Thr286 autophosphorylation and autonomous CaMKII activity in mature brain
The changes in Thr253-αCaMKII phosphorylation in striatum and cortex after MCAo indicate that it is associated with brain region sensitivity to ischemia/excitotoxicity. Furthermore, the ability of tissue slices
As we were interested in rapid phosphorylation responses after excitotoxicity/ischemia we chose to measure changes in the phosphorylation of GluA1 and GluN2B because they are well-characterized substrates for CaMKII that are concentrated at the PSD, where CaMKII is also concentrated. Although overstimulation of most glutamate receptors causes cell death, 36 the calcium-permeable NMDA-Rs appear to be the most sensitive ‘death receptors’. 30 The phosphorylation of the GluA1 subunit of the AMPA-R at Ser831 has been proposed to be a death enhancing effect of CaMKII activation, 30 due to increased single-channel conductance of GluA1 after phosphorylation of this site. 25 By contrast, phosphorylation of the GluN2B subunit of the NMDA-R at Ser1303 desensitizes the NMDA-R, 22 and therefore may inhibit cell death pathways. However, it is not clear whether the CaMKII-mediated phosphorylation of these receptors is involved in the cell death response or whether the key protein targets regulated by CaMKII are downstream of calcium entry.
All animals used in these studies were anesthetized with isoflurane for 1 hour before kill, so that direct comparisons could be made between the
After periods of experimental cerebral ischemia, CaMKII translocates to one of the two sites: (1) the PSD 38 and (2) extrasynaptic clusters. 39 Many CaMKII binding partners, including GluA1 and GluN2B, are present in the PSD, whereas extrasynaptic clustering most likely occurs after aggregation of multiple CaMKII holoenzymes. These translocations are also associated with a reduction in calcium/calmodulin-stimulated CaMKII activity. 38 The precise mechanisms and functional outcomes involved in these translocations/alterations in activity remain to be determined, but highlight the importance of targeting in regulating CaMKII activity and hence function.
Based on our findings, we hypothesize that CaMKII involvement in ischemia-induced neuronal death requires autophosphorylation of CaMKII at two sites: (1) Thr286, which induces CaMKII to become autonomously active, therefore allows CaMKII to continue to phosphorylate key substrates and to maintain sustained activation of cell death pathways; and (2) Thr253 that targets the autonomously active CaMKII to the key substrates necessary to activate these pathways. Furthermore, we hypothesize that brain regions that are inherently more sensitive to excitotoxicity/ischemia express enhanced levels of one or more proteins that bind to CaMKII in a way that allosterically favors autophosphorylation of CaMKII at Thr253 and localizes CaMKII near calcium channels. This would allow a more rapid activation of CaMKII after a rise in intracellular calcium (as occurs after initial occlusion and reperfusion), and may account for the more rapid CaMKII and substrate phosphorylation that was observed in brain regions that are more sensitive to excitotoxicity/ischemia.
Based on strong preclinical data, CaMKII is an attractive target for the development of neuroprotective therapies for brain pathologies involving excitotoxic pathways. However, as CaMKII is expressed in virtually all tissues, existing inhibitors that target CaMKII kinase activity are unlikely to be useful therapeutically, as they are likely to cause many side effects. By contrast, drugs that interfere with CaMKII targeting to specific signaling complexes, such as AMPA- and/or NMDA-Rs, offer the potential for cell and stimulus-specific inhibition that may be beneficial therapeutically, either when administered on their own or in conjunction with existing therapies such as tissue plasminogen activator. Our results suggest that CaMKII targeting mediated by Thr253 phosphorylation may be an attractive focus for such drug development.
DISCLOSURE/CONFLICT OF INTEREST
The authors declare no conflict of interest.
Footnotes
ACKNOWLEDGEMENTS
The authors would like to thank Ms A'qilah Banu Binte Abdul Majeed, Mrs Helen Carpenter, Ms S Phoebe Chung, Ms Sarah McCann, Mr Matthew Morten, Ms Lucy Murtha, Ms Debbie Pepperall, and Ms Amelia Tomkins for their technical assistance on various aspects of this project.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.