
Abstract
Activation of mitochondrial ATP-sensitive potassium (mitoKATP) channels protects the brain against ischemic or chemical challenge. Unfortunately, the prototype mitoKATP channel opener, diazoxide, has mitoKATP channel-independent actions. We examined the effects of BMS-191095, a novel selective mitoKATP channel opener, on transient ischemia induced by middle cerebral artery occlusion (MCAO) in rats. Male Wister rats were subjected to 90 mins of MCAO. BMS-191095 (25 μg; estimated brain concentration of 40 μmol/L) or vehicle was infused intraventricularly before the onset of ischemia. In addition, the effects of BMS-191095 on plasma and mitochondrial membrane potentials and reactive oxygen species (ROS) production in cultured neurons were examined. Finally, we determined the effects of BMS-191095 on cerebral blood flow (CBF) and potassium currents in cerebrovascular myocytes. Treatment with BMS-191095 24 h before the onset of ischemia reduced total infarct volume by 32% and cortical infarct volume by 38%. However, BMS-191095 administered 30 or 60 mins before MCAO had no effect. The protective effects of BMS-191095 were prevented by co-treatment with 5-hydroxydecanoate (5-HD), a mitoKATP channel antagonist. In cultured neurons, BMS-191095 (40μmol/L) depolarized the mitochondria without affecting ROS levels, and this effect was inhibited by 5-HD. BMS-191095, similar to the vehicle, caused an unexplained but modest reduction in the CBF. Importantly, BMS-191095 did not affect either the potassium currents in cerebrovascular myocytes or the plasma membrane potential of neurons. Thus, BMS-191095 afforded protection against cerebral ischemia by delayed preconditioning via selective opening of mitoKATP channels and without ROS generation.
Introduction
Previous studies have provided evidence indicating that the activation of mitochondrial ATP-sensitive potassium (mitoKATP) channels leads to the development of ischemic tolerance in the brain. Unfortunately, research in this area has been hindered by the lack of specific agonists of the mitoKATP channel. Diazoxide, a prototypic mitoKATP channel opener used in the majority of these studies in brain as well as heart (Busija et al, 2005; Kis et al, 2003, 2004; Liu et al, 2002; Nagy et al, 2004; Rajapakse et al, 2003, 2002; Teshima et al, 2003), has also been reported to inhibit the mitochondrial enzyme succinate dehydrogenase (SDH) (Dzeja et al, 2003; Hanley et al, 2002; Kis et al, 2003). Consistent with this evidence, we and others have shown that diazoxide-induced preconditioning in neuronal tissue involves complex mechanisms: direct actions because of activation of mitoKATP channels as well as indirect actions of superoxide anion produced after inhibition of SDH (Kis et al, 2003). To further investigate the role of mitoKATP channel opening in neuronal preconditioning in one previous study, we used BMS-191095, a novel, very selective mitoKATP channel opener in cultured neurons. The administration of BMS-191095 was able to protect cultured neurons against anoxic and chemical stresses via mitoKATP channel activation without other apparent, non-specific effects (Kis et al, 2004). Also, in isolated mitochondria, we have shown that opening of mitoKATP channels with BMS-195095 was associated with depolarization but not with an increase in the production of reactive oxygen species (ROS) (Busija et al, 2005). However, since the degree of mitochondrial depolarization induced in mitochondria by BMS-191095 was less than by diazoxide (Kis et al, 2003, 2004), it is unclear whether BMS-191095 would be effective against ischemic challenge in the intact brain.
The purpose of this study was to examine the ability of BMS-191095 to protect the brain against ischemia. We tested the hypothesis that BMS-191095 would induce both acute and delayed tolerance against transient, focal ischemia in rats. We also examined the effect of BMS-191095 on plasma and mitochondrial membrane potentials and ROS production in cultured neurons. In addition, we determined the effects of BMS-191095 on cerebral blood flow (CBF) and cerebral arterial myocyte potassium currents.
Materials and Methods
Animals
Male Wistar rats weighing 270 to 300 g were used in the experiments. The procedures were approved by the Animal Care and Use Committee of Wake Forest University Health Sciences.
Experimental Design
BMS-191095 (Bristol-Myers Squibb, Princeton, NJ, USA) was dissolved in dimethyl sulfoxide and 15 μL of the solution was infused into the left lateral ventricle under stereotaxic guidance (AP −0.8 mm, lateral 1.5 mm, and dorsoventral 3.7 mm) as described by Nishimura et al (1995), with some modifications.
To examine acute preconditioning, drugs were applied 30 or 60 mins before the induction of ischemia. In the first acute preconditioning group, animals were treated with BMS-191095 (2.5 mg, n = 11, or 25 mg, n = 6) or vehicle alone (n = 17) 30 mins before transient, middle cerebral artery occlusion (MCAO). Assuming that the drug evenly disperses in the brain, the estimated final tissue concentration of BMS-191095 would be 4 and 40 μmol/L, respectively. In a previous study, intraventricularly injected diazoxide produced acute preconditioning against 90 mins MCAO at approximately this dose (Shimizu et al, 2002). Since BMS-191095 induced preconditioning at a 10 times lower concentration in cultured cortical neurons (Kis et al, 2004), we also injected the lower dose of BMS-191095. In the 60 mins preconditioned group, only 25 mg BMS-191095 (n = 7) or vehicle (n = 6) was applied.
To examine delayed preconditioning, animals were treated 24 h before the induction of ischemia with 25 mg BMS-191095 (n = 13) or vehicle (n = 13). In other animals, the mitoKATP channel inhibitor 5-hydroxydecanoate (5-HD; Sigma, St Louis, MO, USA, 40 mg/kg, in sodium chloride 0.9%, i.p.) was administered 30 mins before the administration of 25 mg BMS-191095 (n = 12) or vehicle (n = 10).
Middle Cerebral Artery Occlusion
Animals had free access to food and water before the surgery. Anesthesia was induced with 5% halothane in a 70:30 gas mixture of N2O and O2. Endotracheal intubation was performed, and all the animals were mechanically ventilated with 1.0% to 1.5% halothane in a 70:30 gas mixture of N2O and O2. The right femoral artery was cannulated to monitor mean arterial blood pressure, arterial blood gases, and pH. During surgery, rectal temperature was maintained in a range between 36.5°C and 37.5°C by a heating pad. Transient focal brain ischemia was induced using the MCAO filament method as previously described, with some modifications (Belayev et al, 1996; Longa et al, 1989). In brief, the common carotid artery (CCA), the external carotid artery (ECA), and the internal carotid artery (ICA) were dissected from surrounding connective tissues via a ventral neck midline incision. The branches of ECA were ligated and cut, and the right pterygopalatine artery was ligated. The ECA was then cut between double ligations at the distal portion. Two microvascular clips were placed across the CCA and ICA. A 3–0 monofilament nylon suture with its tip rounded by heating and coated with silicon was introduced into the ECA lumen and advanced into the ICA until some resistance was felt. The suture around the ECA stump was then tightened. The proximal microvascular clip was removed and the incision was closed. After awakening from anesthesia, rats were extubated and kept in the cage during 90 mins of MCAO. The animals were then reanesthetized to remove the suture and to ligate the stump of the ECA. After reperfusion, the rats were kept in a cage and allowed free access to food and water.
Measurement of Infarct Volume
Three days after MCAO, all animals were anesthetized with 5% halothane in O2 and decapitated. The brain was quickly removed and sliced into coronal sections at 2 mm intervals. Each slice was immersed in a 2% solution of 2,3,5-triphenyltetrazolium chloride (Sigma) for 20 mins and then fixed in a 10% formaldehyde solution. The infarct area in each brain slice was determined on the NIH Image program (version 1.62) according to the indirect method proposed by Swanson et al (1990). Infarct volumes were calculated by summation of the infarct area in six brain slices (2 to 14 mm from frontal pole) and integrated by thickness (2 mm) and were expressed in cubic millimeters.
Cerebral Blood Flow Measurement
We measured CBF in anesthetized, spontaneously breathing rats before and after injection of 25 mg BMS-191095 or vehicle using a laser-Doppler probe with a diameter of 0.25 mm (Perimed; Probe 403, Sweden). The head of the animal was secured in a stereotactic frame and the probe was placed over the thinned skull of the right hemisphere 2 mm caudal and 4 mm lateral to bregma. The CBF was determined before injection and then for 30 mins after injection of BMS-191095 or vehicle.
Effect on Whole-Cell K+ Currents in Cerebral Artery Myocytes
Single myocytes were obtained by enzymatic treatment of cerebral arteries as described previously (Katakam et al, 2006). Briefly, isolated cerebral arteries were placed in the dissociation medium (in mmol/L concentration: 110 NaCl, 5 KCl, 2 MgCl2, 0.16 CaCl2, 10 N-2-hydroxyethylpiperazine-N'°-2-ethanesulfonic acid (HEPES), 10 NaHCO3, 0.5 KH2PO4, 0.5 NaH2PO4, 0.49 ethylenediaminetetraacetic acid, 10 taurine, 10 glucose; pH 6.9) containing albumin (4 mg/ml) with papain (1.52 mg/ml) and dithiothreitol (0.54 mg/ml). Cells were dispersed for 30 mins in a shaking water bath at 37°C followed by mild trituration. The solution was centrifuged at 1,000g for 12 mins, and the pellet was resuspended in dissociation medium to obtain freshly dispersed smooth muscle cells.
Only spindle shaped, relaxed cells were used for experiments. The recording chamber was perfused with external solution containing (in mmol/L) 145 NaCl, 4 KCl, 1 CaCl2, 1 MgCl2, 10 HEPES, and 10 glucose (pH 7.4 at room temperature). Pipettes (5 to 10 megaohms resistance) were filled with internal solution containing (in mmol/L) 140 KCl, 0.1 CaCl2, 0.6 EGTA, and 10 HEPES (pH 7.2). The whole cell, voltage clamp configuration was employed by using Axopatch 200B amplifier, Digidata 1322A analog-to-digital converter and pClamp 10.0 software (Axon instruments, Burlingame, CA, USA). Whole-cell K+ currents were elicited with a holding potential of −60 and 10 mV steps from −50 to +110 mV. All experiments were performed at room temperature. The data were analyzed using Clampfit software.
Primary Rat Cortical Neuronal Culture
Primary rat cortical neurons were isolated from Sprague-Dawley rat E18 fetuses as described previously (Kis et al, 2003). Cells were plated at a density of 2 × 105 cells/cm2 onto poly-D-lysine-coated glass coverslips (Becton-Dickinson, San Jose, CA, USA) for confocal microscopic analysis in plating medium consisting of 60% Dulbecco's-modified Eagle medium (Gibco BRL, Grand Island, NY, USA), 20% F-12 HAM (Sigma), and 20% horse serum (Gibco BRL) supplemented with glutamine (Sigma, 0.5 mmol/L). After cell attachment, the plating medium was replaced by Neurobasal medium (Gibco BRL) supplemented with B27 (Gibco BRL, 2%), L-glutamine (Sigma, 0.5 mmol/L), β-mercaptoethanol (Gibco BRL, 55 μmol/L), and potassium chloride (Sigma, 25 mmol/L). Cultures consisted of >98% neurons verified by positive immunostaining for microtubule-associated protein-2, and by negative immunostaining for glial fibrillary acidic protein.
Analysis of Mitochondrial Membrane Potential
Mitochondrial membrane potential (ΔΨm) was monitored using the ΔΨm-sensitive dye, tetramethylrhodamine ethyl ester (TMRE, Molecular Probes, Eugene, OR, USA). Neuronal cultures were loaded in the dark at 37°C in a 5% CO2 incubator with 0.5 μmol/L TMRE in Neurobasal medium for 15 mins. After loading, the cells were washed three times with Dulbecco's phosphate-buffered saline (PBS, Sigma). Experiments were performed at 22°C in PBS. Confocal images of cellular TMRE fluorescence were acquired on a laser scanning microscope (LSM 510; Zeiss, Jena, Germany) using a 63X water immersion objective (Zeiss). Fields of cells were randomly selected. The cells were treated with BMS-191095 (40 μmol/L) with or without prior incubation with 5-HD (2, 5, 10, and 20 mmol/L) and fluorescent images were recorded every 20 secs for 5 mins after treatment (Λex = 543, Λem>560 nm). The average pixel intensity in individual cell bodies was determined using the software supplied by the manufacturer (Zeiss).
Analysis of Plasma Membrane Potential
Plasma membrane potential was monitored using the voltage-sensitive dye aminonaphthylethenylpyridinium (di-8-ANEPPS, Molecular Probes, Eugene, OR, USA). Neuronal cultures were loaded with 1 μmol/L di-8-AN-EPPS in PBS in the dark for 20 mins at 37°C, and then were washed three times with PBS. Confocal images of cellular di-8-ANEPPS fluorescence were acquired using the same laser scanning microscope as was used for ΔΨm measurements. The cells were pretreated with 5-HD (20 mmol/L) to eliminate mitoKATP activity, then fluorescent images (Λex = 488 nm, Λem1 > 650 nm, and Λem2 = 500 to 550 nm) of randomly selected fields were recorded every 20 secs for 10 mins after the application of BMS-191095 (40 μmol/L) or NS-1619 (10 μmol/L), a large conductance calcium-dependent potassium channel opener. The average pixel intensity of individual cell bodies was determined using the software supplied by the manufacturer (Zeiss). Ratio of emissions (Λemi/Λem2) was calculated and data were expressed as a percentage of the starting ratio of the corresponding control culture pretreated with 5-HD.
Analysis of Neuronal Reactive Oxygen Species Formation
Reactive oxygen species generation was assessed using the oxidation-sensitive dye hydroethidine (Molecular Probes), which is oxidized to the fluorescent ethidium by free radicals. Neuronal cultures were preincubated with 5 mmol/L hydroethidine in PBS for 3 mins at 22°C in the dark. Confocal images of cellular hydroethidine fluorescence were acquired using the same confocal microscopy systems used for ΛΨm measurement. BMS-191095 (10, 20, 30, and 40 μmol/L) was applied into the medium and fluorescent images were obtained (Λex = 488, Λem>560 nm). Images were recorded every 20 secs for 10 mins after treatment and the average pixel intensity in individual cell bodies was determined using the software supplied by the manufacturer (Zeiss).
Statistical Analysis
All data are expressed as mean ± s.e.m. One-way analysis of variance (ANOVA) followed by the Fisher's least significant difference test was performed to assess statistical differences for infarct volumes and physiologic parameters. In the case of CBF measurement, values of different time points were compared by one-way repeated measures ANOVA. One way ANOVA followed by Tukeys test (when appropriate) was used to analyze the data obtained from confocal microscopy and patch-clamp experiments. A probability value < 0.05 was regarded as statistically significant.
Results
Physiologic Parameters
The systemic physiologic parameters were within the normal range in all animals studied and showed no statistically significant differences among treatment groups (Table 1).
Physiological parameters of rats before MCAO
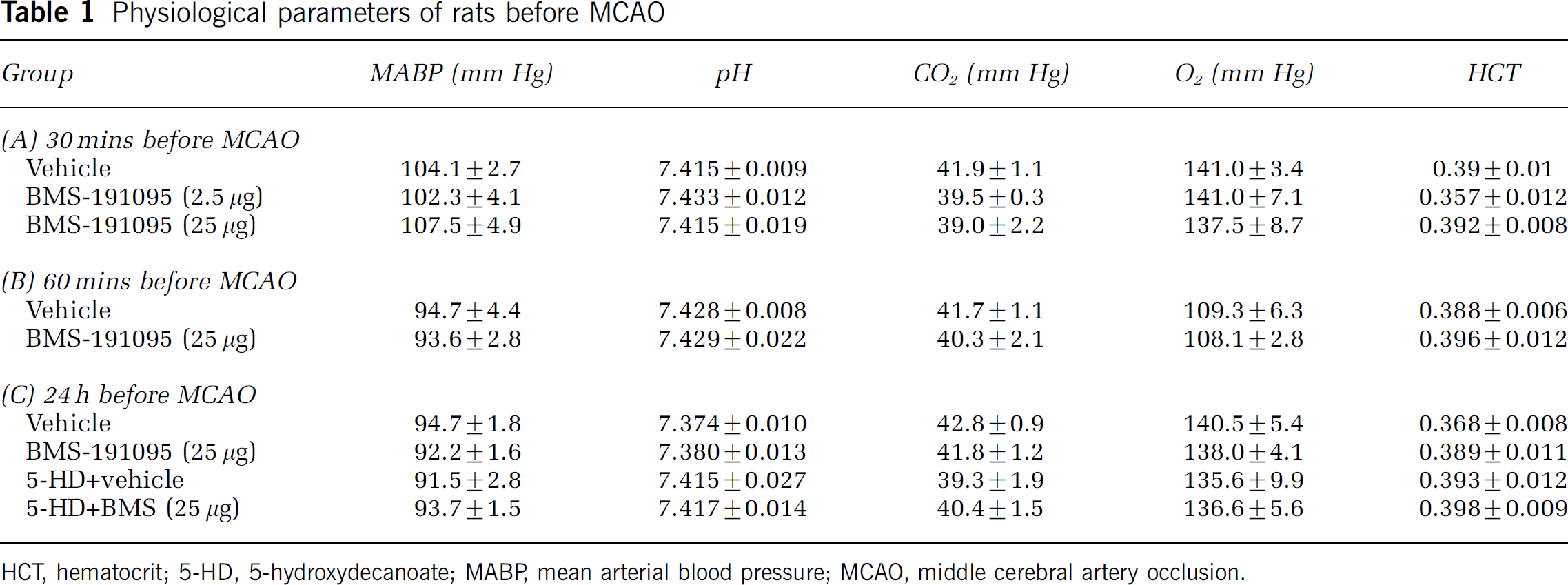
HCT, hematocrit; 5-HD, 5-hydroxydecanoate; MABP mean arterial blood pressure; MCAO, middle cerebral artery occlusion.
Infarct Volume
Treatment with 2.5 and 25 mg BMS-191095 30 mins before MCAO failed to decrease infarct volume compared with the vehicle treatment. Total infarct volumes were 200.8±18.6, 182.9±21.8, and 200.0±33.0 mm3 for the vehicle rats (n = 17), for the 2.5 mg BMS-191095- (n = 11) treated rats, and for the 25 mg BMS-191095- (n = 6) treated rats, respectively. Treatment with 25 mg BMS-191095 60 mins before MCAO did not reduce infarct volume. Total infarct volume was 226.0±24.8 mm3 in the vehicle-treated animals (n = 6) and 248.9±13.3 mm3 in the 25 mg treated rats (n = 7). Cortical and subcortical infarct volumes were not affected by any of the acute BMS-191095 treatments.
However, total infarct volume was reduced from 206.2±16.5 to 141.1±18.1 mm3 in animals pretreated with 25 mg BMS-191095 24 h before MCAO (32% reduction, P < 0.05). This decrease occurred primarily in the cerebral cortex where a 38% reduction of infarct volume occurred (Figure 1). BMS-191095 did not protect the subcortex.
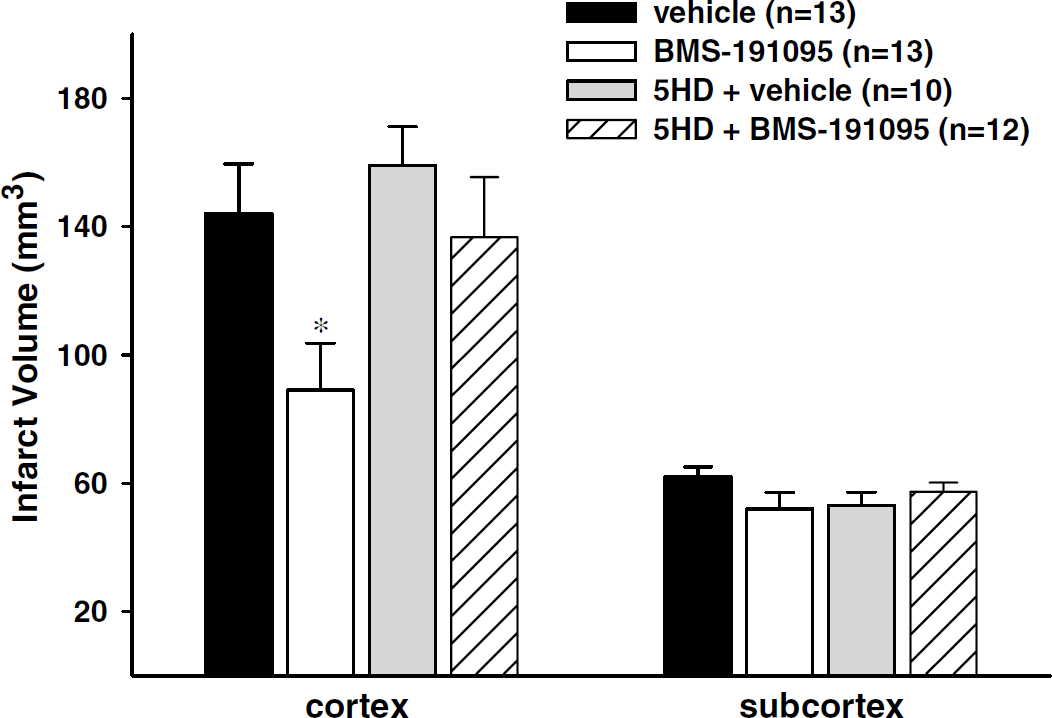
Pretreatment with BMS-191095 protects the brain against MCAO. Cortical and subcortical infarct volumes 72 h after 90 mins of MCAO in animals pretreated with vehicle, 25 mg BMS-191095, 5-HD, or BMS-191095 plus 5-HD 24h before MCAO. BMS-191095 pretreatment reduced cortical infarct volume and this effect was eliminated by co-treatment with 5-HD. Values are mean ± s.e.m. *P < 0.05 versus vehicle.
The administration of 5-HD completely abolished the delayed preconditioning effect of BMS-191095 in cortex. 5-Hydroxydecanoate alone had no significant influence on infarct volumes (Figure 1).
Cerebral Blood Flow Measurement
At every time point examined, the CBF values of animals treated with BMS191095 were not significantly different from those of animals treated with vehicle (Figure 2). However, a slight reduction was found in both groups after injection. Arterial blood pressure, rectal temperature, arterial blood gases, and pH values were similar in both treatment groups and were within normal values for rats (data not shown).
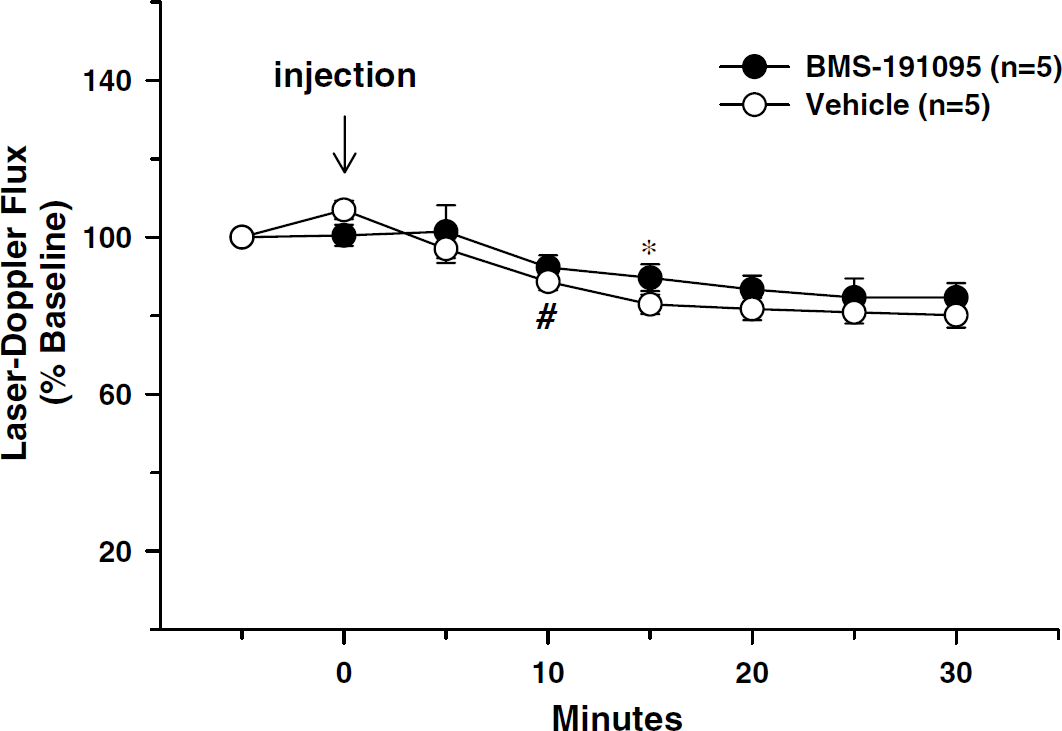
Minimal effects on CBF of BMS-191095. CBF is expressed as percentage of the measured laser-Doppler flux compared with baseline. CBF in BMS-191095-treated animals and vehicle-treated animals were not significantly different from each other, but a minor reduction was found in both treatment groups compared with baseline. Values are shown as mean ± s.e.m. *P < 0.05 versus baseline in the BMS191095 group, #P < 0.05 versus baseline in the vehicle group.
Whole-Cell K+ Currents in Cerebral Artery Myocytes
Application of 40 mmol/L BMS-191095 had no effect on either the current–voltage relationship or peak K+ currents from freshly dissociated cerebral artery myocytes (Figure 3). The peak whole-cell K+ currents normalized to cell capacitance were 591+ 88 pA at baseline and 558±90pA after the administration of 40 μmol/L BMS-191095 (P =NS; n = 5).
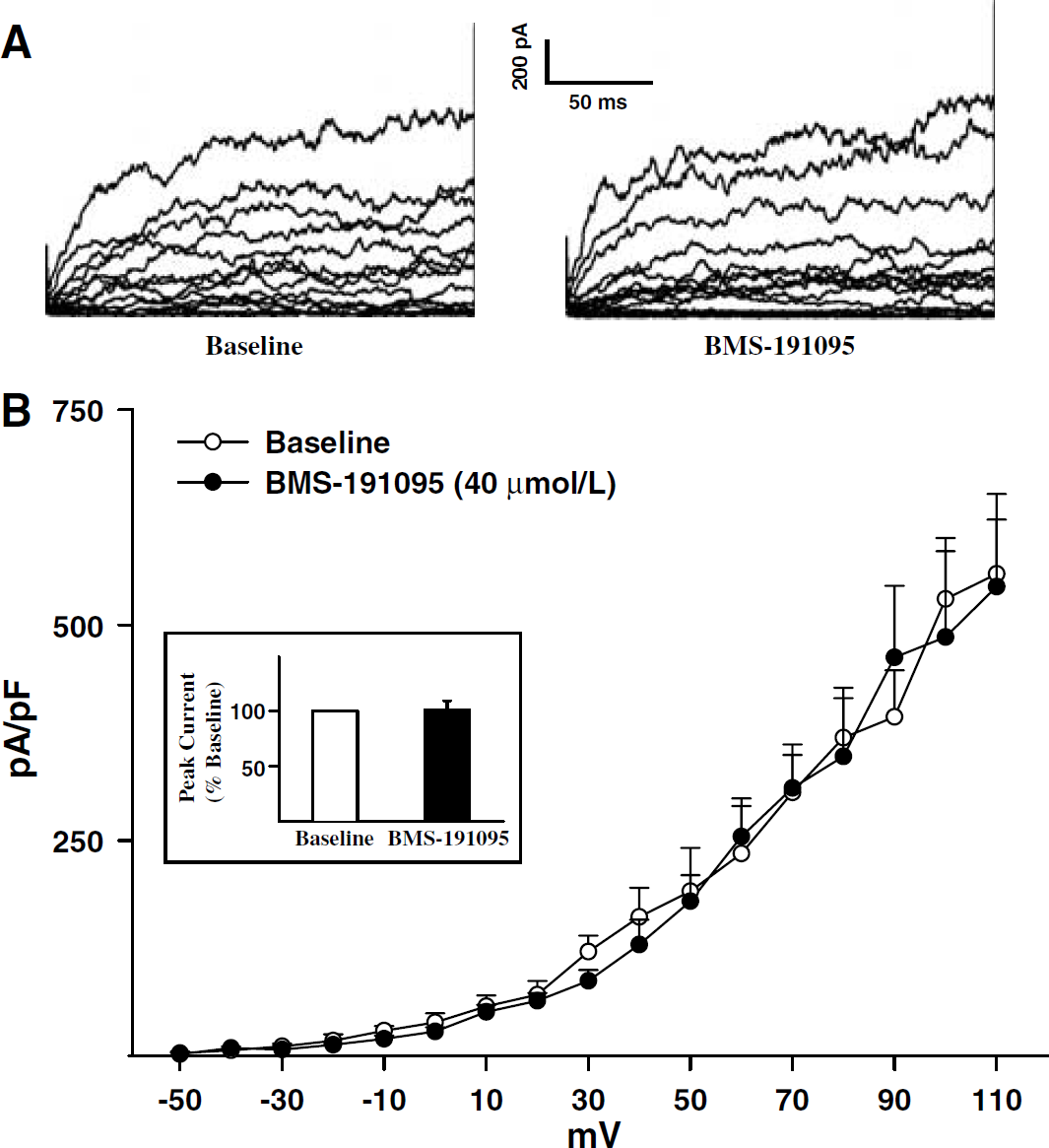
BMS-191095 does not change whole-cell currents. Whole-cell voltage-clamp currents were evoked in freshly dissociated cerebral artery myocytes from Wistar rats by 200 ms, 10 mV steps between −50 and +110 mV from holding potential of −60 mV. Representative traces obtained in the presence and absence of 40 μmol/L BMS-191095 are shown (
Mitochondrial Membrane Potential
Administration of 40 μmol/L BMS-191095 resulted in a rapid mitochondrial depolarization (Figure 4). This effect was significantly and dose dependently inhibited by pretreatment with 5-HD starting at 2 mmol/L. At a concentration of 5 mmol/L, 5-HD almost completely blocked the effect of 40 μmol/L BMS-191095 (Figure 4).
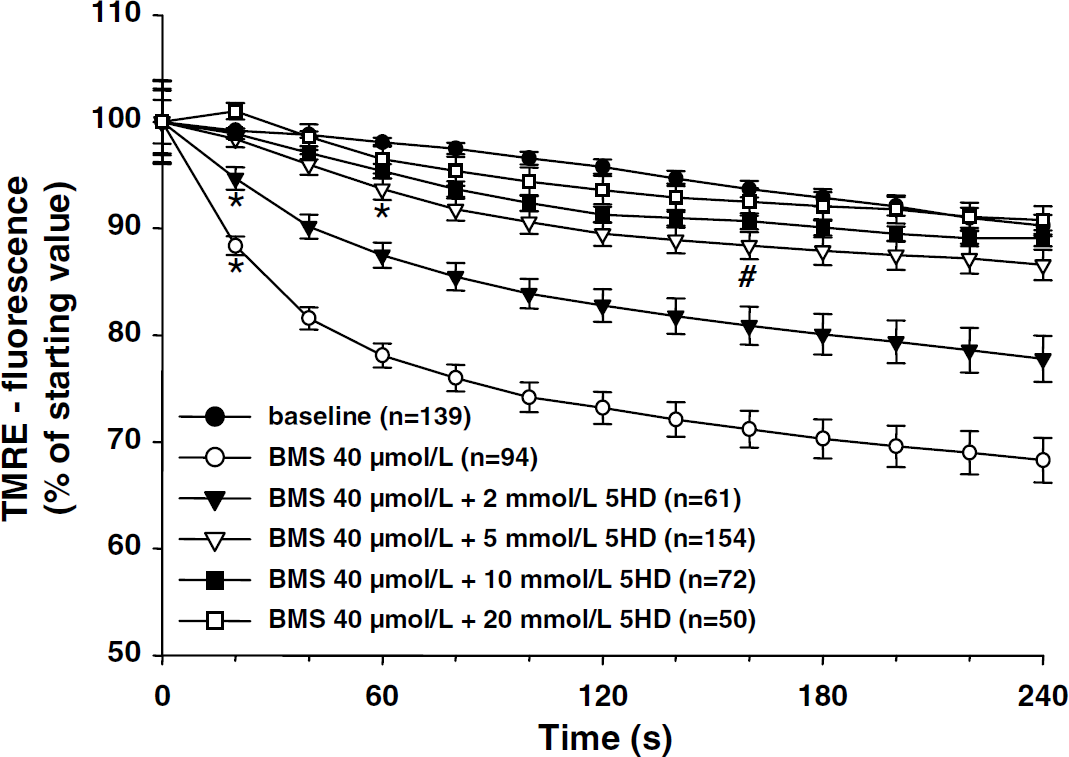
BMS-191095 depolarizes neuronal mitochondria. TMRE fluorescence was measured by confocal microscopy in vehicle- and BMS-191095- (40 μmol/L) treated neurons in the absence and presence of different concentrations of 5-HD (2, 5, 10, and 20 mmol/L). BMS-191095 depolarized mitochondria and this effect was dose dependently reversed by 5-HD. Depolarization is indicated by a decrease in TMRE intensity. Values represent mean ± s.e.m. *P < 0.05, first significant difference compared with baseline. #P < 0.05, last significant difference compared with baseline.
Plasma Membrane Potential
Pretreatment with 20 mmol/L 5-HD alone did not change the fluorescence intensity ratio. Application of 10 μmol/L NS-1619 increased the ratio, which indicates the hyperpolarization of the plasma membrane potential. However, administration of 40 μmol/L BMS-191095 had no significant effect (Figure 5).
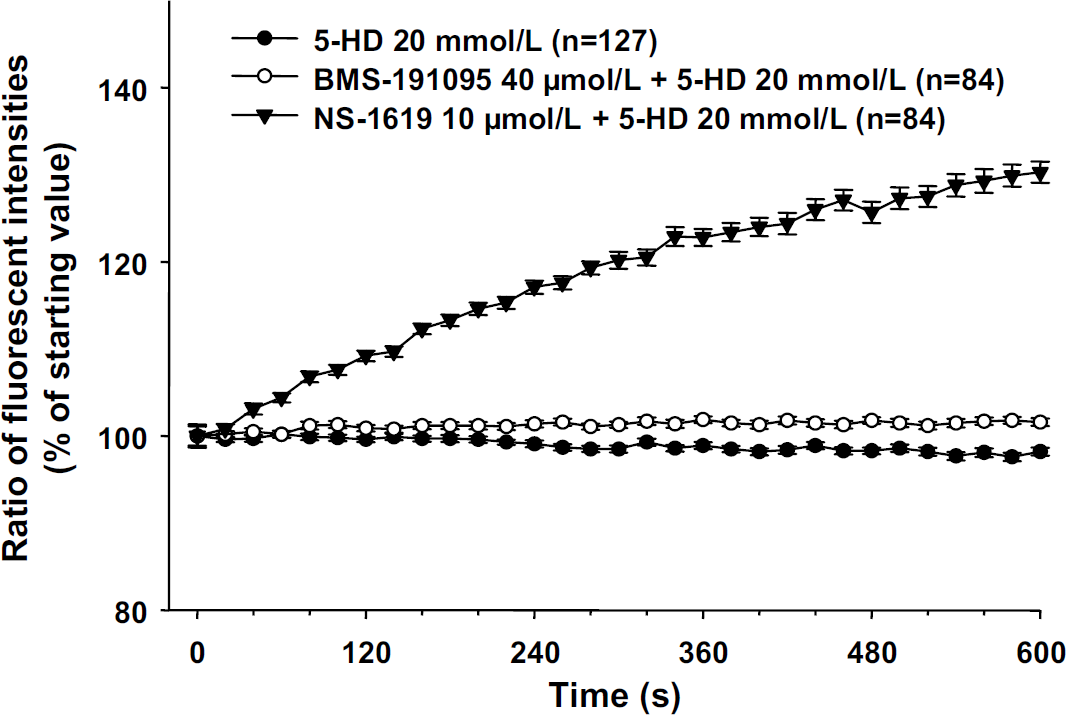
BMS-191095 does not alter plasma membrane potential. The effect of BMS-191095 on plasma membrane potential. Pretreatment with 5-HD (20 mmol/L) had no effect on Di-8-ANEPPS fluorescence ration (emission, >650 nm/500 to 550 nm). NS-1619 (10 μmol/L) hyperpolarized the membrane potential but BMS-191095 (40 μmol/L) had no significant effect.
Reactive Oxygen Species Production
The fluorescence intensity in the control group increased moderately because of normal basal ROS production of the neurons. BMS-191095 did not increase ROS production up to a dose of 40 μmol/L (Figure 6).
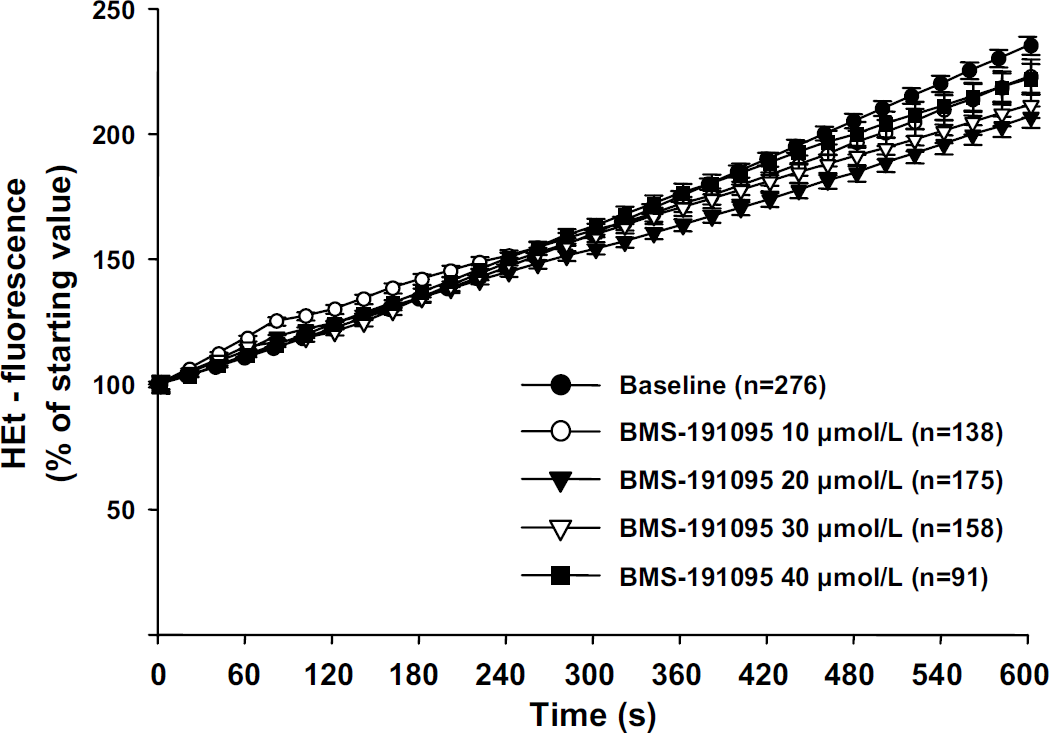
BMS-191095 does not elicit ROS production from neurons. The effect of BMS-191095 on ROS production in cultured cortical neurons. BMS-191095 (40 μmol/L) did not increase ROS production measured with hydroethidine (HEt) at a dose which was sufficient to depolarize mitochondria.
Discussion
In this study, we provide the first evidence that pretreatment with the novel mitoKATP channel agonist BMS-191095 produces a delayed preconditioning effect against transient focal ischemia in the rat brain and that this neuroprotection occurs without initial generation of ROS. In the brain and cerebral circulation, effects of BMS-191095 appear to be selective for the mitoKATP channel. Thus, mitoKATP channel opening alone is sufficient to protect the brain against ischemic injury.
Activation of mitoKATP channels has been proposed to play a pivotal role in neuronal preconditioning. It is well known that there are two temporally distinct types of preconditioning. Acute preconditioning occurs within 2 to 3 h and delayed preconditioning is observed between 24 and 72 h after the initiating event. Diazoxide, a prototype mitoKATP channel opener, induced neuronal preconditioning in different in vitro and in vivo preparations (Domoki et al, 2004; Garcia de Arriba et al, 1999; Kis et al, 2003; Liu et al, 2002; Nagy et al, 2004; Rajapakse et al, 2002; Shake et al, 2001; Shimizu et al, 2002; Teshima et al, 2003). However, diazoxide also inhibits SDH, which leads to increased ROS production. Co-administration of superoxide dismutase with diazoxide blocked the protection in cultured neurons, suggesting that SDH inhibition plays an important role in diazoxideinduced neuroporotection and that the role of the mitoKATP channel in this process remains unclear (Kis et al, 2003, 2004). In addition, while diazoxide is fairly selective for cardiac mitoKATP channels, at relatively high doses it can open surface membrane KATP channels. Diazoxide indeed is a sarcolemmal KATP channel opener in vascular smooth muscle (Garlid et al, 1997) and a smaller dose is needed to reduce arterial blood pressure than to produce a cardioprotective effect (Grover and Atwal 2002). However, although local application to the brain does not dilate surface arteries (Domoki et al, 1999) and affect CBF(Shimizu et al, 2002), when administered systemically, diazoxide increases (Liu et al, 2000) or decreases CBF (Lenzser et al, 2005).
BMS-191095, which apparently does not possess inhibitory effects against SDH, was reported to be highly selective for mitoKATP channels and has potent cardioprotective effects without inducing peripheral vasodilatation or sarcolemmal KATP channel opening (Grover et al, 2001). Thus, investigation of the protective effect of this novel compound allows us to separate the known side effects of diazoxide from the effects of mitoKATP activation. The protective effects of BMS-191095 have been well investigated in the heart (Fischbach et al, 2004; Gross et al, 2003; Grover et al, 2001, 2002; Neckar et al, 2001), but in the brain, there is only one published study with cultured cortical neurons. In cultured neurons, BMS-191095 depolarized the mitochondria in a dose-dependent manner and induced delayed preconditioning against oxygen–glucose deprivation (OGD) or glutamate toxicity probably through a mechanism that involved reduced ROS effects at the time of stress (Kis et al, 2004).
In the present study, we confirm the neuroprotective effect of BMS-191095 in an in vivo ischemia/reperfusion model. We also report the evidence that BMS-191095 selectively activates mitoKATP channels without direct effect on plasma membrane potential in cortical neurons. These results support the preconditioning effects of selective mitoKATP channel activation. The fact that there was no significant difference in the alterations of CBF between BMS-191095-treated and vehicle-injected groups indicates that BMS-191095 has little or no direct effect on cerebrovascular tone. The absence of significant vasodilation in the cerebral circulation also supports the selective activation of mitoKATP channels by BMS-191095 in the brain. It is obvious that the slight decrease found in both treatment groups is not responsible for the mechanism of delayed preconditioning produced by BMS-191095. However, the exact mechanism of the mild reduction is not clear. It could be because of some unrecognized technical aspect of our experimental protocols. To rule out non-specific vascular activity of BMS-191095, we recorded whole-cell K+ currents in myocytes isolated from cerebral arteries. BMS-191095 at the concentration (40 μmol/L) we used in our experiments, had no significant effect on wholecell K+ currents. This is in contrast to the previous report in isolated ventricular myocytes (Grover et al, 2001) where BMS-191095 inhibited delayed rectifier K+ currents. The discrepancy in the observations may be explained by the differences in the tissues studied. Importantly, the lack of effect on plasma membrane K+ currents supports the proposed mitochondrial selectivity of BMS-191095.
BMS-191095 failed to display acute preconditioning in our experiments. In a previous study, diazoxide showed acute preconditioning in rat transient MCAO experiments and the protection occurred in both cortical and subcortical areas (Shimizu et al, 2002). Diazoxide reduced infarct volume up to 50% in the same 90 mins MCAO model (Shimizu et al, 2002), which is moderately greater than the protective effect found in the present study with BMS-191095. Diazoxide also induces greater maximal mitochondrial depolarization compared with BMS-191095 (Kis et al, 2003, 2004). It is likely that the combined effects of diazoxide, involving both mitoKATP channel activation and ROS production, lead to a greater extent of neuroprotection compared with only one of these mechanisms.
In the present study, we provide evidence that the application of BMS-191095, at a concentration which was able to depolarize mitochondria, did not increase ROS production. This result is consistent with a previous finding from our laboratory using isolated mitochondria from piglet brain (Busija et al, 2005). Based on this evidence, we suggest that both diazoxide and BMS-191095 induce preconditioning via mitoKATP channel activation with a mechanism dissociated from ROS generation, and that the effect of diazoxide is probably enhanced by ROS generated by SDH inhibition. The ability of direct SDH inhibition and ROS generation to protect the brain against subsequent ischemic insult, via complex mechanisms probably involving in part ROS-mediated mitoKATP channel activation, have been described previously (Horiguchi et al, 2003).
In the current study, 5-HD, a putative mitoKATP channel blocker completely abolished the neuroprotective effect of BMS-191095. The effect of BMS-191095 on mitochondrial membrane potential in cultured neurons was also completely prevented by pretreatment with 5-HD. These results were consistent with the previous studies that showed 5-HD to completely reverse the cardioprotective effect of BMS-191095 (Grover and Atwal 2002; Grover et al, 2001). In addition, in our recent study with isolated mitochondria from piglet brain, BMS-191095-induced mitochondrial depolarization was completely inhibited by 5-HD (Busija et al, 2005). In cultured rat neurons, however, 1 mmol/L 5-HD failed to inhibit BMS-191095-induced preconditioning against OGD and enhanced the protective effect against glutamate excitotoxicity (Kis et al, 2004). In our present study, 2 mmol/L 5-HD significantly, but not completely, reversed the effect of BMS-191095 on mitochondrial membrane potential. Thus, a higher concentration of 5-HD might be necessary to reverse the protective effect in cultured neurons for reasons that are unclear at this time.
Recent studies have shown that 5-HD is not a direct inhibitor of the mitoKATP channels but rather a metabolic antagonist of diazoxide-induced effects (Das et al, 2003; Dzeja et al, 2003; Hanley et al, 2002, 2003; Kis et al, 2003; Lim et al, 2002). Indeed, a relatively smaller concentration of 5-HD could reverse the effect of diazoxide than used in the present studies with BMS-191095, which indicates the possibility of additional blocking mechanisms of 5-HD specific to diazoxide. At this time, however, we are unaware of other direct cellular effects of BMS-191095. The results acquired from our present study support the view that 5-HD or a metabolite of 5-HD can act directly as an inhibitor of mitoKATP channels, although we cannot rule out possible effects at other sites.
The complete mechanism of neuroprotection after stimulation of mitoKATP channels is not fully understood. The activation of protein kinase C is likely to be involved as a downstream target (Kis et al, 2004). However, the attenuation of ROS production after development of preconditioning during lethal challenges is thought to be one of the major end effectors in this process (Kis et al, 2003, 2004). Higher selectivity of BMS-191095 for mitoKATP channels seems to be useful for further investigation of the role of mitoKATP channel activation in mediating neuronal preconditioning.