
Abstract
Traumatic brain injury (TBI) leads to mossy fiber reorganization, which is considered to be a causative factor in the development of temporal lobe epilepsy. However, the underlying mechanism is not fully understood. Emerging evidence suggests that TrkB–ERK1/2–CREB/Elk-1 pathways are highly related to synaptic plasticity. This study used the rat fluid-percussion injury model to investigate activation of TrkB–ERK1/2–CREB/Elk-1 signaling pathways after TBI. Rats were subjected to 2.0-atm parasagittal TBI followed by 30 minutes, 4 hours, 24 hours, and 72 hours of recovery. After TBI, striking activation of TrkB–ERK1/2–CREB/Elk-1 signaling pathways in mossy fiber organization were observed with confocal microscopy and Western blot analysis. ERK1/2 was highly phosphorylated predominantly in hippocampal mossy fibers, whereas TrkB was phosphorylated both in the mossy fibers and the dentate gyrus region at 30 minutes and 4 hours of recovery after TBI. CREB was also activated at 30 minutes, peaked at 24 hours of recovery, and returned to the control level at 72 hours of recovery in dentate gyrus granule cells. Elk-1 phosphorylation was seen in CA3 neurons at 4 hours after TBI. The results suggest that the signaling pathways of TrkB–ERK1/2–CREB/Elk-1 are highly activated in mossy fiber organization, which may contribute to mossy fiber reorganization seen after TBI.
The mossy fiber in the dentate gyrus (DG) undergoes prominent sprouting and extensive reorganization after traumatic brain injury (TBI), which has been considered to be a causative factor of posttraumatic seizures (Golarai et al., 2001). Human studies have revealed that TBI is a major risk factor in the development of temporal lobe epilepsy (Englander et al., 2003). Hippocampal mossy fibers are composed of excitatory axons derived from DG granule cells, projecting to the CA3 region and forming part of the trisynaptic hippocampal circuit. It is generally held that hippocampal seizures originate from mossy fibers sprouting to develop new abnormal excitatory synapses on DG dendritic spines, resulting in formation of recurrent excitatory circuits in the DG region (Cavazos et al., 2003; Wenzel et al., 2000). Activation of TrkB plays a significant role in seizure-induced mossy fiber sprouting (He et al., 2002; Scharfman et al., 1999, 2002). Phosphorylation of extracellular signal-regulated protein kinase1/2 (ERK1/2) and expression of brain-derived neurotrophic factor (BDNF) are markedly increased in DG granule cells after TBI (Dash et al., 1995 and 2002; Griesbach et al., 2002; Grundy et al., 2000; Mori et al., 2002; Oyesiku et al., 1999; Yang et al., 1996). Transgenic mice overexpressing BDNF exhibit an enhanced response to epileptogenic stimuli (Croll et al., 1999). Elucidating the cellular and molecular mechanisms of epileptogenesis after TBI may provide novel therapeutic approaches aimed at prevention of posttraumatic seizures. However, intracellular signaling events leading to mossy fiber sprouting and reorganization after TBI have not been well studied.
It has been known for several years that cyclic AMP response element-binding protein (CREB) transcription factor plays a key role in synaptic remodeling. However, it has been appreciated recently that the ERK1/2 cascade plays a fundamental role in synaptic plasticity (Sweatt, 2001). Activation of ERK1/2 by BDNF can further activate CREB transcription factor through another kinase to phosphorylate CREB on serine 133 and promotes transcription of genes involving synapse formation (Bonni et al., 1999; Davis et al., 2000; Ernfors and Bramham, 2003). In addition, ERK1/2 activates another transcription activator Elk-1 to induce early- and late-response genes for postsynaptic remodeling (Li et al., 2003a; Thiels et al., 2002, Yordy et al., 2003). Elk-1 is a member of a subfamily of ETS domain proteins called ternary complex factors; i.e., Elk1–SAP1–NET that associates with the serum response factor to regulate gene transcription. Elk-1 can be phosphorylated by ERK1/2 both in vitro and in vivo. Inhibition of ERK1/2 cascades completely blocks the gene expression–dependent long-term potentiation and BDNF-induced neurite outgrowth (Binder et al., 1999; Wu et al., 2001). Expression of BDNF and the tropomyosin-related kinase B (TrkB) is controlled by cyclic AMP–responsive elements, the DNA binding sites for CREB (Shieh et al., 2000). Conversely, BDNF binding to its receptor TrkB activates ERK1/2 and CREB for further BDNF expression (Bonni et al., 1999). It seems, therefore, likely that there is a positive feedback loop—i.e., ERK1/2–CREB–BDNF/TrkB–ERK1/2—for synaptic reorganization.
We have recently found markedly activation of the TrkB–ERK1/2–CREB pathway in DG granule cells after transient cerebral ischemia (Hu et al., 1999, 2000a). In the present study, we investigated the TrkB–ERK1/2–CREB/Elk-1 pathways in hippocampus after TBI. We found that TrkB–ERK1/2–CREB/Elk-1 cascades were strikingly activated predominantly in mossy fiber organization, which may contribute to mossy fiber reorganization after TBI.
MATERIALS AND METHODS
Animal model
Experiments were performed with male Sprague-Dawley rats weighing 270 to 320 g (Charles River Laboratories, Raleigh, NC, U.S.A.). The University of Miami Animal Care and Use Committee approved all experimental procedures. All animals were maintained for at least 7 days before the study in an air-conditioned room with a constant temperature (23–25°C) and a 12-hour light/dark cycle. The rats were fasted but allowed free access to water overnight before surgery. TBI was produced with fluid-percussion (F-P) pressure levels of 2.0 ± 0.2 atm. The basic surgical preparation for F-P brain injury was performed according to previously described methods (Dietrich et al., 1996, 1998; Dixon et al., 1987). The rats were initially anesthetized with 3.0% halothane in a gas mixture of 70% nitrous oxide and 30% oxygen. A femoral artery and vein were cannulated with PE-50 tubing. The rats were then immobilized with pancuronium bromide (0.5 mg/kg, administered intravenously). An endotracheal tube was inserted orally, and the rats were subjected to mechanical ventilation with a mixture of 70% nitrous oxide and 0.5% to 1.5% halothane, with a balance of oxygen. The animals were then placed in a stereotaxic frame, and a 4.8-mm craniotomy was made over the right parietal cortex (3.8 mm posterior to bregma and 2.5 mm lateral to the midline). A plastic injury tube was placed over the exposed dura and fixed with adhesive dental acrylic, which was permitted to harden; the scalp was then sutured. Before TBI, blood gases, and mean arterial blood pressure were monitored and maintained at physiologic levels. Brain temperatures were measured with a thermistor probe placed in the left temporal muscle, whereas core temperatures were determined with a rectal thermometer. Brain temperature was maintained at 37°C with a heating lamp during surgical procedures. Blood gases, blood glucose, and hematocrit values were also measured 15 minutes after TBI.
Four experimental groups were used in this study: a sham-operated group and groups subjected to 2.0 atm F-P TBI followed by 30 minutes, 4 hours, and 24 hours of recovery. Each experimental group consisted of at least four rats. Sham-operated rats were subjected to the same surgical procedures but without TBI. For the 30-minute recovery case, animals were maintained under anesthesia for the entire post-TBI period, after which time the brains were collected. For longer periods of recovery, halothane was discontinued after TBI, all wounds were sutured, and animals were returned to their cages. The animals were reanesthetized, tracheotomized, and artificially ventilated at 4 and 24 hours of recovery after TBI and the brains were collected. Tissue samples for biochemical studies were obtained by freezing the brains in situ with liquid nitrogen while the animal's respiration was maintained with a respirator. Hippocampi were dissected out in a glove box freezer at a temperature of −15°C as described previously (Hu et al., 1999). For confocal microscopy, the brains were perfused with ice-cold 4% phosphate-buffered paraformaldehyde while the animal's respiration was maintained with a respirator. The brains were sectioned with a Leica vibratome (Germany) at a thickness of 50 μm.
Confocal microscopy
An affinity-purified phospho-specific Trk antibody (pY490) directed against a synthetic phospho-tyr490 peptide corresponding to residues 485 to 493 (IENPQY*FSD) of human TrkA was obtained from Cell Signaling Inc. (Beverly, MA, U.S.A.). This sequence is highly conserved among the TrkA, TrkB, and TrkC neurotrophin receptors among rat, mouse, and human. The immunostained phospho-Trk in the mossy fiber pathway and CA3 stratum oriens of the hippocampus was recently identified as phospho-TrkB (He et al., 2000; Merlio et al., 1993). In the present study, we therefore used phospho-TrkB to describe phospho-Trk antibody immunostaining in the mossy fiber region. A monoclonal antibody against phospho-ERK1/2 and polyclonal antibodies against phospho-CREB and phospho–Elk-1 were also obtained from Cell Signaling. A mouse monoclonal antibody against synaptophysin was obtained from Sigma (St. Louis, MO, U.S.A.).
Double-label fluorescence confocal microscopy was performed on coronal brain sections (50 μm) from sham-operated control animals and from animals subjected to 2.0-atm TBI followed by 30 minutes, 4 hours, and 24 hours of recovery. At least four brain sections prepared from four different animals in each experimental condition were used for confocal microscopy. The brain sections were washed twice in phosphate-buffered saline (PBS) for 5 minutes at room temperature and then in PBS containing 0.2% Triton X-100 (TX-100) for 30 minutes. Brain sections were heated for 1 minute with a microwave oven for antigen retrieval. Non-specific binding sites were blocked in 3% bovine serum albumin (BSA) in PBS/0.2% TX-100 for 30 minutes. The primary antibodies were diluted 1:200 in PBS/0.1% TX-100 and 1% BSA. After incubation (overnight at 4°C) with the phospho-Trk primary antibody, the sections were washed three times in PBS containing 0.1% TX-100 for 10 minutes at room temperature. The sections were then incubated for 2 hours at 4°C with the monoclonal phospho-ERK1/2 or synaptophysin antibodies, respectively, both at dilution of 1:400 in PBS/0.1% TX-100 and 1% BSA, followed by three washes for 10 minutes at room temperature. Fluorescent secondary antibodies, fluorescein-labeled anti-rabbit and lissamine rhodamine-labeled anti-mouse, were diluted 1:200 in PBS containing 0.1% TX-100 and 1% BSA, and applied for 1 hour at room temperature. Sections were washed several times in PBS/0.1% TX-100, mounted and coverslipped on glass slides. For double immunostaining of phospho-ERK1/2, phospho-CREB, phospho-Elk-1 and total CREB with propidium iodide, brain sections were incubated first with the primary antibodies, and then with a mixture of fluorescein-labeled secondary antibodies and 15 μg/mL propidium iodide. The slides were analyzed on a Zeiss laser-scanning confocal microscope.
Preparation of subcellular fractions
The subcellular fractions were prepared according to one of our previous publications with modifications (Hu et al., 1999). In the original protocol, P1 and P2 were prepared separately by centrifugation at 800 g and 10,000 g, respectively. P1 contains nuclei and cell debris whereas P2 includes cell membranes, synaptosomes, and mitochondria. In this study, we had limited amount of tissue from injured dorsal hippocampus so that we could prepare only one fraction containing both P1 and P2 by centrifugation at 10,000 g. Briefly, tissues were obtained from sham-operated rats and rats subjected to TBI followed by 30 minutes, 4 hours, and 24 hours of recovery. Dorsal hippocampal tissue from the injured hemisphere was dissected and cut into small pieces in a −15°C glove box freezer, and they were homogenized on ice with a Dounce homogenizer in 15 volumes of homogenization buffer containing 15 mmol/L Tris base/HCl pH 7.6, 1 mmol/L DTT, 0.25 mol/L sucrose, 1 mmol/L MgCl2, 1.25 μg/mL pepstatin A, 10 μg/mL leupeptin, 2.5 μg/mL aprotinin, 0.5 mmol/L PMSF, 2.5 mmol/L EDTA, 1 mmol/L EGTA, 0.1 mol/L Na3VO4, 50 mmol/L NaF, and 2 mmol/L sodium pyrophosphate. The homogenates were then centrifuged at 10,000g at 4°C for 10 minutes to obtain pellets and supernatants. The pellets containing nuclei, mitochondria and synaptosomes were washed with homogenization buffer containing 1% Triton and 0.4 mol/L NaCl for 30 minutes on ice and centrifuged at 25,000g for 10 minutes at 4°C to get another pellet (Pp), and supernatant (Ps). The Pp contains cell membranes as well as mitochondrial and nuclear matrix, and was used for Western blot of membrane proteins such as TrkB. The Ps contains nuclear extracts, as well as synaptosomal (synaptosol) and mitochondrial supernatants, was used for Western blot of transcription factors such as CREB. The protein concentration of these samples was determined by the Lowry method (Lowry et al., 1951).
Western blot analysis
Western blot analysis was carried out on 8% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Samples separately prepared from each individual rat contained 25–50 μg of protein and were separately applied to a SDS-PAGE slab gel. At least four samples prepared from four different animals in each experimental condition were used for Western blot analyses. Following electrophoresis, proteins were transferred to an immobilon-P membrane. The immobilon-P membrane was incubated with a primary monoclonal antibody against phospho-ERK1/2 at a dilution of 1:2,000, and polyclonal antibodies against phospho-TrkB, phospho-CREB, and phospho-Elk-1, all at a dilution of 1:1,000, overnight at 4°C. The membranes were then incubated with horseradish-peroxidase conjugated anti-rabbit or anti-mouse secondary antibodies for 45 minutes at room temperature. The blots were developed using the ECL detection method. Western blot bands were quantified with Kodak 1D gel analysis software.
RESULTS
Rats were subjected to sham operation and to 2.0-atm TBI followed by 30 minutes, 4 hours, 24 hours, and 72 hours of recovery, respectively. Physiologic parameters in experimental animals were measured and are shown, in part, in Table 1. P
Physiologic parameters in animals during or after TBI
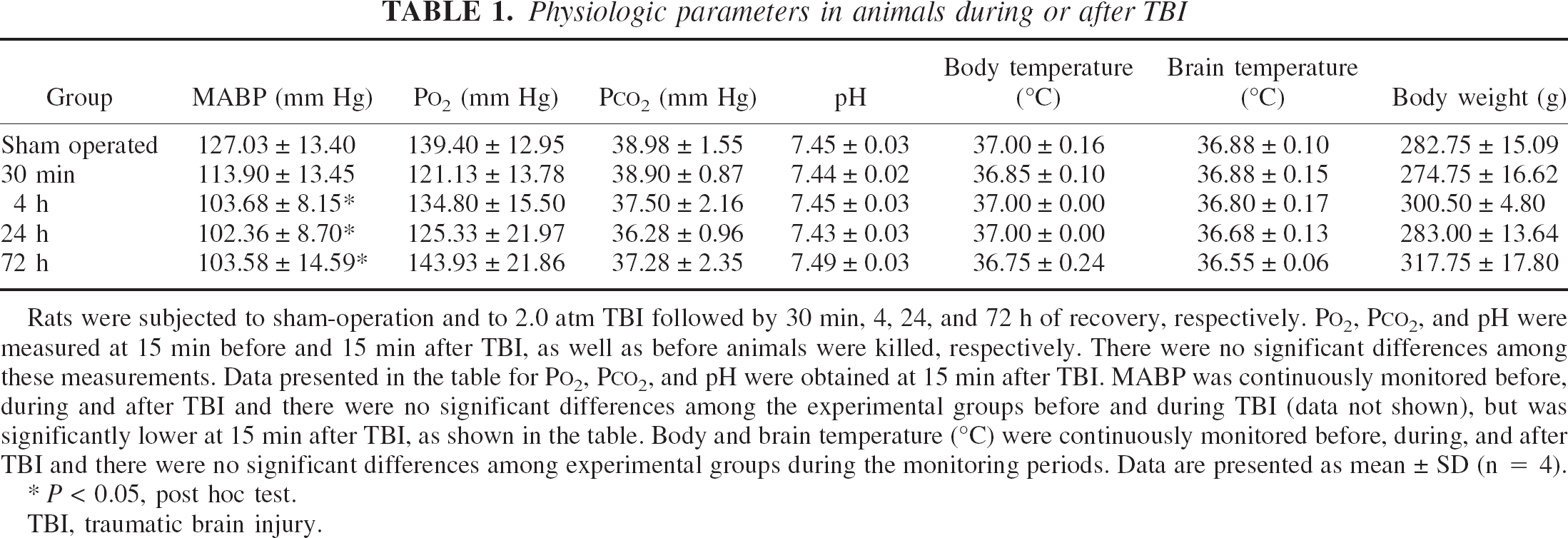
Rats were subjected to sham-operation and to 2.0 atm TBI followed by 30 min, 4, 24, and 72 h of recovery, respectively. P
P < 0.05, post hoc test.
TBI, traumatic brain injury.
Brain sections from sham-operated control rats and rats subjected to 2.0-atm F-P TBI followed by 30 minutes, 4 hours, and 24 hours of recovery were double-stained with a monoclonal antibody against phospho-ERK1/2 and a nucleic acid dye, propidium iodide (PI). These were examined by laser-scanning confocal microscopy. In Figs. 1 and 2, the red color represents PI staining. PI is often used as a dye to stain necrotic cells in culture because it is unable to pass normal cell membrane. In fixed brain sections, however, cell membranes are treated with fixatives and sliced so that PI is able to penetrate into and stain all cells on brain section. PI staining can somewhat distinguish normal neurons from glia and endothelial cells, as well as differentiate normal from damaged neurons by nuclear size and shape, as well as regional distribution. In sham-operated brain sections, neuronal nuclei stained with PI are large in size and round in shape relative to glia. Endothelial cells are elongated and are aligned with vessels. In comparison with sham-operated controls, TBI-damaged nuclei stained with PI were found in CA3 and became severely deformed (Fig. 1B, red, large arrows). This is somewhat different from the morphology of ischemia-damaged CA1 neurons, which are mostly shrunken and polygonal shaped, as well as dimly stained with PI under confocal microscopy (Hu et al., 2000b). The green color indicates specific antibody staining (Figs. 1–2). Phospho-ERK1/2 (green), representing active ERK1/2, was markedly increased in the mossy fiber territories and moderately upregulated in the CA1 region at 30 minutes after TBI (Fig. 1A-B, green). The alteration of ERK in the CA1 region might be due to the development of secondary ischemia in this region. Strikingly, in contrast with nuclear translocation of phospho-ERK1/2 after brain ischemia (Hu et al., 2000a,b), increase in phosph-ERK1/2 immunoreactivity was not prominent in the cytoplasm and nuclei of hippocampal neurons but was predominantly located in mossy fiber axons and neuropil areas (Figs. 1A and B, green). Furthermore, the increase in phospho-ERK1/2 in mossy fibers declined to a certain degree but was still above the levels of sham-operated control at 4 hours after TBI (Fig. 1B, green). Occasionally, phospho-ERK1/2 immunoreactivity was observed in the CA3 dendritic region at 4 hours after TBI (Fig. 1B, 4h, green, small arrows). Consistently, both 42-kd phospho-ERK2 and 44-kd phospho-ERK1 were significantly increased mainly in the synaptosolic fraction on Western blots, peaked at 30 minutes, and gradually declined thereafter, whereas total ERK1/2 was unchanged in that subcellular fraction after TBI (Figs. 1C and 1D). There were no significant changes in phospho-ERK1/2 in other subcellular fractions (data not shown). In comparison with the hemisphere contralateral to TBI, phospho-ERK1/2 was upregulated in neuronal cell bodies as well as in the neuropils of the contused cortical region, but predominantly in the mossy fiber axons of the hippocampal region after TBI (Fig. 2). Relative to sham-operated controls, no obvious changes were observed in phospho-ERK1/2, phospho-CREB, phospho-TrkB, or phospho-Elk-1 in the contralateral brain regions after TBI (data not shown).
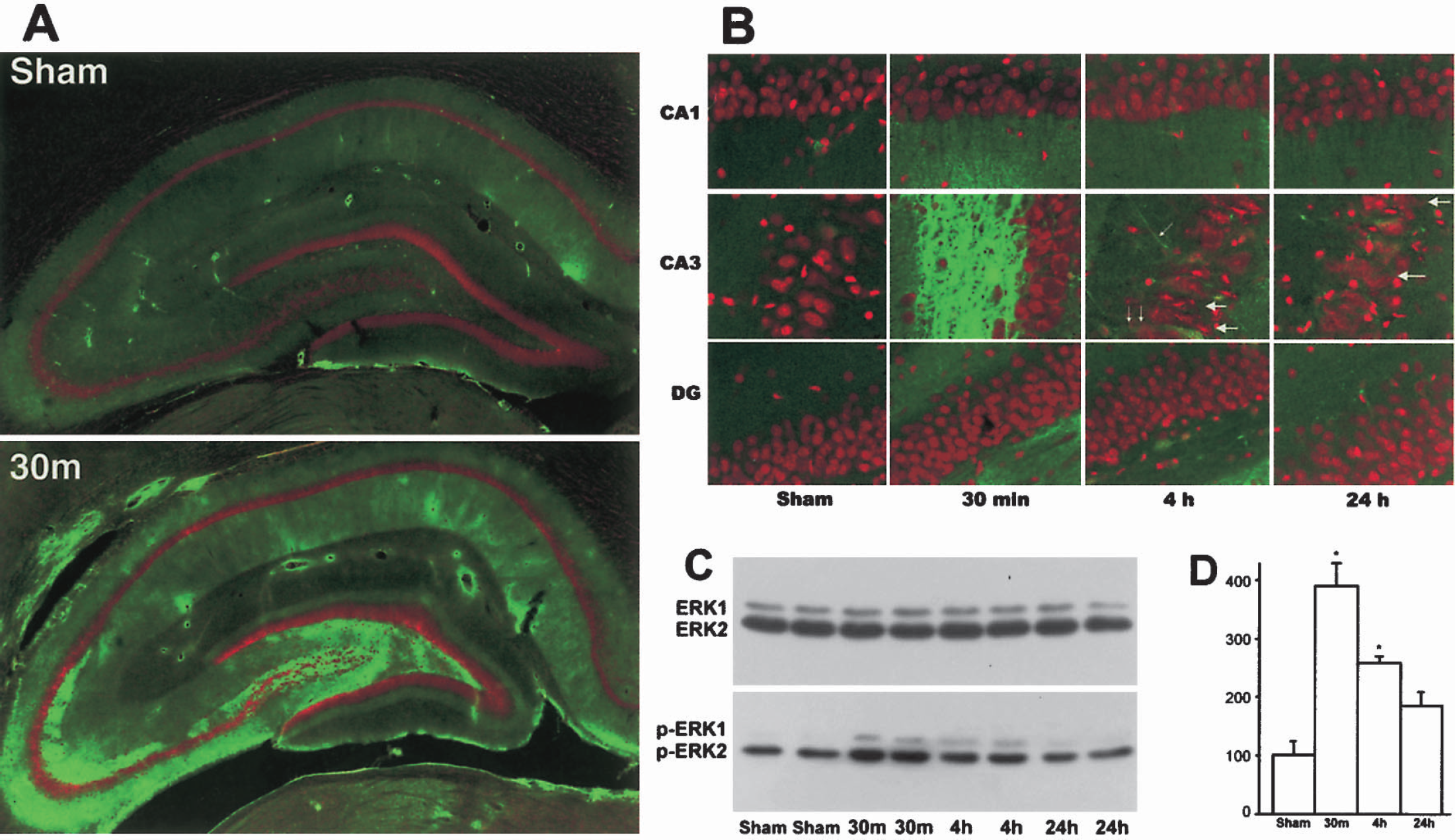
(
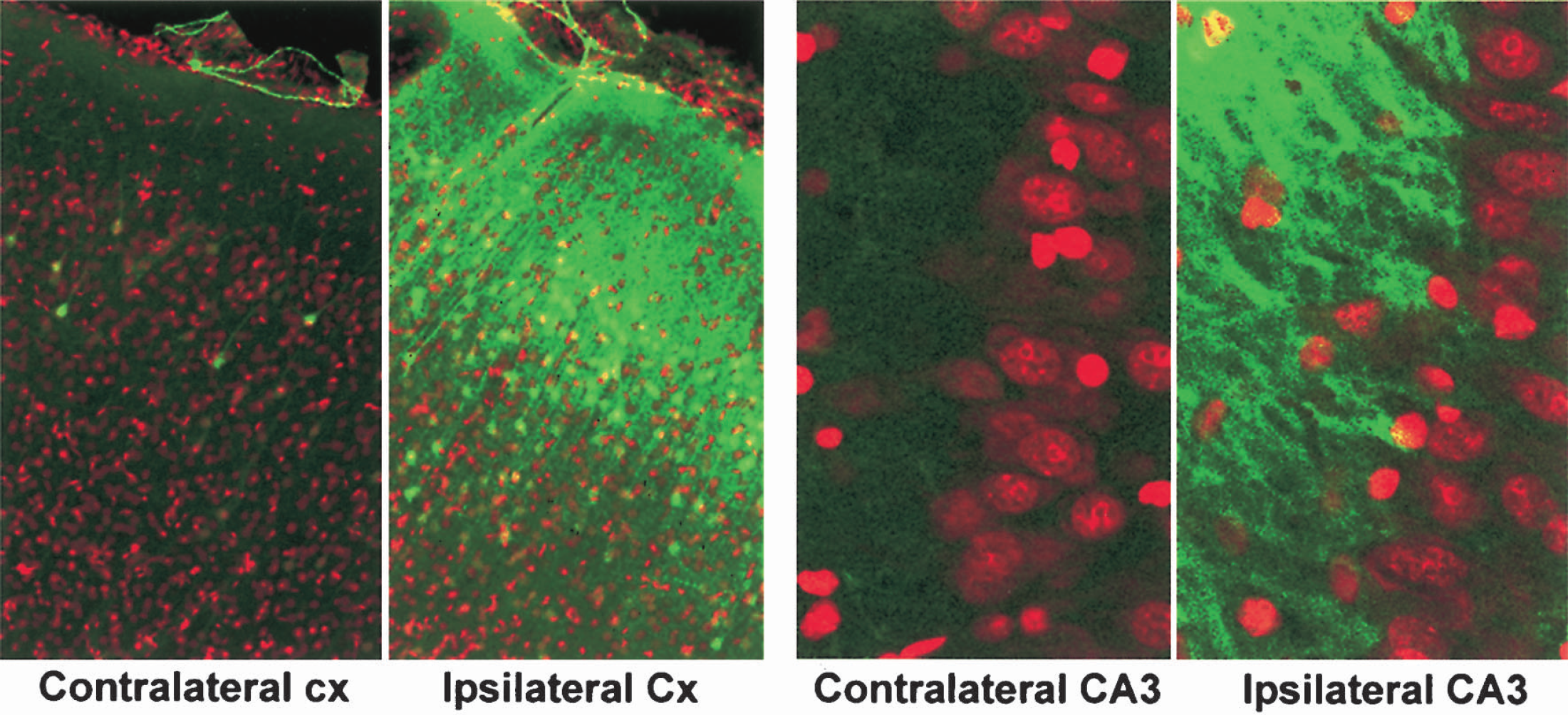
Confocal microscopic images of phospho-ERK1/2 (green) and PI (red) on cortical (Cx) and hippocampal sections from sham-operated control rats and rats subjected to 2.0-atm TBI followed by 30 minutes of recovery. PI stains all nuclei as red color. Phospho-ERK1/2 immunostaining (green) was markedly increased in the cell bodies, dendrites, and neuropils in the TBI-contused cortical region, but was uniquely increased in the mossy fiber projections in the hippocampus at 30 minutes of recovery after TBI.
TrkB is an ERK1/2 upstream neurotrophin receptor in the hippocampus (He et al., 2002). To study whether TrkB was activated and colocalized with phospho-ERK1/2 after TBI, double-staining of phospho-TrkB and phospho-ERK1/2 was performed on brain sections from sham-operated control rats (C) and rats subjected to TBI followed by 30 minutes, 4 hours, and 24 hours of recovery (Fig. 3A). The brain sections were examined by confocal microscopy. Similar to the increase in phospho-ERK1/2 (red), phospho-TrkB (green) was markedly upregulated in ipsilateral hippocampal regions (Fig. 3A) as well as in the contused cortical area (data not shown) at 30 minutes of recovery after TBI. Immunoreactivity of phospho-TrkB (green) was not totally overlapped with phospho-ERK1/2 (red) in the mossy fiber region (Fig. 3A, 30 minutes, CA3, arrows). Furthermore, phospho-TrkB immunoreactivity declined after 30 minutes of recovery but was still higher than the control level at 4 hours after TBI (Fig. 3A, green). Consistently, phosphorylation of TrkB was significantly increased on Western blots, peaked at 30 minutes, and gradually declined thereafter in the hippocampal membrane fractions (Figs. 3B and 3C).
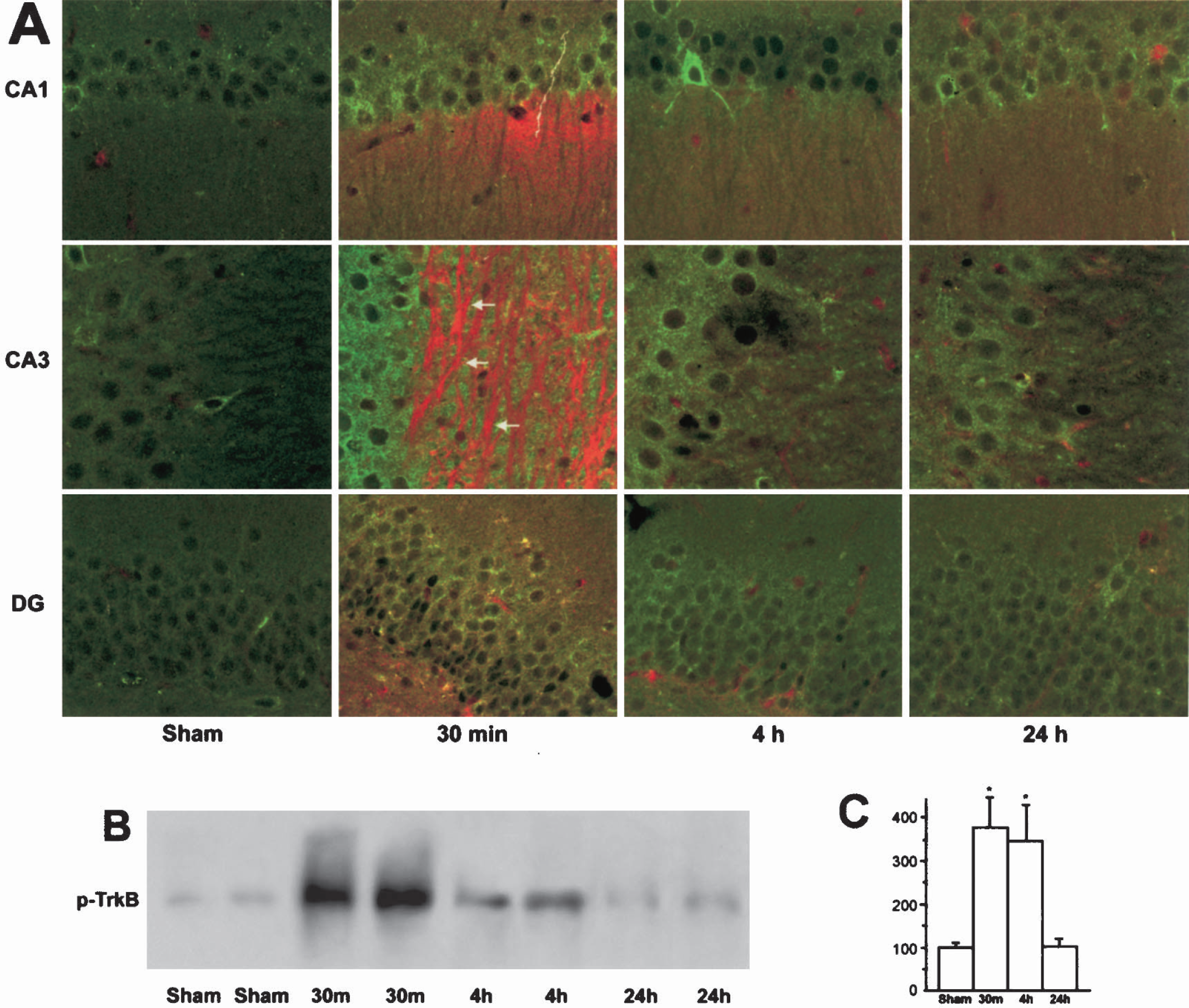
(
To confirm further subcellular distribution of phospho-TrkB after TBI, double-labeling experiments using antibodies against synaptophysin, a nerve-terminal marker, and phospho-TrkB were performed. Figure 4 illustrates high-resolution confocal microscopic images of a double-labeling experiment in which synaptophysin is displayed in red, phospho-TrkB appears green, and areas with overlapping immunoreactivity appear orange color. Phospho-TrkB (green) was not only colabeled with synaptophysin (red) in the nerve terminals (phospho-TrkB + synaptophysin, orange color, arrows), but also collocated with the dendritic plasmalemma (D, green), inside nerve terminals that were stained with anti-synaptophysin (red), in the mossy fiber area (Fig. 4, arrows).
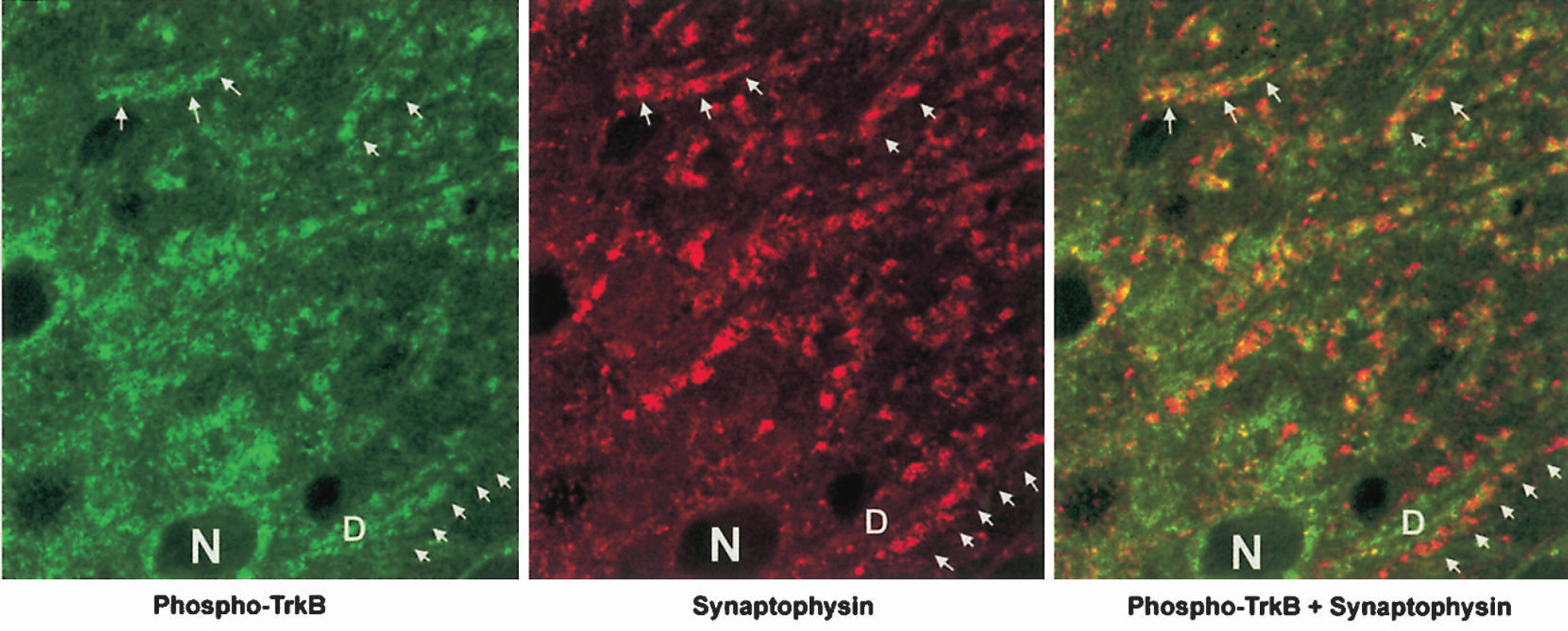
High-resolution confocal microscopic images of double-labeling of phospho-TrkB (green) and synaptophysin (red) in the CA3 dendritic region (stratum lucidum). Brain sections were from a sham-operated control rat and a rat subjected to 2.0 F-P TBI followed by 30 minutes of recovery. The images were enlarged threefold with the confocal zoom function. Phospho-TrkB (green, arrows) was not only colabeled with synaptophysin (red, arrows), but also colocated with the dendritic plasmalemma (D, green) facing the nerve terminals stained with anti-synaptophysin antibody in the double-labeled image (Phospho-TrkB + Synaptophysin, arrows). N indicates nuclei of the CA3 neurons.
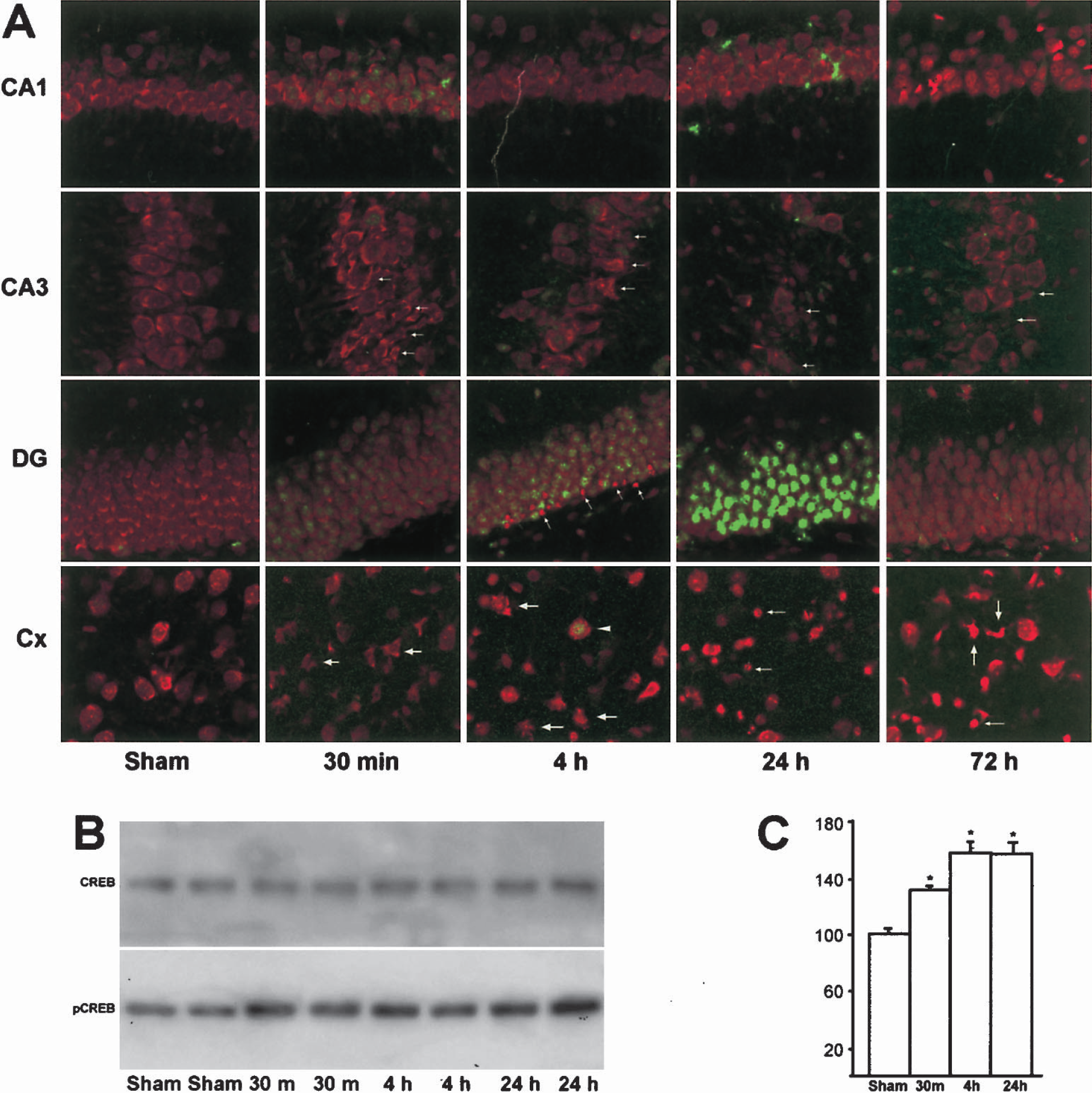
Transcription factors CREB and Elk-1 are ERK1/2 downstream effectors. To investigate whether CREB and Elk-1 are activated after TBI, double-labeling of phospho-CREB and PI (Fig. 5A), or phospho-Elk-1 and PI (Fig. 6) was performed on brain sections. These brain sections were from sham-operated control rats (C) and rats subjected to TBI followed by 30 minutes, 4 hours, 24 hours, and 72 hours of recovery. Phospho-CREB was moderately upregulated in a few neurons scattered in the hippocampus at 30 minutes after TBI (Fig. 5A, green). Thereafter, immunoreactivity of phospho-CREB declined in both CA1 and CA3 neurons, but peaked at 24 hours after TBI in the nuclei of DG granule cells (Fig. 5A, green). Increase in phospho-CREB was also seen in the survival cortical neurons in the contused region mainly at 4 hours after TBI (Fig. 5A). Western blots confirmed that phospho-CREB was increased, whereas total CREB was unchanged in the hippocampal cell extracts after TBI (Figs. 5B and 5C). No obvious change in PI staining (red) was found on these brain sections after TBI, but TBI-damaged nuclei were clearly visible in some PI-stained CA3 and cortical neurons (Fig. 5A, arrows). Some PI-stained dense-nuclei can also be observed in the inner layer of DG granule cells after TBI (Fig. 5A, arrows). The morphology of these condensed DG nuclei appeared similar to the dead granule cell nuclei after hypoglycemia (Ouyang and Hu, 2000), but the origins of these condensed nuclei remain to be elusive. In comparison with CREB, phospho-Elk-1 was moderately upregulated in the nuclei of CA3 neurons only at 4 hours after TBI (Fig. 6, green).
(
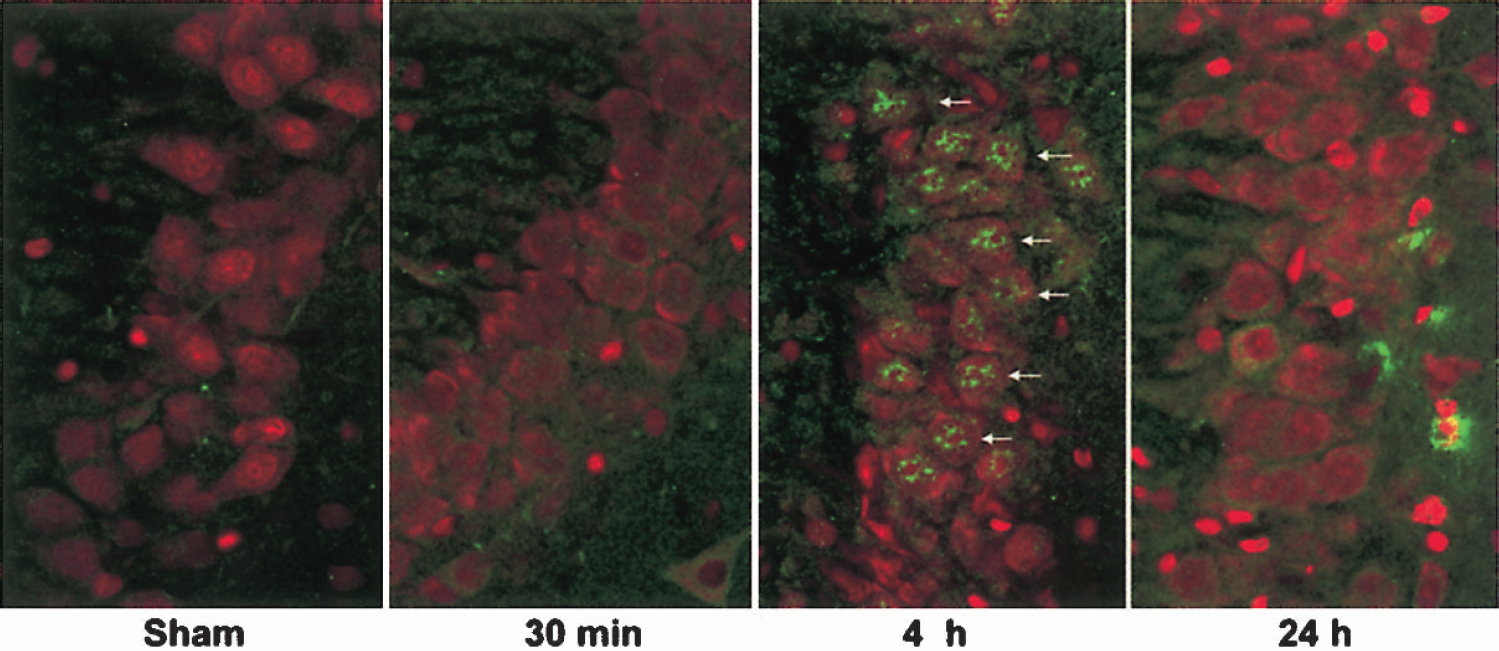
Confocal microscopic images of phospho-Elk-1 (green) and PI (red) on hippocampal sections of sham-operated control and 30 minutes, 4 hours, and 24 hours after TBI. Phospho-Elk-1 (green) was increased in the nuclei of CA3 neurons at 4 hours of recovery after TBI (arrows).
DISCUSSION
Traumatic brain injury often leads to mossy fiber sprouting and posttraumatic seizures, but the underlying intracellular events are not well understood. This study demonstrated striking activation of TrkB–ERK1/2–CREB/Elk-1 pathways predominantly in mossy fiber organization, including DG granule cells, mossy fibers, and CA3 pyramidal neurons after TBI. In normal hippocampus, DG granule cells send out excitatory axons to form mossy fibers projecting to other hippocampal regions such as the dendritic region of CA3. However, in seizure-prone hippocampus, mossy fibers sprout their terminals to form new abnormal excitatory synapses on dendritic spines of DG granule cells (Cavazos et al., 2003). This reorganization of mossy fibers results in formation of recurrent excitatory circuits in the DG region and increases seizure susceptibility (Cavazos et al., 2003; Wenzel et al., 2000). TBI induces mossy fiber sprouting and is a major risk factor for human epilepsy (Englander et al., 2003). The rat model of F-P TBI used in this study develops seizure susceptibility in DG neurons within days that persists for many weeks after injury (Golarai et al., 2001). The epileptic susceptibility in this rat model coincides very well with the reorganization of hippocampal mossy fibers after TBI (Golarai et al. 2001). Based on the results of the present study, one may speculate that overactivation of TrkB–ERK1/2–CREB/Elk-1 in mossy fiber organization may contribute to the mossy fiber sprouting and seizure susceptibility after TBI. This assumption is consistent with the well-defined roles of TrkB–ERK1/2–CREB/Elk-1 pathways in remodeling of synaptic circuits and synaptic plasticity (Hardingham et al., 2001; Ernfors and Braham, 2003; Li et 2003b; Mazzucchelli and Brambilla, 2000; Sweatt, 2001; Ying et al., 2002). It should be noted that ERK phosphorylation appeared to be more massive than TrkB phosphorylation at 30 minutes after TBI. This might be because ERK may also be activated by other extracellular stimuli such as glutamate release after TBI (Miller et al., 1990).
Activation of ERK1/2 after TBI has recently been reported (Dash et al., 1995 and 2002; Otani et al. 2002; Raghupathi et al., 2003). In a lateral controlled cortical impact model in rats, phosphorylation of ERK1/2 is markedly upregulated in neurons and neuropils of both ipsilateral and contralateral hemispheres after TBI (Dash et al., 2002). In a rat F-P model with a severity of 3.5 to 4.0 atm, Otani et al. (2002) reported that phospho-ERK1/2 was increased in neuronal cell bodies and astrocytes. In comparison with these studies, the present study showed a marked activation of ERK1/2 predominantly in mossy fiber organization, but without obvious changes in cell bodies or in the contralateral hippocampal neurons after TBI. The discrepancy among these reports and the present study is likely due to different injury severities and animal models. Metabolic stress and energy failure due to secondary brain ischemia (i.e., mean CBF < 0.25 mL · g−1 · min−1) ensue in both the controlled cortical impact model (Dash et al., 2002) and the F-P model (Otani et al. 2002; Lee et al., 1999). The controlled cortical impact model also reduces CBF globally, with 85% and 49% reductions in a contused cortical region and contralateral cortex at approximately 3 hours after TBI (Hendrich et al., 1999). However, moderate TBI (1.8 to 2.2 atm) in the parasagittal F-P model used in the present study does not lead to severe metabolic stress and energy failure in hippocampus (Dietrich et al., 1998). Striking activation of ERK1/2 mainly in DG cell bodies and their nuclei were reported after transient cerebral ischemia (Hu et al., 2000a). Therefore, metabolic stress and brain hypoperfusion secondary to severe TBI could contribute to the activation of ERK1/2 in neurons and astrocytes, whereas moderate TBI without severe metabolic stress may activate ERK1/2 mainly in axons such as mossy fiber axonal projections. This explanation is in line with the fact that phosphorylation of ERK1/2 also occurred in neurons and their dendrites in the contused cortical region (see Fig. 3) where moderate metabolic stress ensues after moderate TBI (Dietrich et al., 1996; Truettner et al., 1999). Therefore, the unique activation of ERK1/2 in axons is consistent with prominent axonal change after moderate TBI (Povlishock and Kontos, 1992). In addition to the unique phosphorylation of ERK1/2 in hippocampal mossy fibers, this study provides additional evidence that ERK1/2-mediated upstream and downstream events, TrkB, CREB, and Elk-1 phosphorylations are increased in mossy fiber organization after TBI.
Activation of TrkB-ERK1/2-CREB/Elk-1 pathways in mossy fibers would be expected to have functional significance after TBI. It is generally believed that hippocampal seizures originate from the formation of abnormal recurrent excitatory circuits by newly formed synapses between mossy fibers and DG dendritic spines (Li et al., 2003b; Scharfman et al., 2002). In the F-P animal model, TBI induces mossy fiber reorganization and increases seizure susceptibility (Golarai et al., 2001; Hamm et al., 1995; Santhakumar et al., 2001). Activation of TrkB in both presynaptic and postsynaptic membranes plays a significant role in seizure-induced mossy fiber sprouting (Binder et al., 1999; Gall, 1993; He et al., 2002; Li et al., 2003b; Scharfman et al., 1999). Overactivation of TrkB–ERK1/2–CREB/Elk-1 has been implicated in causing hippocampal seizures (Binder et al., 2003; Davis et al., 2000; Li et al., 2003b; Thiels et al., 2002; Ying et al., 2002). Blockade of the TrkB–ERK1/2 pathway with a synthetic peptide inhibitor prevents mossy fiber sprouting and seizure activity in a kindling model in rat (Li et al., 2003b). High levels of BDNF expression in DG granule cells are associated with seizure susceptibility and enhance the efficacy of excitatory mossy fiber synapses onto CA3 pyramidal neurons (Scharfman et al., 2002). Evidence suggests that overactivation of TrkB–ERK1/2–CREB/Elk-1 pathways may contribute to posttraumatic mossy fiber reorganization and increased seizure susceptibility. Further analyses of the regulatory and structural synaptic genes and proteins by which TrkB–ERK1/2–CREB/Elk-1 pathways may influence seizure susceptibility after TBI could provide novel concepts for the treatment of posttraumatic seizures. However, TrkB–ERK1/2–CREB/Elk-1 pathways are recognized as cell growth-related events to repair damaged axons (Dash, et al., 2002; Ernfors and Bramham, 2003; Hu et al., 2000a,b; Mori et al., 2002). An intriguing question arises: are pathways of TrkB–ERK1/2–CREB/Elk-1 activated too much, resulting in formation of abnormal synapses that increase posttraumatic seizure susceptibility after TBI? Or is the formation of new abnormal synapses regulated by different cellular events from that of synaptic repair? Many synaptic functional changes have been reported in hippocampal regions after TBI (Reeves et al., 1997a,b; Zanier et al., 2003). Therefore, dissecting the cellular and molecular mechanisms underlying normal synaptic plasticity and epileptogenesis after TBI may provide new therapeutic avenues for promoting posttraumatic axonal repair but preventing posttraumatic epilepsy.