
Abstract
Systemic preconditioning with the TLR9 ligand CpG induces neuroprotection against brain ischemic injury through a tumor necrosis factor (TNF)-dependent mechanism. It is unclear how systemic administration of CpG engages the brain to induce the protective phenotype. To address this, we created TLR9-deficient reciprocal bone marrow chimeric mice lacking TLR9 on either hematopoietic cells or radiation-resistant cells of nonhematopoietic origin. We report that wild-type mice reconstituted with TLR9-deficient hematopoietic cells failed to show neuroprotection after systemic CpG preconditioning. Further, while hematopoietic expression of TLR9 is required for CpG-induced neuroprotection it is not sufficient to restore protection to TLR9-deficient mice that are reconstituted with hematopoietic cells bearing TLR9. To determine whether the absence of protection was associated with TNF, we examined TNF levels in the systemic circulation and the brain. We found that although TNF is required for CpG preconditioning, systemic TNF levels did not correlate with the protective phenotype. However, induction of cerebral TNF mRNA required expression of TLR9 on both hematopoietic and nonhematopoietic cells and correlated with neuroprotection. In accordance with these results, we show the therapeutic potential of intranasal CpG preconditioning, which induces brain TNF mRNA and robust neuroprotection with no concomitant increase in systemic levels of TNF.
Keywords
INTRODUCTION
Despite progress in endovascular and surgical procedures for patients with coronary-artery disease and atherosclerosis, there is significant peri-procedural morbidity because of ischemic brain injury. The use of diffusion-weighted imaging has recently confirmed that a substantial number of patients undergoing cardiac surgeries such as coronary artery bypass grafting, cardiac valve replacement, and aortic repair, display evidence of peri- or post-operative cerebral ischemic lesions.1–4 These incidences of brain ischemia-reperfusion can be anticipated before they occur; thus, prophylactic treatment would be highly beneficial for these patients. Research investigating prophylactic stroke therapies has identified Toll-like receptor ligands as preconditioning agents that provide robust protection against ischemic injury.
Systemic low-dose administration of the TLR9 agonist unmethylated cytosine-phosphate-guanine-rich DNA oligonucleotides (CpG) before stroke induces neuroprotection against subsequent ischemic damage in rodent and nonhuman primate models.5,6 CpG preconditioning requires tumor necrosis factor (TNF) to protect against stroke injury, as shown by the failure of TNF-deficient mice to show neuroprotection after CpG preconditioning. 6 However, the target of TLR9 engagement and subsequent role of TNF is unclear. TLR9 is expressed on a wide variety of cell types both in the brain (microglia, neurons, astrocytes, cerebral vascular endothelium) and on hematopoietic cells in the periphery.7–11 Thus, it is unknown whether CpG preconditioning activates TLR9 on cells within the brain or whether TLR9 signaling in systemic circulating cells is required to elicit the neuroprotective phenotype.
After systemic administration of CpG, circulating hematopoietic cells are activated resulting in cell state changes that affect cytokine production12–14 and endothelial reactivity.
7
Hematopoietic cell activation can induce changes in parenchymal tissues including the brain, resulting in a secondary cascade of events.
15
In addition, it is possible that CpG administered systemically crosses the blood–brain barrier (BBB) to directly affect the brain. We have previously shown that mouse mixed cortical cells pretreated with CpG have significantly decreased cell death in an
To determine whether hematopoietic cells are required for CpG preconditioning, we used TLR9-deficient (TLR9KO) and wild-type (WT) mice to generate bone marrow chimeric mice expressing TLR9 exclusively on hematopoietic cells (WT → TLR9KO) or parenchymal cells (TLR9KO → WT). The term ‘parenchymal cells’ refers to cells of the recipient that are radiation resistant and of nonhematopoietic origin, e.g., endothelial and epithelial cells, neurons, astrocytes, microglia, and smooth muscle cells. We show that hematopoietic expression of TLR9 is required, but not sufficient for systemic CpG preconditioning-induced neuroprotection. In addition, we provide evidence that although TNF is required for CpG preconditioning, systemic TNF levels did not correlate with the protective phenotype. However, induction of cerebral TNF mRNA required expression of TLR9 on both hematopoietic and parenchymal cells and correlated with neuroprotection. This suggests that induction of TNF in the brain is associated with CpG-induced neuroprotection. These results underscore the importance of understanding the mechanism associated with CpG-induced neuroprotection to further the development of candidate therapeutics for ischemic injury to the brain.
MATERIALS AND METHODS
Animal Care and Criteria
C57Bl/6J mice and the congenic mouse strain B6.SJL-
Generating Chimeric Mice
The TLR9KO mice backcrossed onto C57Bl/6 have the CD45.2 allelic version of the CD45 leukocyte common antigen expressed on all leukocytes. To distinguish between recipient and donor cells in the chimeric animals, we used the congenic strain B6.SJL-
Determination of Reconstitution Efficiency
After 4 to 6 weeks of reconstitution, chimerism was verified using flow cytometry to determine the percent blood leukocytes positive for CD45.1 (TLR9 replete) versus CD45.2 (TLR9KO). Mice were deeply anesthetized with isoflurane, and blood was collected via cardiac puncture in sodium heparin and red blood cells were lysed using two volumes of 1 × RBC lysis buffer (Biolegend, San Diego, CA, USA). Samples were washed using cell staining buffer (Biolegend) and were incubated with Fc-Block (BD Biosciences, Franklin Lakes, NJ, USA) for 10 minutes. Cells were stained with mouse anti-mouse CD45.1 (eBiosicences, San Diego, CA, USA) and CD45.2 antibodies (BD Biosciences) conjugated with PeCy7 and FITC, respectively, for 20 minutes at 4°C. Results were acquired using an LSRII cytometer (BD Biosciences) with no fewer than 10,000 total events collected per sample. Post acquisition analysis was performed using FlowJo 9.1 (Tree Star Inc., San Carlos, CA, USA).
Drug Treatments
Mice were administered a subcutaneous injection of CpG ODN 1826 (CpG; Invivogen, San Diego, CA, USA), lipopolysaccharide (LPS) (
Ischemia-Reperfusion Model
Focal cerebral ischemia was induced by middle cerebral artery occlusion (MCAO) as described previously. 6 In brief, MCAO was performed in anesthetized mice (1.5% to 2% isoflurane) by threading a 7–0 silicon-coated nylon surgical filament (Docol, Redlands, CA, USA) through the external right carotid artery to the internal carotid artery, blocking blood flow at the bifurcation into the middle cerebral artery and anterior cerebral artery. After 45 or 60 minutes of occlusion, the monofilament was removed and blood flow was restored. The duration of MCAO was optimized to obtain consistent baseline infarct sizes across studies between surgeons. Cerebral blood flow was monitored throughout the procedure by laser Doppler flowmetry (Transonic System Inc., Ithaca, NY, USA) and animals were excluded if blood flow was not reduced by 80% or greater during occlusion. Body temperature was maintained at 37°C during the surgery.
Evaluation of Infarct Size
Twenty-four hours after MCAO, mice were deeply anesthetized with isoflurane and perfused with ice-cold saline containing 2 U/mL sodium heparin. Brains with olfactory bulbs removed were sectioned into 1 mm slices beginning from the rostral end, for a total of seven slices. The infarct area was visualized by incubating the sections in 1.5% 2,3,5-triphenyltetrazolium chloride (TTC; Sigma-Aldrich) in phosphate-buffered saline for 10 minutes at 37°C before transferring to 10% formalin (Sigma-Aldrich). Sections were then imaged and the infarct was measured by an investigator blinded to the experimental conditions using ImageJ software (NIH Image, Bethesda, MD, USA). Infarct volume was calculated using the indirect method ((contralateral live – ipsilateral live)/contralateral live × 100) to account for the effects of edema, and the results are normalized to genotype-matched vehicle-treated controls.
Analysis of Plasma Tumor Necrosis Factor Levels
Mice were deeply anesthetized with isoflurane and blood was collected via cardiac puncture in sodium heparin (2 U/mL). The blood was then centrifuged (380 × g for 20 minutes) and the plasma was removed and stored at −80°C. ELISA was used to quantify plasma TNF (R&D Systems, Minneapolis, MN, USA). Samples were run in duplicate.
Tissue Processing and Quantitative Real-Time PCR
Total RNA was isolated from the cerebral cortex using the Qiagen Rneasy Lipid Mini Kit (Qiagen, Valencia, CA, USA). RNA was reverse transcribed using an Omniscript Reverse Transcription kit (Qiagen). Quantitative PCR was performed using the TaqMan Gene Expression Assays (Applied Biosystems, Carlsbad, CA, USA) on the Applied Biosystems Step One Plus PCR system. Results were normalized to β-actin expression and are represented as delta cycle threshold (dCT) values.
Statistical Analysis
Statistical testing was performed using Prism 5 software (GraphPad, LaJolla, CA, USA). Values are given as group means ± standard error of the mean (s.e.m.). Data were analyzed by Student's
RESULTS
CpG Preconditioning Requires TLR9
While CpG is known to activate TLR9 signaling, prior studies had not examined whether CpG preconditioning
Figure 1). Thus, CpG-induced neuroprotection
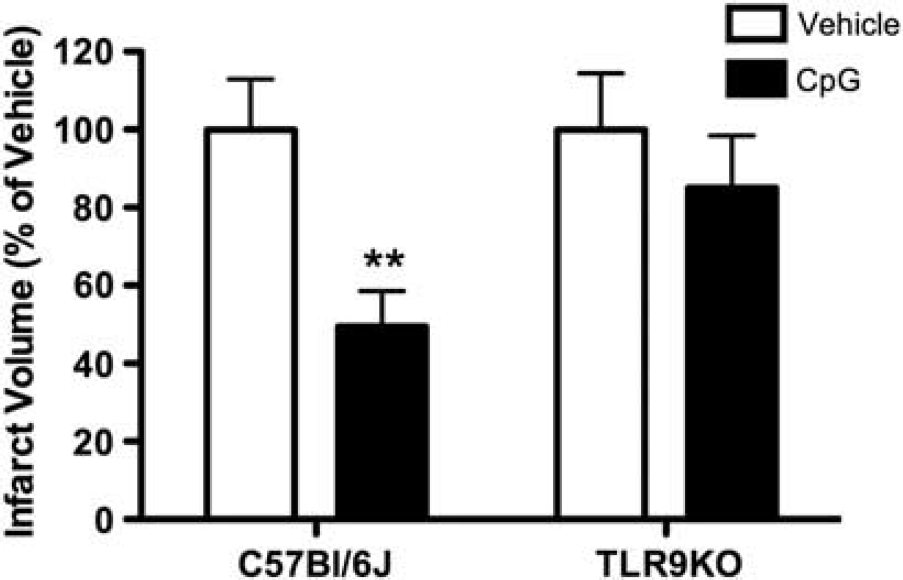
CpG preconditioning requires TLR9. C57Bl/6J or TLR9KO mice were administered subcutaneous injection of CpG (1.6 mg/kg;
TLR9 Is Required in Both the Hematopoietic and Parenchymal Compartments for CpG-Induced Neuroprotection Against Brain Ischemia
TLR9 is expressed on multiple cell types within the body including circulating hematopoietic cells and brain cells.7–11 To address the role of hematopoietic cells in response to systemic CpG preconditioning, we generated radiation-induced reciprocal bone marrow chimeras from WT (CD45.1 haplotype) and TLR9KO (CD45.2 haplotype) mice. We generated three groups of chimeric mice: (1) mice lacking TLR9 on hematopoietic cells (TLR9KO → WT), (2) mice lacking TLR9 on parenchymal (nonhematopoietic) cells (WT → TLR9KO), and (3) control mice replete with TLR9 (WT → WT). To confirm chimerism, we stained peripheral blood from these animals with antibodies specific to the CD45.1 (WT) and CD45.2 (TLR9KO) leukocyte common antigens to distinguish between donor and recipient cells, and then analyzed these samples using flow cytometry. Four to six weeks after bone marrow transfer, >80% of the hematopoietic cells were donor derived in the chimeric mice (Figure 2).
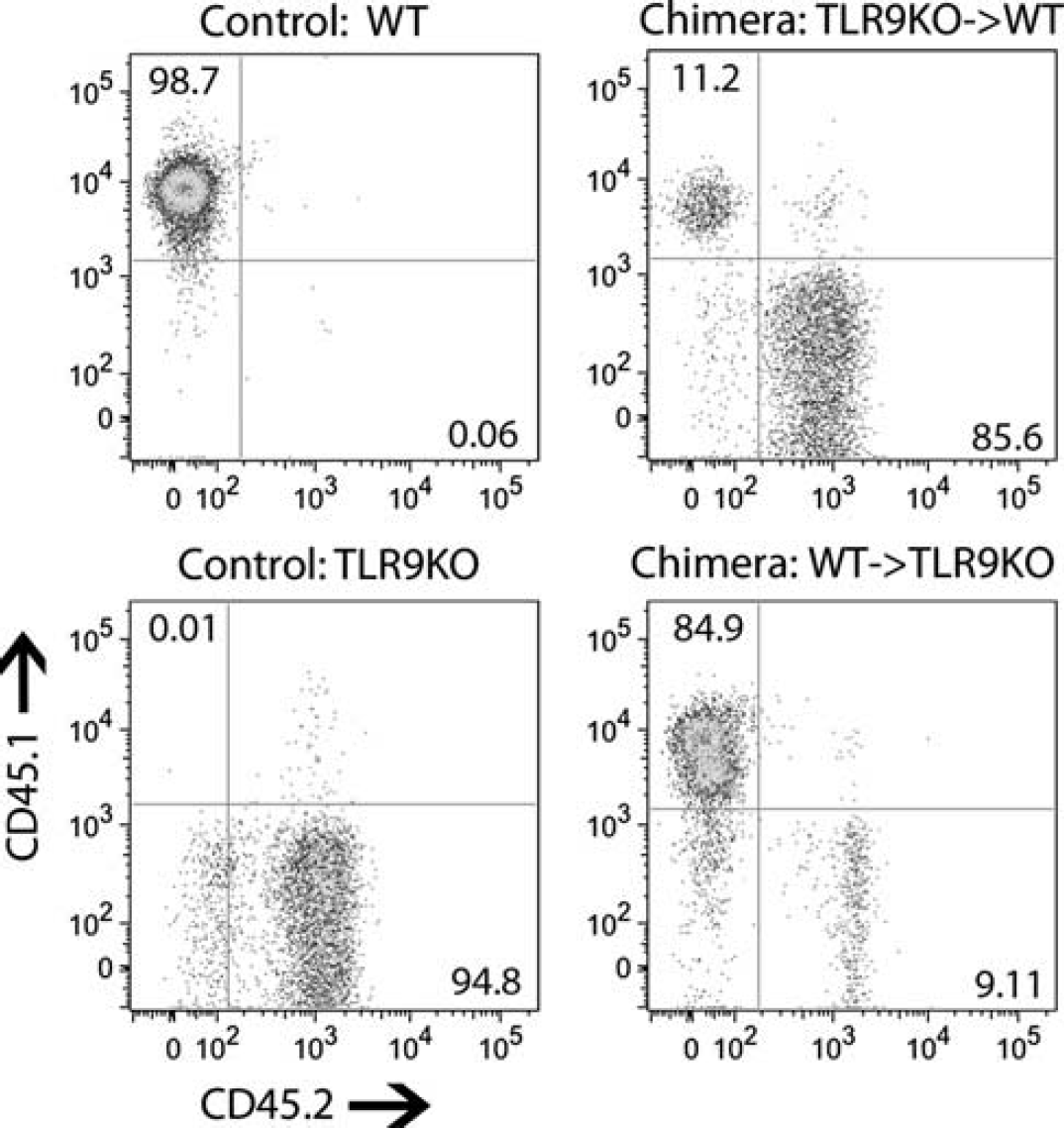
Generation of TLR9 reciprocal bone marrow chimeric mice. Mice were irradiated with a split dose of 12 grays then given intravenous injection of donor bone marrow cells. TLR9KO → WTand WT → TLR9KO chimeric mice were generated. To confirm chimerism, different allelic forms of the CD45 leukocyte common antigen are used to distinguish WT cells (CD45.1 positive; y axis) and TLR9KO cells (CD45.2 positive; x axis) in the systemic circulation by flow cytometry. Four to six weeks after bone marrow transfer, >80% of the hematopoietic cells were donor derived in the chimeric mice. Representative plots for each group are shown. WT, wild type.
To determine whether TLR9 expression in the hematopoietic compartment has a role in CpG-induced neuroprotection, chimeric mice were preconditioned with CpG (1.6 mg/kg; subcutaneously) or vehicle 72 hours before MCAO, and ischemic damage was evaluated 24 hours after occlusion. As expected, WT → WT mice administered CpG exhibited significant protection from ischemic injury compared with vehicle-treated controls (infarct size 65.66 ± 8.34% of vehicle;
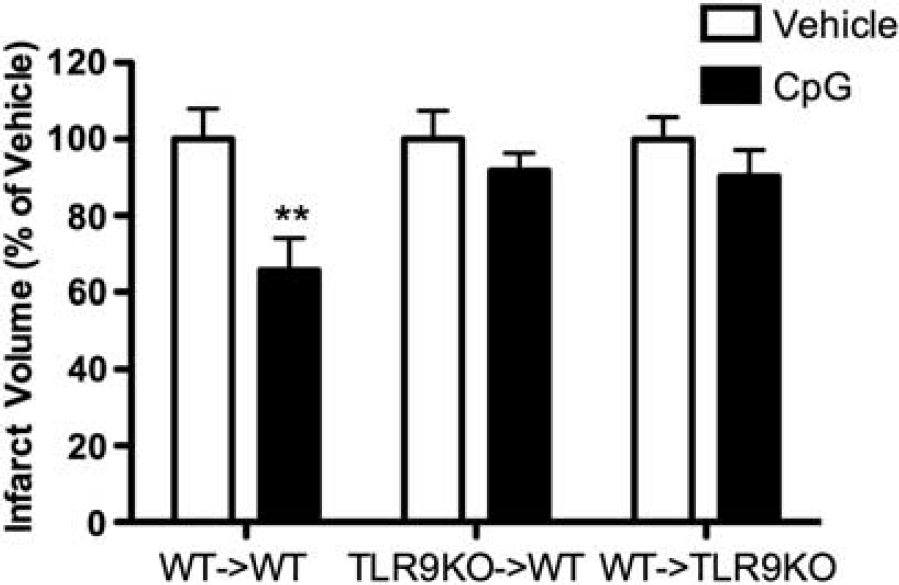
TLR9 is required in hematopoietic and parenchymal compartments to achieve CpG-induced neuroprotection against brain ischemic injury. TLR9KO → WT, WT → TLR9KO, and WT → WT mice were injected subcutaneously with CpG (1.6 mg/kg; subcutaneously) or vehicle 72 hours before middle cerebral artery occlusion (MCAO) (
TLR9 on Hematopoietic Cells Is Not Required to Elicit a Peripheral Tumor Necrosis Factor Response to CpG
To further understand the absence of a CpG preconditioning effect in mice that do not express TLR9, we measured circulating plasma levels of TNF, a critical mediator of CpG-induced neuroprotection. We have shown previously that systemic CpG preconditioning fails to protect TNF-deficient mice against ischemic brain injury and that CpG induces an increase in plasma TNF within 1 hour after administration.
6
These results suggest that systemic induction of TNF may be required for the neuroprotective effect of CpG preconditioning. Thus, we tested whether TLR9 expression on hematopoietic cells was required for CpG-mediated induction of systemic TNF levels using mice deficient in hematopoietic TLR9 (TLR9KO → WT). We found that TNF was induced in the plasma in response to CpG (1.6 mg/kg; subcutaneously) in the TLR9KO → WT mice (442.39 ± 63.8 pg/mL;
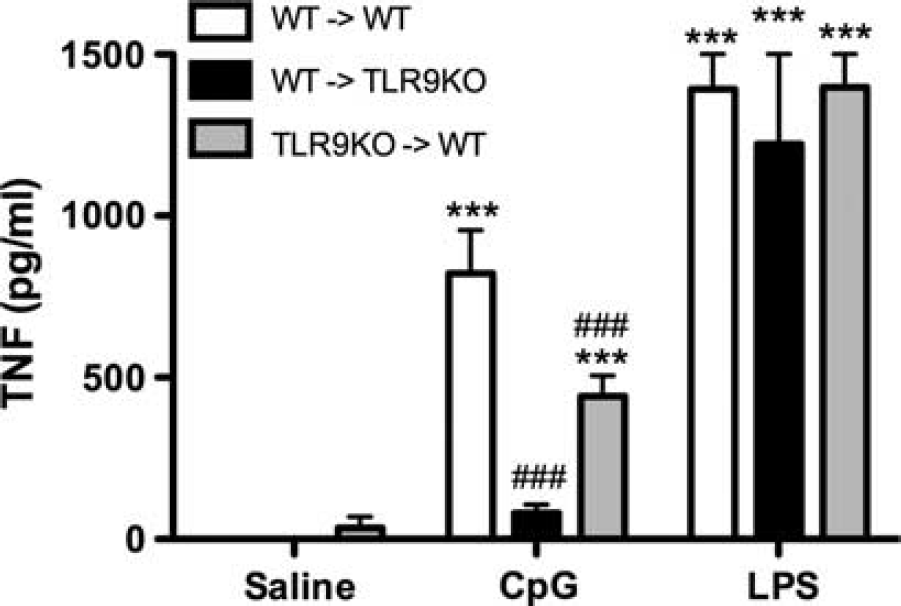
Expression of TLR9 on hematopoietic cells is not necessary to induce tumor necrosis factor (TNF) in the systemic circulation. TLR9KO → WT, WT → TLR9KO, and WT → WT mice were injected subcutaneously with CpG (1.6 mg/kg), lipopolysaccharide (LPS) (1 mg/kg), or vehicle (
In addition, we examined whether TLR9 expression on hematopoietic cells was sufficient to produce TNF in response to CpG using WT → TLR9KO chimeric mice, which only express TLR9 on hematopoietic cells. We found that TNF levels failed to increase in response to CpG administration despite the presence of TLR9 on the hematopoietic cells (Figure 4). Together, these data suggest that parenchymal cells may be major contributors to the CpG-mediated induction of plasma TNF, and that TLR9 expression on hematopoietic and parenchymal cells is necessary for optimal induction of TNF. Of note, all chimeras, irrespective of genotype, induced TNF levels that were equivalent to WT mice in response to the TLR4 ligand, LPS (1 mg/kg; Figure 4), showing that the differences were specific to CpG/TLR9-mediated TNF induction.
CpG Induction of Brain Tumor Necrosis Factor Requires TLR9 Expression in Both the Hematopoietic and Parenchymal Compartments
Systemic LPS administration has been shown to induce TNF in the brains of chimeric mice that have been reconstituted with TLR4-deficient hematopoietic cells
16
; thus, we hypothesized that systemic CpG preconditioning would also induce TNF in the brains of mice deficient in TLR9 expression on hematopoietic cells. Therefore, we examined TNF expression in the brains of mice lacking TLR9 on hematopoietic cells (TLRKO → WT) or parenchymal cells (WT → TLR9KO) 3 hours after CpG (1.6 mg/kg; subcutaneously) or vehicle administration. We found that TNF mRNA in the brain was increased in WT → WT control mice in response to systemically administered CpG (13.9 ± 0.4 dCT;
Brain TNF mRNA expression 3 hours after drug administration
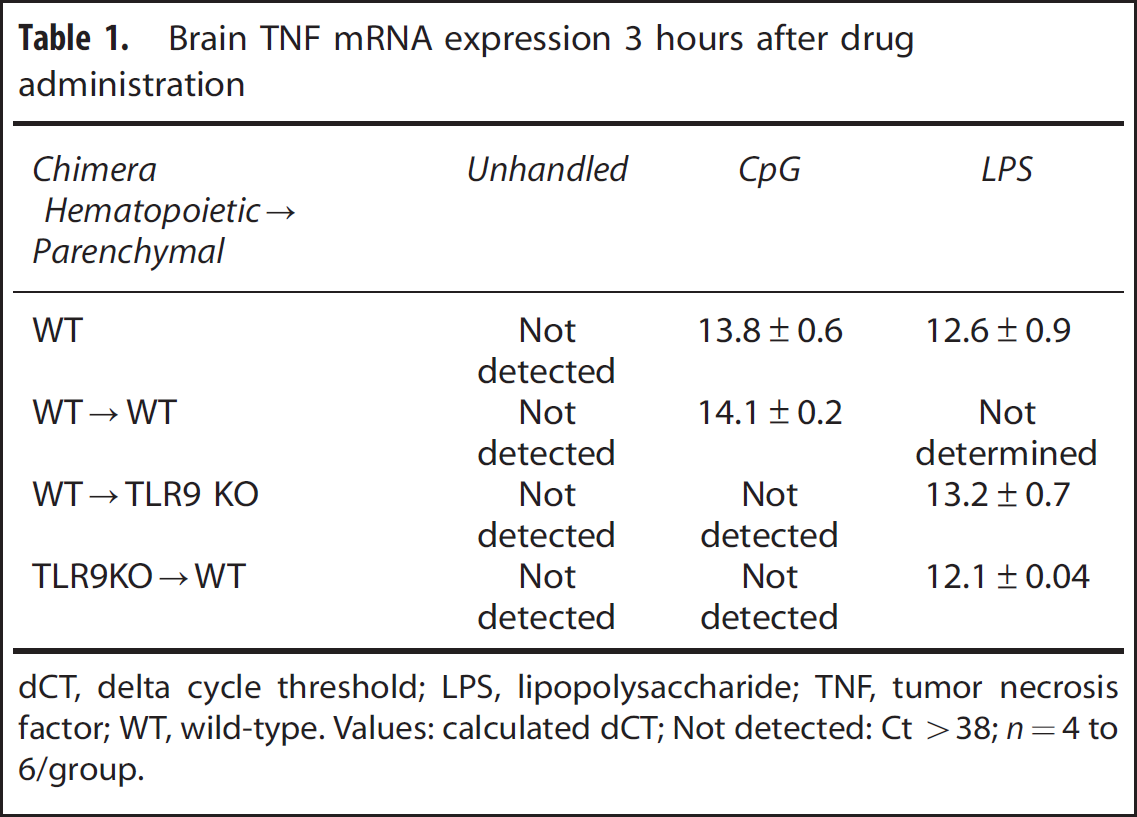
dCT, delta cycle threshold; LPS, lipopolysaccharide; TNF, tumor necrosis factor; WT, wild-type.
Values: calculated dCT; Not detected: Ct >38;
Intranasal CpG Preconditioning Provides Robust Neuroprotection in the Absence of Increased Plasma Tumor Necrosis Factor
We next determined whether CpG preconditioning by intranasal administration could elicit brain TNF expression and subsequent neuroprotection without the induction of circulating plasma TNF. Ross
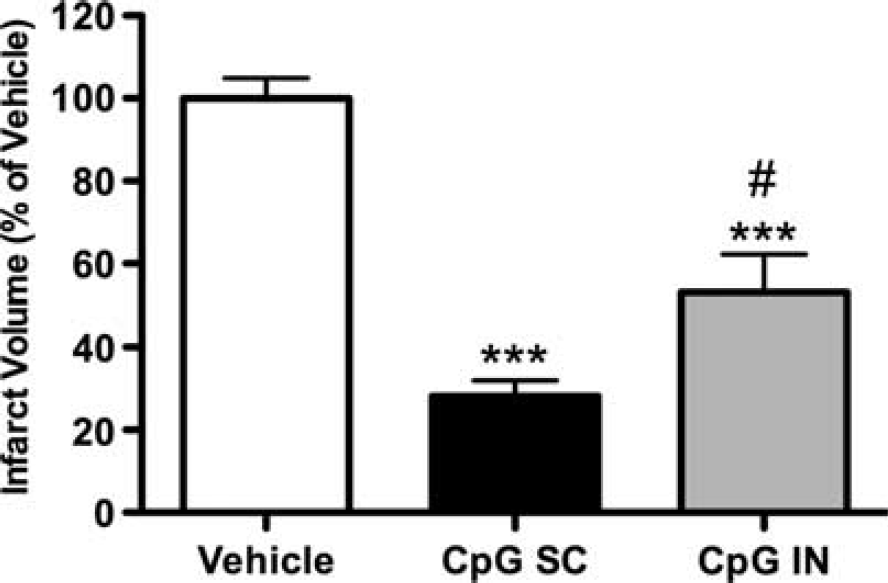
Intranasal administration of CpG preconditions mice against subsequent ischemic injury. Mice were administered CpG or vehicle via intranasal (IN) (CpG 3.2 mg/kg;
DISCUSSION
Preconditioning via systemic administration of CpG induces robust neuroprotection against subsequent cerebral ischemia. We show that the protective effect of CpG requires TLR9, as CpG preconditioning in mice deficient in TLR9 fails to protect against stroke injury. We created TLR9KO bone marrow chimeric mice to evaluate the target for CpG-induced neuroprotection against stroke. We found that the neuroprotective effect of systemic CpG preconditioning was absent in mice deficient in either hematopoietic or parenchymal expression of TLR9. This indicates that the cells from both compartments interact to orchestrate the neuroprotective response to CpG that limits ischemic damage. The requirement for both hematopoietic and parenchymal cells to respond to a stimulus was also reported in adjuvant studies using the immune modulator poly-IC. Reciprocal bone marrow chimeric mice created with mice deficient in the poly-IC receptors, TLR3 and Melanoma differentiation associated protein 5 did not mount a primary T-cell adjuvant response to poly-IC. 19 In this case, receptor expression was needed in both compartments to produce a response to poly-IC that was equivalent to WT → WT controls. These results indicate that hematopoietic cells alone are not sufficient for immune activation and that other cell types must interact to elicit a WT response.
We have shown previously that neuroprotection induced by systemic CpG preconditioning requires TNF and that TNF is induced in the plasma after CpG preconditioning.
6
Here, for the first time, we show that CpG preconditioning also induces TNF mRNA in the brain after CpG preconditioning. Using the TLR9 bone marrow chimeric strategy, we investigated the source and importance of CpG-mediated induction of TNF in the plasma and in the brain. We found that CpG treatment in mice with TLR9 only on parenchymal cells (TLR9KO → WT), but not on hematopoietic cells, produced a significant yet attenuated peripheral TNF response compared with WT mice. However, the systemic TNF response was completely absent when TLR9 was expressed only on hematopoietic cells (WT → TLR9KO) and not on parenchymal cells. This finding was not anticipated given that hematopoietic cells are known to express TLR9.12,13 In support of our finding, another study showed that primary macrophages expressing TLR9 did not secrete TNF or IL-6 in response to CpG uptake in culture.
20
Several recent studies support the concept that nonhematopoietic cells are critical to inflammatory responses. For example, a study that used TLR4 chimeric mice in a model of LPS-induced uveitis found that, although macrophages and dendritic cells are prevalent in the eye, TLR4-mediated responses on parenchymal cells (e.g., endothelial and epithelial cells) were responsible for the induction of the inflammatory disease state.
21
In addition, Longhi
In contrast, induction of TNF mRNA in the brain was absent in both the WT → TLR9KO and TLR9KO → WT mice, suggesting that both hematopoietic and parenchymal cells are required for the cerebral induction of TNF via systemic CpG preconditioning. Of note, WT → WT controls preconditioned with CpG-induced cerebral and systemic TNF levels equivalent to C57BL/6 mice and were protected against cerebral ischemia, validating that this irradiation model does not compromise the innate immune response in the brain, blood, nor the response to ischemia-reperfusion injury.23,24 Our findings show that TLR9 expression in both the hematopoietic and parenchymal compartments is required for CpG preconditioning-induced expression of TNF in the brain and induction of neuroprotection. Thus, CpG-mediated neuroprotection correlates with TNF induction in the brain before stroke. We failed to see a similar correlation between systemic levels of TNF and neuroprotection. Although TNF is a proinflammatory mediator several studies have shown that TNF can protect neurons from excitotoxic and ischemic injury.25–30 Of particular importance to our findings is that preconditioning with TNF via intercisternal administration protects against stroke; however, systemic preconditioning with TNF does not provide protection.
28
In addition, Lambertsen
Taken together, our findings suggest that systemic administration of CpG requires the interaction between TLR9-expressing hematopoietic and parenchymal cells for neuroprotection and induction of brain TNF mRNA. Communication between immune signals and the brain is referred to as the neuroimmune axis and can occur via multiple pathways including afferent vagal nerve fibers, circumventricular organs, the BBB, and the hypothalamicpituitary–adrenal axis. 22 Since it has been shown previously that endothelial cells become activated by CpG and interact with hematopoietic cells, 7 we propose that the BBB serves as the critical site of communication for systemic CpG preconditioning and the subsequent induction of TNF in the brain based on the following models (Figure 6). Systemic CpG activates endothelial cells and circulating hematopoietic cells, such as leukocytes, causing them to adhere to the endothelium. These cell–cell interactions propagate endothelial and hematopoietic cell activation and cause the release of inflammatory cytokines, which produces the rise in systemic TNF levels. The endothelial activation may increase BBB permeability, allowing for CpG to cross the BBB and directly activate TLR9-expressing CNS cells, such as microglia, astrocytes, and neurons. In addition, evidence suggests that oligodeoxynucleotides of similar size can cross the BBB via saturable transporters and enter the CNS. 31 We have shown previously that systemic CpG preconditioning causes upregulation of genes related to TLR signaling in the brain. 32 Further, direct stimulation of astrocytes and microglia with CpG has been shown to produce TNF.11,33,34 The activated endothelium may also signal CNS cells indirectly, possibly by abluminal secretion or transport of inflammatory cytokines into the perivascular space or through interaction with adjacent cells such as pericytes and astrocytes, to promote an inflammatory response in the brain.15,22,35,36 Once activated, neurons, microglia, and astrocytes can produce TNF.37–39 These processes may be responsible for promoting TNF production in the CNS after preconditioning, which correlates with neuroprotection against stroke.
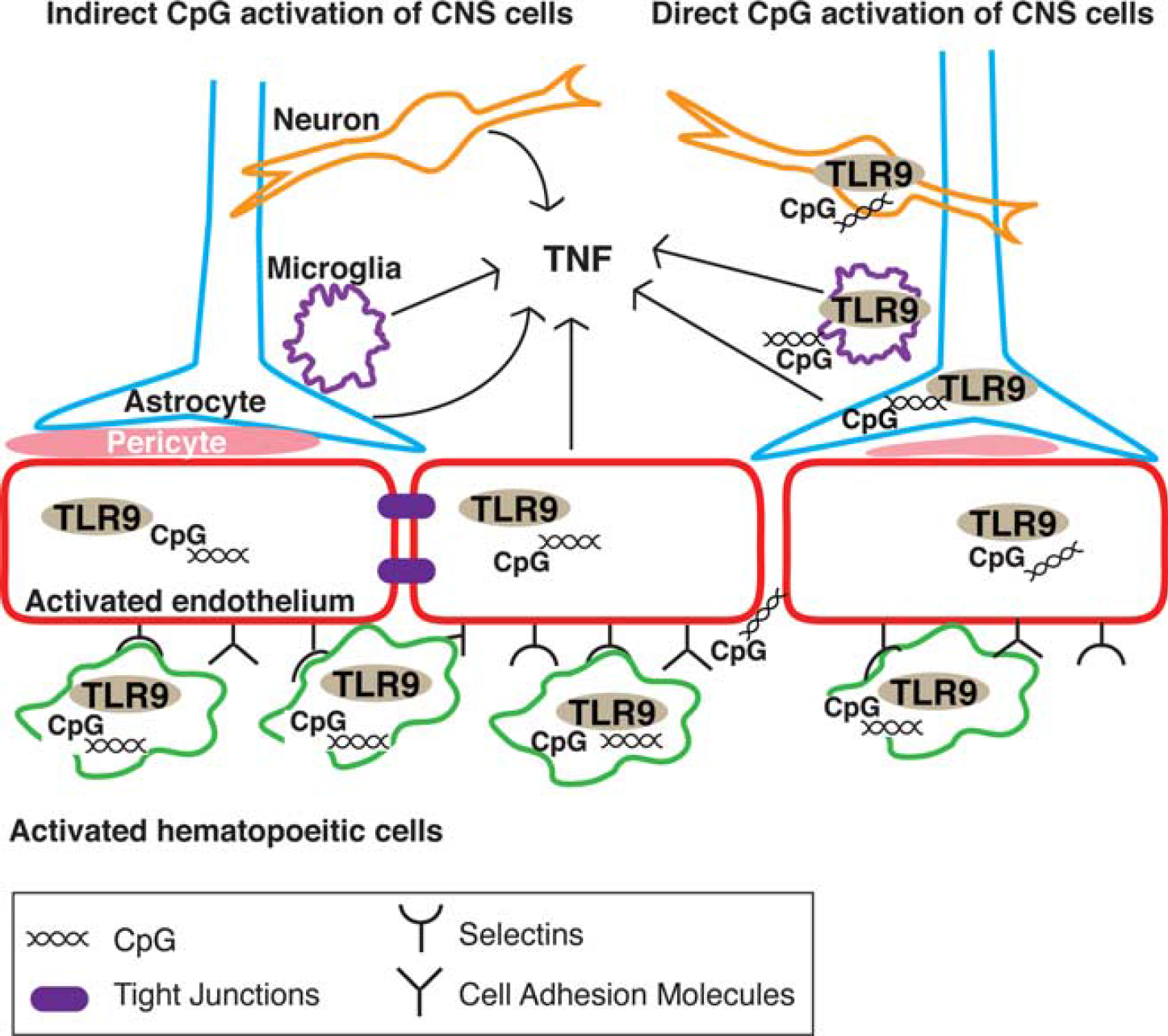
Proposed cellular mechanism of cerebral tumor necrosis factor (TNF) induction by systemic CpG preconditioning. Systemic CpG preconditioning requires activation of both hematopoietic and parenchymal cells to induce cerebral TNF and neuroprotection against stroke. Systemic CpG preconditioning may elicit brain TNF via indirect or direct activation of central nervous system (CNS) cells. Indirect activation (left side): systemic CpG preconditioning activates leukocytes (green) and endothelial cells (red) via TLR9. Endothelial cells and leukocytes interact via adhesion molecules (CAMs, selectins, integrins), which signal the brain to induce TNF, either through adjacent cells such as pericytes or astrocytes or through proinflammatory cytokines. Direct activation (right side): CpG stimulation of leukocytes and endothelial cells increases the permeability of the blood–brain barrier (BBB) allowing CpG to cross and directly activate TLR9 in CNS cells to induce TNF. Both mechanisms likely occur simultaneously
Our findings suggest that CpG-stimulated hematopoietic and parenchymal cells cooperate to elicit a TNF response in the brain before stroke to promote neuroprotection. Importantly, we found that the hematopoietic compartment can be bypassed by intranasal administration of CpG preconditioning, which induced TNF mRNA in the brain and resulted in robust reduction in infarct size without any detectable TNF in the plasma. This finding supports the importance of TNF expression in the brain before stroke and that neuroprotection does not require a systemic TNF response. Further, the absence of the systemic inflammatory response is therapeutically relevant because, in human patients, systemic TNF can lead to severe side effects such as hypotension, hepatotoxicity, and leukopenia, and TNF is known to have a deleterious role in septic shock. 40 The intranasal route of administration is a noninvasive method to target the CNS and has shown success in clinical trials for the treatment of cognitive diseases such as Alzheimer's and schizophrenia. 41 Thus, from a therapeutic perspective, intranasal administration of CpG offers a dosing strategy with superior clinical relevance for the prophylactic treatment of patients at high risk of suffering from cerebral ischemia, such as patients undergoing cardiovascular surgery.
These studies are the first to address the role of hematopoietic versus parenchymal cells in neuroprotection induced by systemic CpG preconditioning and to delineate the role of TNF expression in the periphery versus the brain. We show that TLR9-dependent hematopoietic and parenchymal cell responses to CpG are required to induce TNF expression in the brain and to provide neuroprotection. Importantly, neuroprotection induced by CpG preconditioning is independent of peripheral TNF expression. Further, we show that intranasal CpG preconditioning provides robust protection against stroke and results in cerebral TNF expression without induction of peripheral TNF levels, thus providing a superior route of administration for clinical applications of CpG as a prophylactic stroke treatment. These results advance the understanding of neuroimmune signaling involved in preconditioning-induced neuroprotection and provide insight for the development of potential stroke therapeutics by establishing that circulating TNF levels are not requisite.
DISCLOSURE/CONFLICT OF INTEREST
The authors declare no conflict of interest.
Footnotes
ACKNOWLEDGEMENTS
The authors would like to thank Valerie Conrad, Tao Yang, and Delfina Homen for their superior technical support.