
Abstract
The direction of the chemical reaction of ATP synthetase is reversible. The present study was designed to determine whether mitochondria produce or consume ATP during ischemia. For this purpose, changes in mitochondrial membrane potential were measured in vivo at the site of a direct current (DC) electrode using a potentiometric dye, 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide (JC-1), and a rat model of focal ischemia. Two μL of dye (control group) or dye with oligomycin, an ATP synthetase inhibitor (oligomycin group), was injected into the parieto-temporal cortex through the DC electrode. With the initiation of ischemia, a decrease in mitochondrial potential was observed within 20 seconds in the oligomycin group (earlier than the onset of DC deflection, P = 0.02). In contrast, in the control group, mitochondrial potential was maintained at 91 ± 5% of the pre-ischemia level for 118 ± 38 seconds before showing full depolarization simultaneously with DC deflection. During the period of ischemia, the mitochondrial potential was higher in the control group (66 ± 9%) than in the oligomycin group (46 ± 8%, P < = 0.0002), whereas DC potential was lower in the control group (–18 ± 3) than in the oligomycin group (–15 ± 2 mV, P = 0.04). These observations suggest that mitochondria consume ATP during ischemia by reversing ATP synthetase activity, which compromises cellular membrane potential by consuming ATP.
Keywords
It is well known that mitochondria play a role as a generator of ATP by increasing the proton gradient across the inner mitochondrial membrane and by coupling the proton gradient to phosphorylation with ATP synthetase. Mitochondria are also key regulators of cytosolic calcium concentration by activating an inward flow via the uniporter in the inner mitochondrial membrane and by outward flow via the Ca2+/Na+ exchanger and an Na+-independent mechanism in the inner mitochondrial membrane (Gunter and Pfeiffer, 1990). Thus maintenance of mitochondrial function coupled to mitochondrial membrane potential (ψm) is crucial for cell survival.
Recently, much interest has been shown in changes in ψm after cerebral ischemia because of two important discoveries. One is the mitochondrial permeability transition (Fiskum, 2000; Mattiasson et al., 2003), which is initiated by excessive uptake of calcium into mitochondria during periods of ischemia and reperfusion (Griffiths, 2000; Zoratti and Szabo, 1995) and has been suggested to cause apoptosis and necrosis by the release of cytochrome c and by the cessation of ATP production, respectively (Kroemer et al., 1998; Schild et al., 2001). The second important discovery is the possible involvement of the mitochondrial K+ATP channel in the development of ischemic preconditioning (Fryer et al., 2000; Shimizu et al., 2002). However, the mechanism by which activation of the mitochondrial K+ATP channel protects neuronal cells remains to be discovered.
Despite the recent interest in ψm during cerebral ischemia, changes in ψm have been observed only in vitro studies, including studies using isolated mitochondria (Di Lisa et al., 1995), cultured neurons (Iijima et al., 2003; Yamauchi et al., 2003), and brain slices (Sick and Perez-Pinzon, 1999). Changes in ψm have not been observed in vivo.
Unlike the cellular membrane potential that is mainly generated by an Na+ and K+ gradient across the cellular membrane with the function of Na+,K+-ATPase, as shown in Fig. 1A, ψm is generated by a hydrogen gradient with the function of the respiratory chain (activities of complexes I, III, and IV) on the inner mitochondrial membrane mainly using oxygen and NADH. With the forward activity of ATP synthetase (complex V), ATP is produced by consuming the proton gradient across inner mitochondrial membrane.
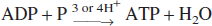
However, in the absence of respiratory chain function (such as anoxia, resulting in decrease in proton gradient), as shown in Fig. 1B, the ATP synthetase may reverse and may start to pump up the hydrogen to maintain ψm by consuming ATP (Jennings et al., 1991; Rouslin et al., 1986).
A: (Normal condition) The mitochondrial potential (180 mV) (Kamo et al., 1979) is produced by an H+ gradient generated by the activities of the respiratory chain (complexes I, III, and IV) on the inner mitochondrial membrane by consuming oxygen and NADH. The H+ gradient is consumed by ATP synthetase (complex V) to generate ATP for the hosting neuron. The Na+, K+-ATPase on the cellular membrane consumes ATP that is produced by mitochondria and by anaerobic glycolysis to maintain cellular membrane potential (73 ± 6 mV) (Gao et al., 1999). B: (Hypothesis 1, energy consumption by mitochondria) During ischemia, oxygen supply is not sufficient to maintain mitochondrial potential. To complement insufficient mitochondrial potential, ATP synthetase starts to reverse its activity and generates an H+ gradient by consuming ATP produced by anaerobic glycolysis.
It is known that there is 8% of co-lateral blood flow even in the ischemic core of focal ischemia (3-vessel occlusion model) (Buchan et al., 1992). In the ischemic core, cerebral glucose utilization remains at 40% of that in the contralateral hemisphere by the use of glucose in the co-lateral blood flow (Back et al., 1995). It is likely that ATP is produced by anaerobic glycolysis in the ischemic core, even though the total amount of ATP production is not sufficient for cell survival.
It is not known whether mitochondria in cells in the ischemic core become organelles that consume ATP produced by anaerobic glycolysis in the cytosol or still produce ATP during ischemia by using oxygen from the co-lateral blood flow. In the former case, as shown in Fig. 1B (hypothesis 1), mitochondria complement insufficient ψm (hydrogen gradient) by reversing ATP synthetase and by consuming ATP in the cytosol. In this case, use of an ATP synthetase inhibitor (oligomycin, which is an inhibitor for both forward and reversed activities of ATP synthetase) should reduce the onset time of mitochondrial depolarization and should deteriorate ψm during ischemia. In the latter case, as shown in Fig. 1C (hypothesis 2), mitochondria sacrifice their insufficient ψm to supply ATP for the hosting neuron. In this case, use of an ATP synthetase inhibitor (oligomycin) should prolong the onset time of mitochondrial depolarization and should ameliorate ψm during ischemia.
The direction of chemical reaction of ATP synthetase during cerebral ischemia is an important factor determining ψm and availability of ATP in the cytosol. The main goals in the present study were to determine the dynamic changes in ψm and the direction of the chemical reaction of ATP synthetase during focal ischemia in vivo. The effects of ATP synthetase on ψm and cellular membrane potential were evaluated by using oligomycin, a specific inhibitor of ATP synthetase activity.
MATERIALS AND METHODS
Sprague-Dawley rats (Charles River, Wilmington, MA, U.S.A.), weighing 294 ± 18 g, were used in this study. The animals had free access to water and were fasted overnight before the experiments. Animal protocols were approved by the University of Miami's Animal Care and Use Committee. All chemicals were obtained from Sigma (St. Louis, MO, U.S.A.) unless otherwise noted.
General procedures
Anesthesia was induced with a mixture of 5% halothane in oxygen. Following oral tracheal intubation, anesthesia was maintained by artificial ventilation (7025; Ugo basile, VA, Italy) with 1% halothane in 30% oxygen balanced with nitrous oxide. A polyethylene catheter (PE50) was placed in the right femoral artery for continuous mean arterial blood pressure monitoring and blood sampling. Blood gases were measured (ABL 50; Radiometer, Copenhagen, Denmark) and maintained within normal ranges during the experimental period. After placement in a stereotaxic apparatus, a large cranial window was made on the left parietal-temporal bone (Fig. 2). The dura was kept intact. To monitor the cortical depolarization associated with the initiation of focal ischemia, two glass microelectrodes (less than 5 μm in tip diameter) for monitoring direct current (DC) potential were inserted in the cranial window at a depth of 750 μm through dural incisions (Fig. 2). The positions of the electrodes were 5 mm posterior to the bregma and 5 mm and 7 mm lateral from the sagittal line. Brain surface temperature was maintained at 37.0 ± 0.5°C with a gentle flow of warmed saline (38.0 ± 0.5°C, monitored and controlled) perfusing over the skull surface. Rectal temperature was monitored and maintained at 37.0 ± 0.5°C using a heated water blanket.
The shaded area (orange) represents the large cranial window made on left parietal-temporal bone. The left middle cerebral artery was observed and occluded inside the cranial window. DC1 and DC2 represent the locations of direct-current recording sites. DC, direct current.
Measurement of changes in ψm
Mitochondrial potential was measured using a potentiometric dye, JC-1 (Molecular Probes, Eugene, OR, U.S.A.), which accumulates in a potential-dependent manner in mitochondria and subsequently forms J-aggregates from monomers. Because J-aggregates and monomers fluoresce red (590 nm) and green (530 nm), respectively, with excitation light (485 nm), the ratio of red:green indicates ψm. Dye loading was performed by injecting 2 μL of dye (JC-1, 6 μM; cyclodextrine, 20%; DMSO, 0.5%; in physiologic saline) into the parieto-temporal cortex through the DC electrode over a period of 2 minutes. During the dye loading, DC potential was stable, and no spreading depression was observed. In a pilot study, it was confirmed that no histologic change (hematoxylin-eosin staining) was induced during a period of 24 hours after the injection of JC-1 dye. The excitation light (2 seconds exposure in each emission) was produced by a 150-watt xenon lamp and was conducted to the cortical surface through a liquid optical fiber (diameter, 5 mm). An iris was placed between the xenon lamp and the optical fiber to prevent photooxidation of the dye. The intensities of red fluorescence and green fluorescence in an area (470 × 470 μm) adjacent to the DC-recording site were measured every 20 seconds using an electrically cooled charge-coupled device (CCD) camera (ST-9XE; SBIG, CA, U.S.A.) mounted on a macro scope (x 1.0 objective) with green and red bandpass filters. The ratio of red:green fluorescence, indicative of ψm, was normalized to that of the control value of the red:green ratio.
Focal ischemia
Focal cerebral ischemia (three-vessel occlusion) was initiated according to the method of Buchan and colleagues (1992) with slight modification. In the present experiment, we used Sprague-Dawley rats instead of Wistar rats, which were used in the original method. Bilateral common carotid arteries (CCAs) were exposed, and loose ligatures were placed around them. As shown in Fig. 2, the left middle cerebral artery (MCA) was accessed by piercing dura inside the cranial window 0–1 mm above the rhinal fissure. Ischemia was initiated by tightening ligatures around the bilateral CCAs and occluding the left MCA by lifting the vessel to 0–1 mm above the rhinal fissure with an 80-μm stainless-steel hook attached to a micromanipulator. The adequacy of recirculation upon release of the MCA/CCAs occlusion was assessed by direct visual observation using a surgical microscope. Three rats underwent a sham operation in which the hook was placed under the MCA and removed without lifting the artery; it showed no histologic damage 24 hours later.
RESULTS AND DISCUSSION
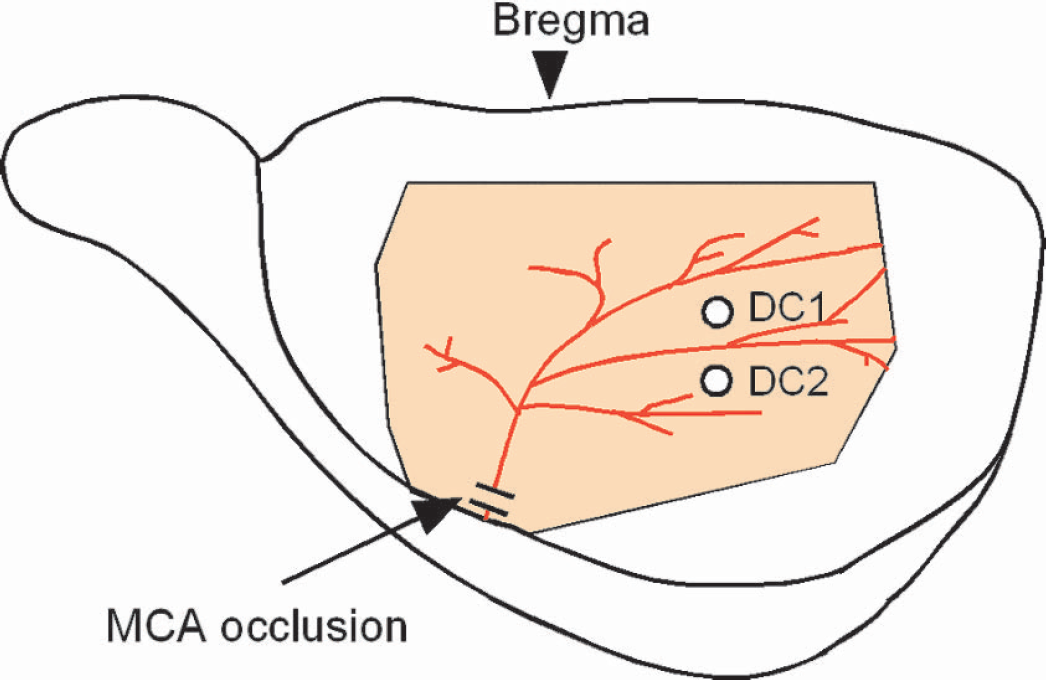
JC-1 has been widely used for the measurement of changes in ψm in isolated mitochondria (Di Lisa et al., 1995), isolated cells (Sharikabad et al., 2001), cultured neurons (Iijima et al., 2003; Yamauchi et al., 2003), and brain slices (Sick and Perez-Pinzon, 1999) but has not been used to measure ψm in vivo. Two studies were performed to evaluate the signals of JC-1 in the current experimental setting during normal conditions. First, the effects of hemoglobin (Hb) concentration in the cortical tissue on the red:green ratio were evaluated because Hb may absorb excitation light and each emission light to different degrees and Hb concentration is expected to change during ischemia/reperfusion. To attenuate Hb concentration without affecting ψm, physiologic saline saturated with oxygen was flushed into the bilateral common carotid arteries from the external carotid arteries at a pressure greater than the systolic arterial pressure for 10 seconds. Although, as shown in Fig. 3A, the intensities of red and green fluorescence were increased, corresponding to the attenuation of Hb concentration, the red:green ratio was stable during the infusion (99% of the control level). This observation indicates that the red:green ratio is minimally affected by changes in Hb concentration.
: Effects of hemoglobin concentration in cortical tissue upon red and green fluorescence and upon the red:green ratio were evaluated by infusion of oxygen-saturated saline into the bilateral common carotid arteries. Triangles (red), rectangles (green), and circles (blue) represent changes in intensities of red and green fluorescence and in the red:green ratio, respectively. Note that intensities of red and green fluorescence were increased by lowering the hemoglobin concentration, but the red:green ratio was stable during the infusion.
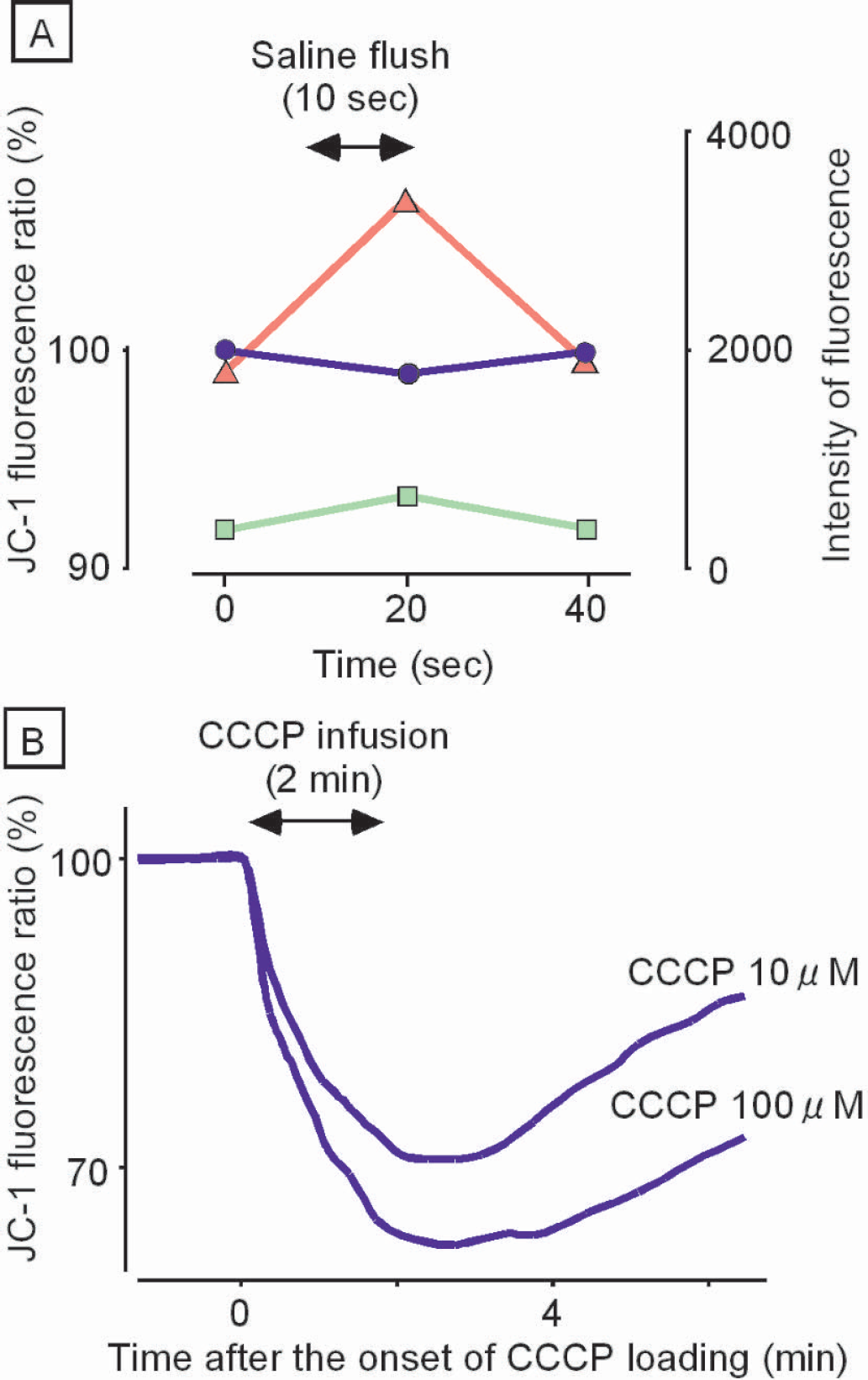
Second, the semiquantitative response of the red:green ratio to changes in ψm was evaluated by collapsing ψm using carbonyl cyanide m-chlorophenylhydrazone (CCCP), which is a strong uncoupling agent of mitochondria. CCCP (0.5 mL) was infused into the subdural space from the fronto-parietal region over a period of 2 minutes. The infusion pressure was monitored and was maintained at less than 10 mmHg by simultaneous withdrawal of CCCP from another slit made on the dura in the temporal region. As shown in Fig. 3B, the red:green ratio was decreased to 70% and 62% of the control level by infusing 10 μM and 100 μM of CCCP, respectively, suggesting that the red:green ratio of JC-1 dye is a semiquantitative indicator of ψm in vivo.
Measurements of changes in ψm and DC potential in vivo
Changes in ψm and DC potential during a 10-minute period of focal ischemia were observed in 14 rats. In 7 animals, dye was injected into cortical tissue to observe ψm (control group). In the other 7 animals, oligomycin (10 ng, 5 μg/ml) was co-injected with the dye to observe ψm without ATP synthetase activity (oligomycin group). The values of physiological parameters obtained before the onset of focal ischemia are shown in Table 1. The values of all parameters were maintained within normal limits during the experimental period.
Physiological variables measured before the onset of ischemia
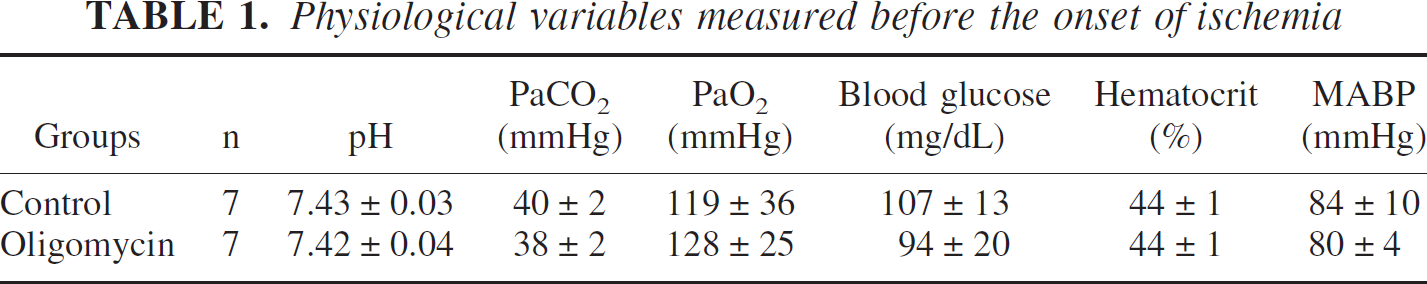
Within 20 seconds after the onset of ischemia, ψm in the control group decreased to 91 ± 5% of the pre-ischemia level (Fig. 4A), and this level of ψm was maintained for 118 ± 38 seconds until the occurrence of another decline. The second decline in ψm was observed at the same time as a negative deflection in DC potential, indicating the occurrence of anoxic depolarization in the cellular membrane. In contrast to the bi-phasic changes in ψm in the control group, the oligomycin (ATP synthetase inhibitor) group showed a single phasic change (Fig. 4B). In the oligomycin group, ψm started to decrease within 20 seconds after the onset of ischemia. Negative shift of DC potential (44 ± 26 seconds after the onset of ischemia) was observed later than the depolarization of ψm (P = 0.02). Because it has been reported that tissue oxygen tension decreases rapidly after the onset of ischemia (from 17.9 ± 10.2 mmHg to 6.6 ± 9.6 mmHg at the rate of 6.41 ± 4.75 mmHg/10 seconds) (Raffin et al., 1991), it is thought that the rapid decrease in ψm in the oligomycin group is related to attenuated activity of the proton pump in the mitochondria because of the depletion of oxygen in cortical tissue.
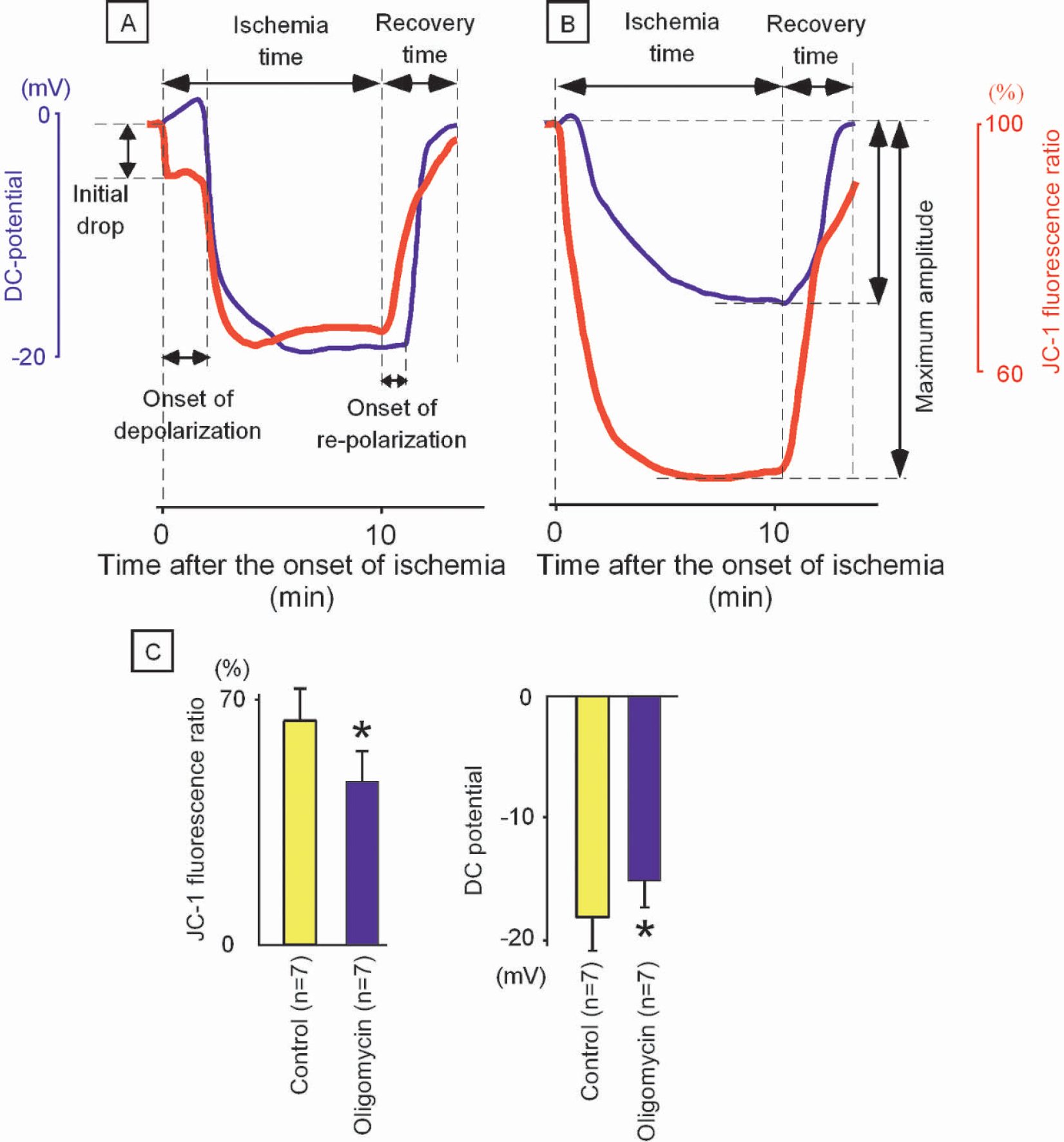
A: Changes in ψm (red line) and DC potential (blue line) during a 10-minute period of focal ischemia in the control group. Note that ψm showed a bi-phasic change with the initiation of ischemia. The second decline in ψm was observed at the same time as depolarization in DC potential.
The onset time of DC deflection in the oligomycin group was significantly shorter than that in the control group (44 ± 26 seconds vs. 118 ± 38 seconds, P = 0.002). Because it has been reported that ATP content is decreased to 69% and 45% of the control level when astrocytes are incubated with oligomycin for 2 and 20 minutes, respectively (Juthberg and Brismar, 1997), it is assumed that the pre-ischemic level of ATP in the oligomycin group was attenuated because of inhibition of the forward activity of ATP synthetase and was depleted in the early stage after the onset of ischemia.
Because forward activity of ATP synthetase consumes the hydrogen gradient and decreases ψm, to produce ATP, if ATP synthetase was producing ATP in the ischemic core (Fig. 1C), inhibition of ATP synthetase by oligomycin should have prolonged the onset of mitochondrial depolarization. In the oligomycin group, however, mitochondrial depolarization was observed within 20 seconds after the onset of ischemia and was faster than DC depolarization. This observation suggests that ATP synthetase in mitochondria was not making ATP but was consuming ATP to complement its insufficient membrane potential in the ischemic core (Fig. 1B).
In the control group, ψm was maintained at 91 ± 5% for 118 ± 38 seconds, and the second decline in ψm was observed at the same time as depolarization of cellular membrane potential. It is thought that ψm was maintained at 91 ± 5% by consuming ATP in the cytosol with reversed activity of ATP synthetase. The fact that ψm and cellular membrane potential were simultaneously lost in the control group indicates that both ATP synthetase on the mitochondrial membrane and Na+, K+-ATPase on the cellular membrane used the same energy source in the cytosol (Fig. 1B).
It is thought that mitochondria at this point were not a generator of energy but had become one of the consumers of cytosolic ATP. The symbiotic relationship between mitochondria and the neuronal cell could be terminated by a critical decrease in blood supply. In support of this notion, it has been reported that the rate of ATP depletion in heart muscle after the onset of ischemia is delayed by 90% by the use of oligomycin, suggesting that ATP is consumed by the reversed activity of ATP synthetase in mitochondria (Jennings et al., 1991; Rouslin et al., 1986).
During the period of ischemia, DC potential was lower in the control group (–18 ± 3) than in the oligomycin group (–15 ± 2 mV, P = 0.04), whereas ψm was higher in the control group (66 ± 9%) than in the oligomycin group (46 ± 8%, P = 0.0002). This observation also supports the hypothesis that mitochondria consume ATP to complement insufficient ψm during ischemia. Because the binding constant of ATP synthetase for ATP (Km = 0.1 nM) (Kozlov and Skulachev, 1982) is 10,000 times lower than that of Na+, K+-ATPase in the cellular membrane (Km = 1 μM) (Robinson, 1983; Yamaguchi and Tonomura, 1980), ATP in the cytosol is expected to be predominantly used by the reversed activity of ATP synthetase. It is thought that cellular membrane potential in the control group was compromised by the depletion of ATP level caused by the consumption by mitochondria.
The reversed activity of ATP synthetase during ischemia (not only at the onset of ischemia) may have detrimental effects upon cell survival because of the following mechanisms. First, an increase in DC deflection that is caused by the depletion of ATP results in acceleration of calcium influx into the cytosol. Second, because calcium uptake is facilitated by the electrochemical gradient in mitochondria, maintenance of ψm during cellular membrane depolarization accelerates calcium uptake into the mitochondria and may lead to mitochondrial dysfunction and to the initiation of permeability transition. In this context, Belisle and colleagues (2002) found that diazoxide, an opener of the mitochondrial K+ATP channel, is capable of inhibiting reversed activity of ATP synthetase without attenuating forward activity of ATP synthetase. Diazoxide decreases ψm and calcium uptake into the mitochondria during anoxia without attenuating ATP content during the reperfusion period. The neuroprotective effects of diazoxide (Shimizu et al., 2002) could be related to inhibition of the reversed activity of ATP synthetase during ischemia.
CONCLUSION
ψm is maintained for 118 ± 38 seconds after the onset of ischemia by reversing ATP synthetase activity. During ischemia, mitochondria behave as organelles that consume ATP in the cytosol. The reversed activity of ATP synthetase seems to continue during ischemia and appears to compete with the cellular membrane Na+, K+-ATPase for ATP in the cytosol. This reversed activity of ATP synthetase compromises cellular membrane potential and may facilitate calcium influx into the cytosol and mitochondria during ischemic depolarization, leading to the uncontrolled activation of numerous enzymes and to dysfunction of mitochondria, respectively. Attenuation of the reversed activity of ATP synthetase without inhibiting the forward activity of ATP synthetase could be beneficial for neuroprotection during ischemia.