
Abstract
Elimination of corticosteroids after ischemia, by removal of the adrenals, has been reported to preserve neuronal integrity later. To establish the therapeutic potential of this observation, the authors address two questions: first, whether clinically more relevant steroid manipulations after ischemia exert similar protective effects, and second, whether changes in synaptic functioning occur along with structural alterations. To test this, the authors treated animals immediately after hypoxia—ischemia with (1) the steroid synthesis inhibitor metyrapone, (2) the synthetic glucocorticoid receptor agonist dexamethasone, (3) the selective glucocorticoid antagonist RU 38486, or (4) corticosterone. Metyrapone, but none of the other compounds, attenuated the occurrence of seizures immediately after ischemia, Twenty-four hours after hypoxia—ischemia, CA1 hippocampal field potentials in response to stimulation of fibers were found to be reduced. The attenuation of synaptic transmission was partly prevented by metyrapone. None of the other experimental treatments influenced the impaired synaptic function, Gross morphologic analysis revealed no differences in the loss of neuronal structure between the experimental groups at this time point. Taken together, these data suggest that metyrapone preserves neuronal functioning despite loss of neuronal structure. The authors tentatively conclude that preventing the ongoing production of steroids shortly after ischemia can delay and attenuate the appearance of ischemia-related pathology.
Corticosteroid hormones are secreted from the adrenal glands, readily cross the blood—brain barrier, and bind to both corticosteroid receptor types in the brain: the mineralocorticoid receptor (MR) and the glucocorticoid receptor (GR), which are co-localized in hippocampal neurons (McEwen et al., 1986; de Kloet, 1991), Because of a difference in affinity of these receptor types for corticosterone, low levels of corticosterone predominantly occupy MR, whereas GR are progressively occupied with higher hormone levels (De Kloet, 1991). On binding of corticosterone to MR or GR, the hormone modulates signal transduction, cellular viability, and hippocampal structure (Joëls and De KIoet, 1994; McEwen and Sapolsky, 1995).
Earlier studies provide evidence that elevated glucocorticoid levels increase vulnerability of hippocampal neurons in response to challenges such as ischemia (Sapolsky and Pulsinelli, 1985; Morse and Davis, 1990), Conversely, treatment with steroid synthesis inhibitors before the ischemic insult, to prevent the production of corticosteroids during ischemia, preserves neuronal integrity and prevents the occurrence of seizures (Krugers et al., 1995; Smith-Swintosky et al., 1996; Krugers et al., 1998), Both in vitro and in vivo studies point to the GR as a mediator of these damaging effects of corticosterone (Koide et al., 1986; Packan and Sapolsky, 1990; Adachi et al., 1998).
An important question from the therapeutic point of view is whether modulation of plasma corticosterone levels after ischemia is as effective as treatment before the ischemic insult. Preliminary evidence suggests that removal of the adrenal glands (adrenalectomy) after ischemia preserves neuronal integrity later on (Sapolsky and Pulsinelli, 1985; Morse and Davis, 1990), In the current study, we elaborate these preliminary findings: First, we studied whether treatment with the steroid synthesis inhibitor metyrapone or with the GR-antagonist RU 38486 after the ischemic insult—a clinically more relevant alternative for adrenalectomy—can delay or attenuate postischemic pathology. We also tested the efficacy of the synthetic GR-agonist dexamethasone. Dexamethasone is hampered in its transport across the intact blood—brain barrier by the mdr- la protein (Meijer et al., 1998); consequently, it indirectly suppresses production of endogenous corticosterone by a negative feedback action at the level of the pituitary but lacks GR agonistic actions in the brain. Therefore, we predict that dexamethasone suppresses the corticosterone output, which could potentially reduce postischemic pathology. Second, we studied the effect of these postischemic steroid treatments not only on structural, but also on functional parameters in the hippocampus, a region that is known to display severe ischemia-induced pathology. To this purpose, we recorded field potentials in the hippocampal CA1 area in vitro in response to stimulation of Schaffer collateral/commissural fibers, 24 hours after hypoxia—ischemia. At the same time point, we determined morphologic alterations in the hippocampus using Nissl staining and immunohistochemical analysis for the structural protein, microtubule-associated protein 2 (MAP2).
MATERIALS AND METHODS
Experimental animals
All experiments were approved by the Committee on Animal Bio-Ethics of the University of Amsterdam. Male Wistar rats (325 to 375 g) were housed individually at a light—dark regime (8 hours/20 hours) and fed ad libitum. Surgical procedures were performed between 9 and 11 hours (when basal plasma corticosterone levels are low). Immediately after hypoxia—ischemia, 11 animals (ischemia plus vehicle [Isch + Veh]) received an injection with sesame oil (0.25 mL), whereas 12 animals (ischemia plus corticosterone [Isch + Cort]) received an injection with corticosterone at a dose (40 mg/kg subcutaneously) known to elevate steroid levels over a prolonged period (Sapolsky and Pulsinelli, 1985; Krugers et al., 1998). Ten animals (Isch + Met) received an injection with metyrapone (150 mg/kg subcutaneously), which inhibits the rise in steroid levels (Krugers et al., 1998), and 13 animals (Isch + RU 38486) received an injection with RU 38486 at a dose (25 mg/kg, subcutaneously) known to block the GR (Ratka et al., 1989). Seven animals were injected with the synthetic GR agonist dexamethasone (10 μg/kg), which blocks stress-induced pituitary corticotropin release, resulting in reduced adrenal corticosterone output (De Kloet et al., 1975; Meijer et al., 1998). Moreover, eight animals were exposed to normoxia (norm + Veh) and received an injection with vehicle. These animals were controls for surgery.
Hypoxia—ischemia procedure
Hypoxia—ischemia was induced using a modified Levine model in which temporary unilateral carotid artery ligation is combined with hypoxic ventilation (Dijkhuizen et al., 1998; Krugers et al., 1998). This procedure reduces CBF at the occluded side during hypoxia—ischemia, thereby inducing a unilateral global hemispheric ischemia (Dijkhuizen et al., 1998). Animals were anesthetized using halothane (2%) in a mixture of 30% O2 and 70% N20. The right arteria femoral is was cannulated for continuous blood pressure monitoring during the normoxia and hypoxia—ischemia periods (Hewlett Packard Pressure transducer, Boeblingen, Germany). Animals were intubated and subsequently ventilated (Infant ventilator MK2, Loosco, Amsterdam, The Netherlands) for 10 minutes with 30% O2 and 70% N2O (first normoxia period), next for 20 minutes with 10% O2 and 90% N2O (hypoxia), and subsequently for 5 minutes with 30% O2 and 70% N2O (second normoxia period). During the hypoxia period, the right carotid artery was occluded, thereby inducing hypoxia—ischemia. The rectal temperature was maintained between 36.0° and 37.0°C. Blood samples were taken immediately after the second normoxia period to measure plasma corticosterone levels. The blood samples were transferred to centrifuge tubes containing 10 mL of heparin solution (500 U/mL) and centrifuged for 20 minutes at 3500 g. The supernatant was stored at −20°C for analysis of plasma corticosterone levels using a radioactive immunoassay.
Seizures
Hypoxia—ischemia evokes convulsions after the insult (Krugers et al., 1995; Krugers et al., 1998). To establish whether modulation of steroid levels or blockade of the GR affects the incidence of these convulsions, we recorded the number of animals per group that displayed visible seizures from the period after hypoxia—ischemia until perfusion 24 hours later using a video camera. In addition, the animals were subdivided into three categories: (1) no seizures, (2) I to 50 seizures, and (3) more than 50 seizures during the first 4 hours after ischemia.
Electrophysiologic procedures
Twenty-four hours after hypoxia—ischemia, animals were decapitated, and trunk blood was collected to determine plasma corticosterone levels. The brains were quickly removed and chilled in carbogenated (95% O2 and 5% CO2) artificial cerebrospinal fluid (aCSF) at 4°C containing (in millimolar amounts): NaCl (120), KCl (3.5), MgSO4 (1.3), NaH2PO4 (1.25), CaCl2 (2.5),
Schaffer collateral/commissural fibers were stimulated by a bipolar stainless steel stimulation electrode (60 μm diameter) that delivered 150-microsecond pulses (Krugers et al., 1997). The evoked field potentials were recorded with aCSF-filled glass electrodes (impedance approximately 2 MΩ). The population spike (PS) and field excitatory postsynaptic potential (fEPSP) were recorded in stratum pyramidale and stratum radiatum, respectively. The amplitude of the PS and slope of the fEPSP were determined as shown in Figs. 1A and 1B.
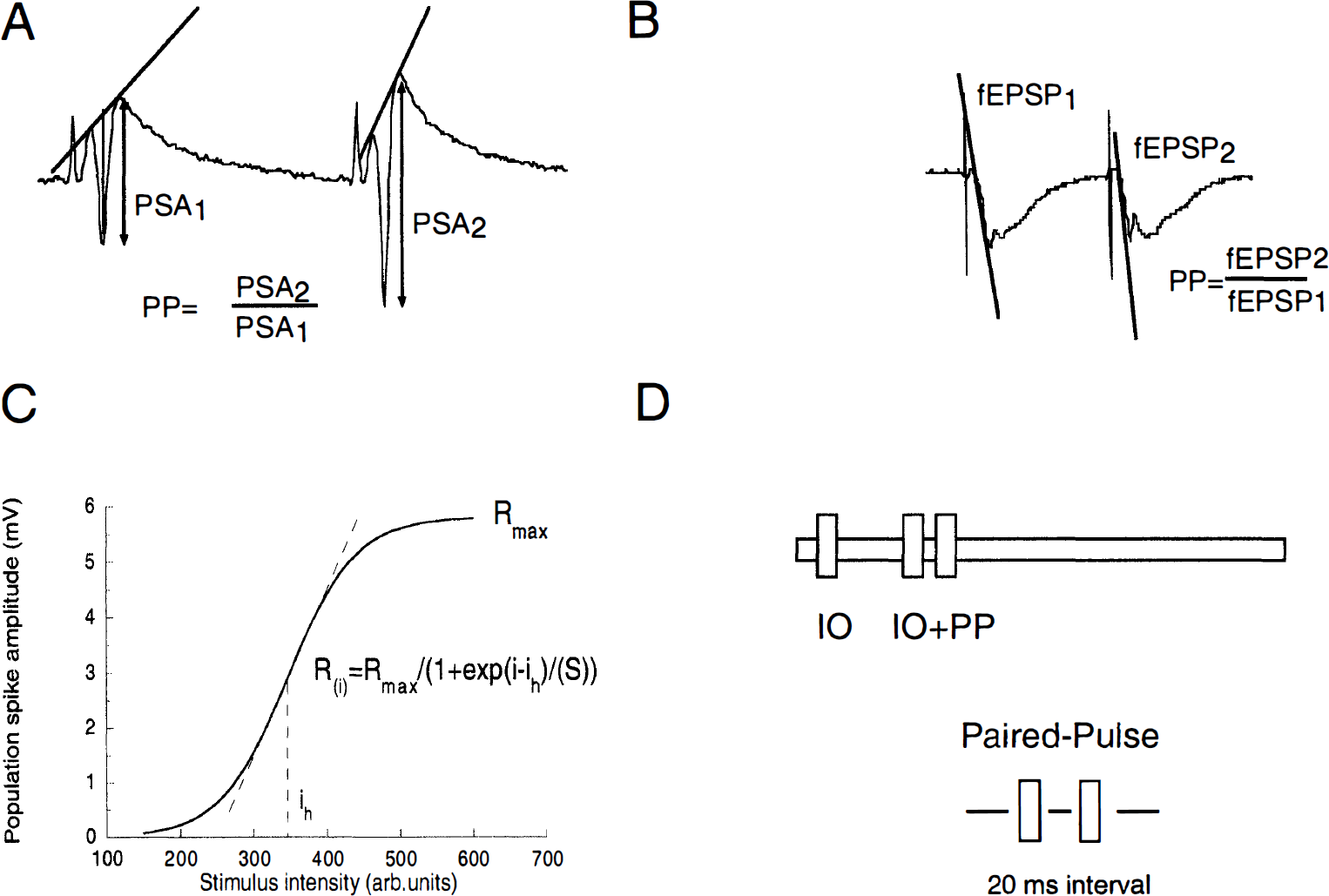
The population spike amplitude
The stimulation protocols, data acquisition, and analysis of the evoked potentials were performed with an Atari computer with in-house developed software. A schematic overview of the experimental protocols is illustrated in Fig. 1D.
Recording of field potentials was started at least 15 minutes after placing the stimulation and recording electrodes. To determine the stimulus intensity that evokes half-maximal and maximal field potential responses, an input—output (IO) curve was constructed that comprised 10 stimulus intensities (interval of 10 seconds) ranging from threshold to maximal stimulus intensities. The relation between stimulus intensity and the evoked response was fit by a sigmoidal function: R(i) = Rmax/(1 + exp(i – ih)/(S)), where R(i) is the response at intensity (i), Rmax, is the maximal response, ih is the intensity at which half-maximal response is observed, and S represents an index proportional to the slope of the IO curve (Fig. 1C). During the experiment, stimuli were delivered with half-maximal stimulus intensity determined at the start of the experiment unless stated otherwise. Next, field potentials were recorded during 10 minutes with stimuli delivered at an interval of 30 seconds. In all experiments discussed in this study, PS amplitudes and fEPSP varied by less than 20% during this period. After this period, a second IO curve was followed by the response to paired-pulse (PP) stimulation, delivered at half-maximal stimulus intensity and maximal stimulus intensity with an interstimulus interval of 20 milliseconds. The ratio of the PS amplitudes or slope of the tEPSP measured after PP stimulation was determined as indicated in Figs. 1A and 1B.
Histology
In addition to the slices used for electrophysiologic recordings, four slices (i.e., two 400-μm sections from both the ipsilateral and contralateral side) were kept in 4% paraformaldehyde in 0.05 mol/L phosphate buffer (pH 7.4) for 24 hours to determine morphologic alterations. After overnight fixation, one slice from the ipsilateral and contralateral side was transferred to 0.1 mol/L phosphate-buffered saline (pH 7.4) containing 0.1% sodium azide. In these sections, we studied changes in the immunocytochemical distribution of the cytoskeletal protein MAP2. The two remaining sections were used for Nissl staining. The 400-μm sections were cryoprotected by overnight storage in 30% sucrose in 0.1 mol/L phosphate buffer (pH 7.4), cut to 30-μm thick coronal sections at −15°C using a cryostat microtome, and collected in 4% paraformaldehyde in 0.05 mol/L phosphate buffer (pH 7.4) for cresylviolet staining, or in 0.1 mol/L phosphate-buffered saline (PBS) containing 0.1% sodium azide for MAP2 immunocytochemical study. Immunocytochemical staining for the microtubule associated protein MAP2 was performed on free-floating sections as described later, all steps being identical for all animals. The tissue sections were preincubated for 20 minutes in 0.1% H2O2 in 0.05 mol/L PBS, subsequently rinsed in PBS, and immersed in 5% normal sheep serum (Sigma Chemical Co., St. Louis, MO, U.S.A.) in PBS for 30 minutes to reduce background staining. Next, the sections were incubated with the first antibody (monoclonal mouse anti-MAP2, clone HM-2, Sigma) and diluted 1 :500 in PBS and 1% normal sheep serum overnight at 4°C under gentle movement of the incubation medium. After the primary antibody incubation, sections were rinsed in PBS and again preincubated with 5% normal sheep serum for 30 minutes before exposure to biotinylated sheep anti-mouse immunoglobulin G (Amersham, diluted 1:200) in PBS for 2 hours at room temperature. Thereafter, the sections were thoroughly rinsed in PBS and incubated in streptavidin-horseradish peroxidase (Zymed, diluted 1:200) in PBS for 2 hours at room temperature. Finally, after subsequent rinsing in PBS and 0.05 mol/L Tris-buffer (pH 7.4), the sections were processed by the diaminobenzidine-H2O2 reaction (30 mg of diamininobenzidine and 0.01% H2O2/100 mL 0.05 mol/L Tris-buffer), guided by a visual check.
The current histologic procedures did not allow accurate quantitative analysis of the observed damage. We settled for a conservative qualitative analysis in which two investigators examined the Nissl-stained and MAP2 immunostained sections of each animal in parallel, in a blind fashion. Histologic sections were assigned to one of the following classes:
No damage: The histologic analysis of the ipsilateral side did not reveal any difference with the contralateral side of the same animal and with control sections from normoxia-treated animals.
Moderate damage: Nissl staining revealed loss of integrity in the dentate gyrus (DG), although no apparent irregularly shaped pyramidal CA1 neurons are present compared with the contralateral side.
Severe damage: Nissl staining showed pyknotic cells in the DG and irregularly shaped pyramidal CA1 cells; MAP2 immunostaining was absent in (part of) the DG and faint in the CA1 area.
Statistics
All data are expressed as mean ± SD. Blood pressure and rectal temperature were analyzed using analysis of variance with repeated measures. The other parameters (plasma corticosteroid levels, maximal values of the PS amplitude, slope of the fEPSP, slope factor S, and half-maximal stimulation intensity of the IO curves and PP responses) were analyzed by one-way analysis of variance followed by Fisher's test. Values of P < 0.05 were considered to be significant.
RESULTS
Physiologic values and plasma corticosterone levels
Blood pressure was stable during the first normoxia period (Fig. 2A). During hypoxia—ischemia, however, blood pressure dropped significantly compared with the animals that were exposed to normoxia but returned to pre—hypoxia—ischemia levels during the second normoxia period. No differences were present between the different groups exposed to hypoxia—ischemia. The rectal temperature, which was not significantly different between the experimental groups, remained stable during the surgical procedures (Fig. 2B).
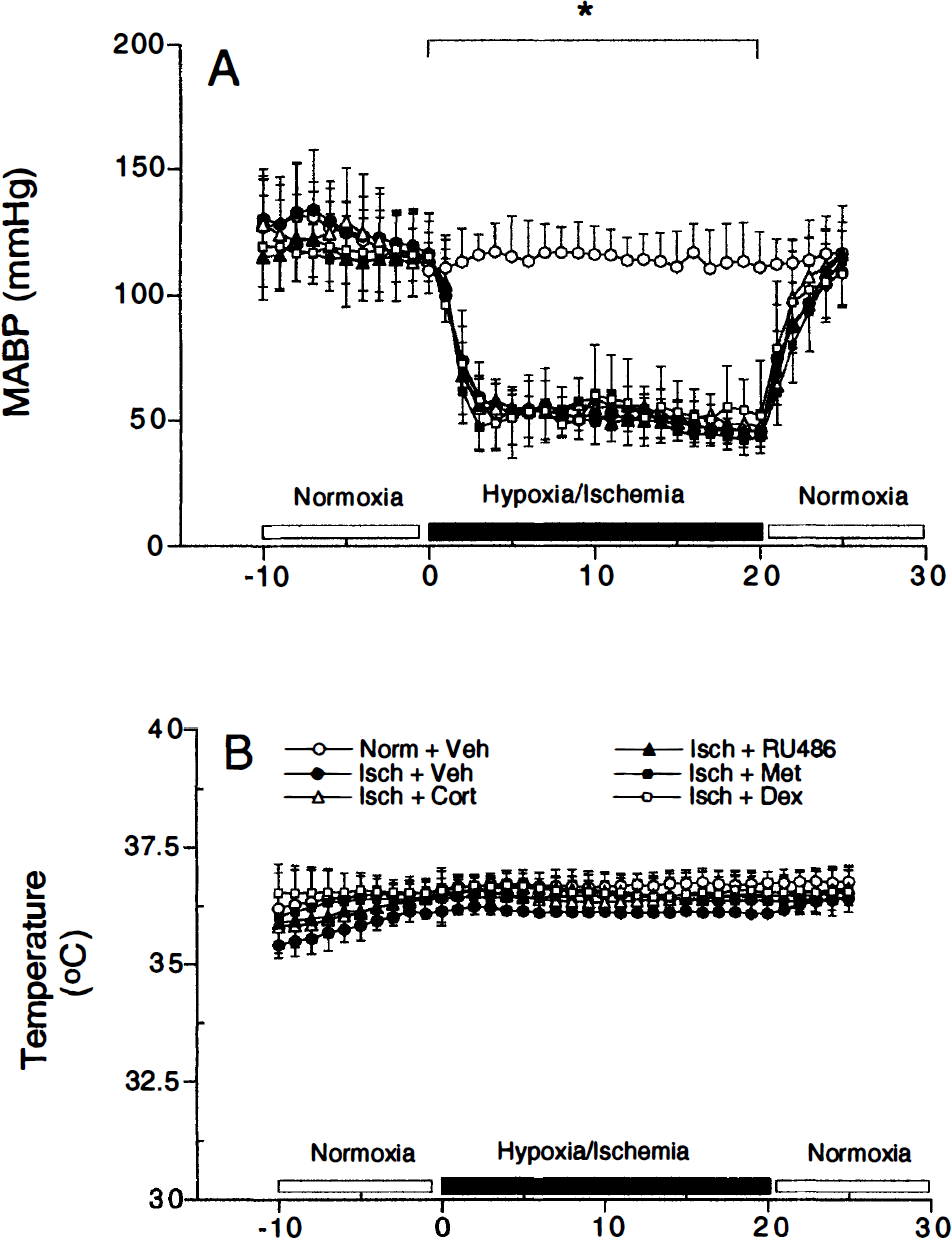
Mean arterial blood pressure (MABP)
Blood samples were taken immediately after hypoxia—ischemia and after decapitation to measure plasma corticosteroid levels. Analysis of the corticosterone levels of all animals exposed to hypoxia—ischemia did not reveal statistically significant differences between the experimental groups at the two time points studied (Figs. 3A and 3B). Analyzing the levels of the animals included for electrophysiologic measurements yielded significantly elevated steroid levels immediately after decapitation in corticosterone-treated animals over normoxia-treated, as well as vehicle-, metyrapone-, and dexamethasone-treated ischemic animals ([F(5,42) = 2.565; P = 0.04], data not shown).
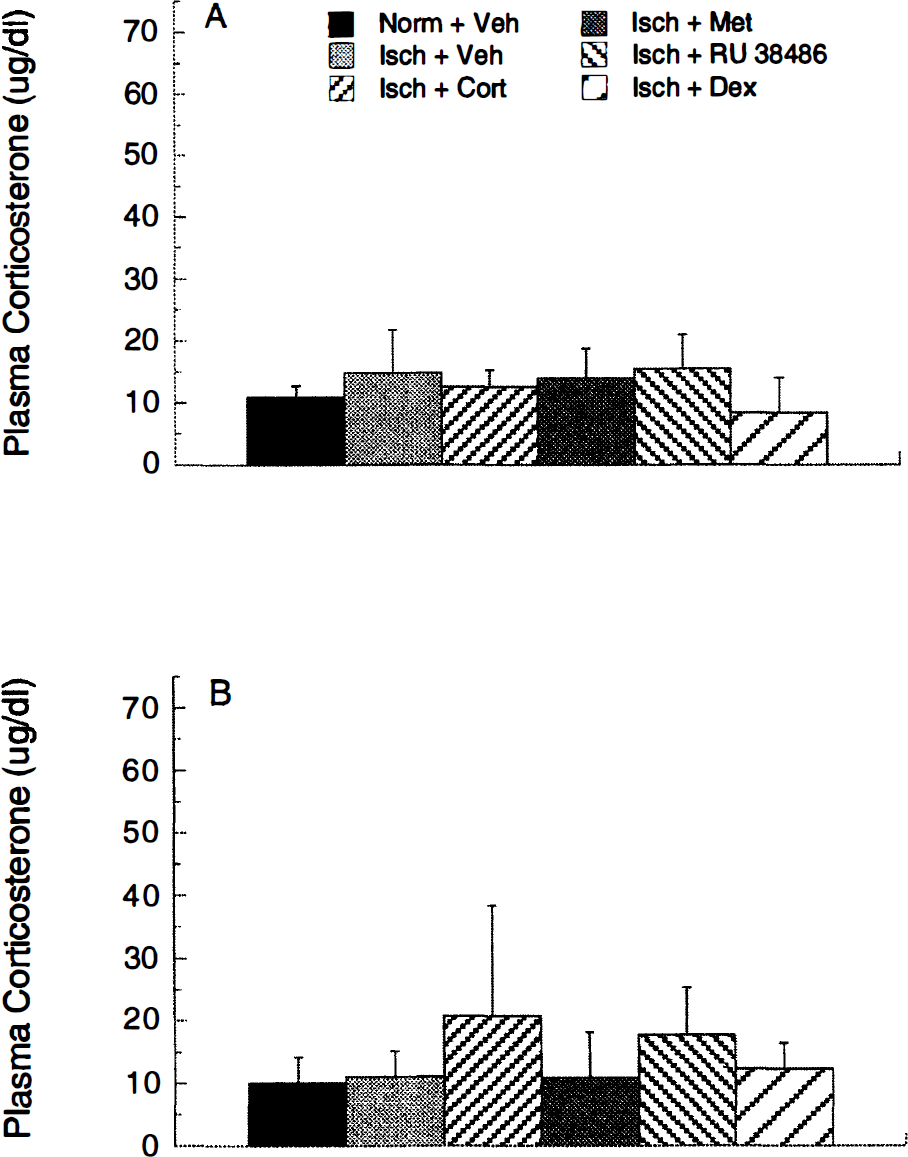
Plasma corticosteroid levels measured immediately after hypoxia—ischemia
Seizures
Hypoxia—ischemia elicited visible seizures as reported previously (Krugers et al., 1995; Krugers et al., 1998). These seizures were characterized by spinning of the animal around its body axis, jerking movements, or clonic contractions of the paws. In general, these seizures were intermittent and lasted less than 30 seconds. Over the entire 24 hours after ischemia, the incidence of these seizures was comparable between the experimental groups (Table 1). However, during the first 4 hours after ischemia, the lowest incidence of seizures was found in the animals treated with metyrapone (Table 1). Detailed analysis of the number of seizures during these first 4 hours after ischemia reveals that only one metyrapone-treated animal showed more than 50 seizures (Fig. 4), whereas most of the metyrapone-treated animals did not display seizures during the first hours after ischemia, pointing to a temporary beneficial effect of metyrapone.
Body weight (mean ± SD) and the number of animals displaying visible seizures after hypoxia—ischemia
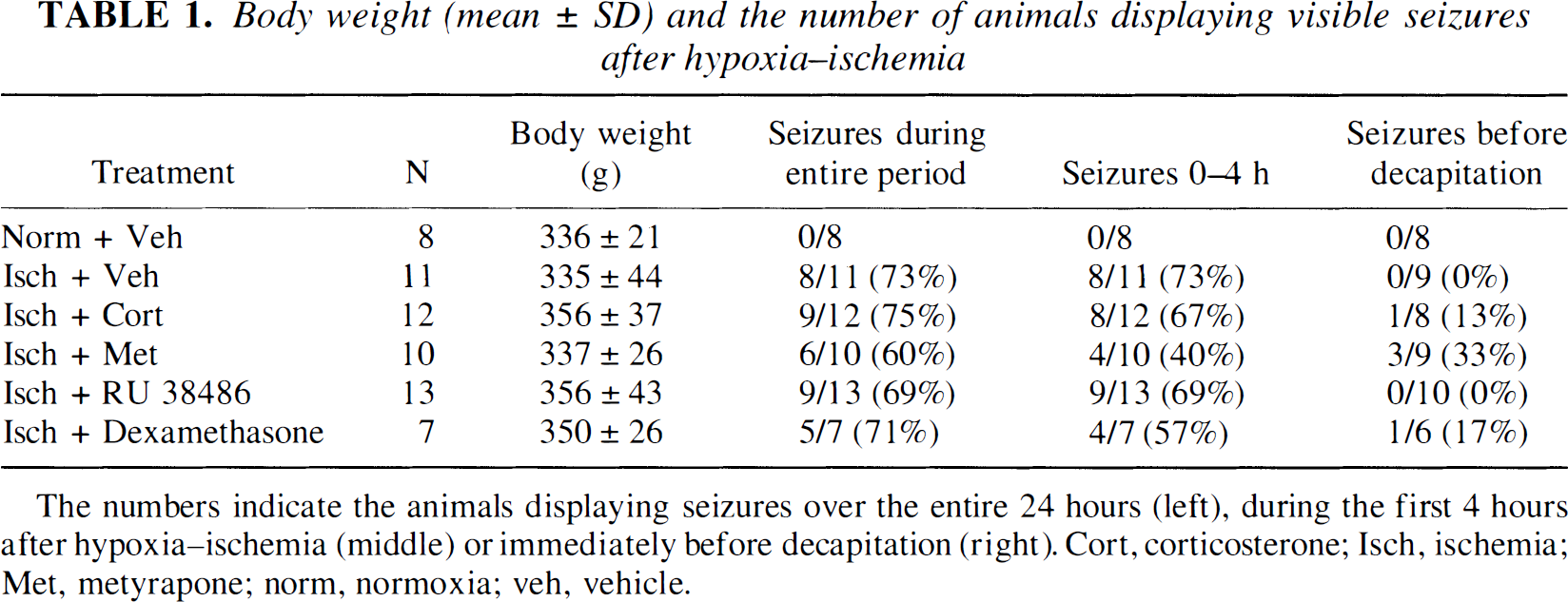
The numbers indicate the animals displaying seizures over the entire 24 hours (left), during the first 4 hours after hypoxia—ischemia (middle) or immediately before decapitation (right). Cort, corticosterone; Isch, ischemia; Met, metyrapone; norm, normoxia; veh, vehicle.
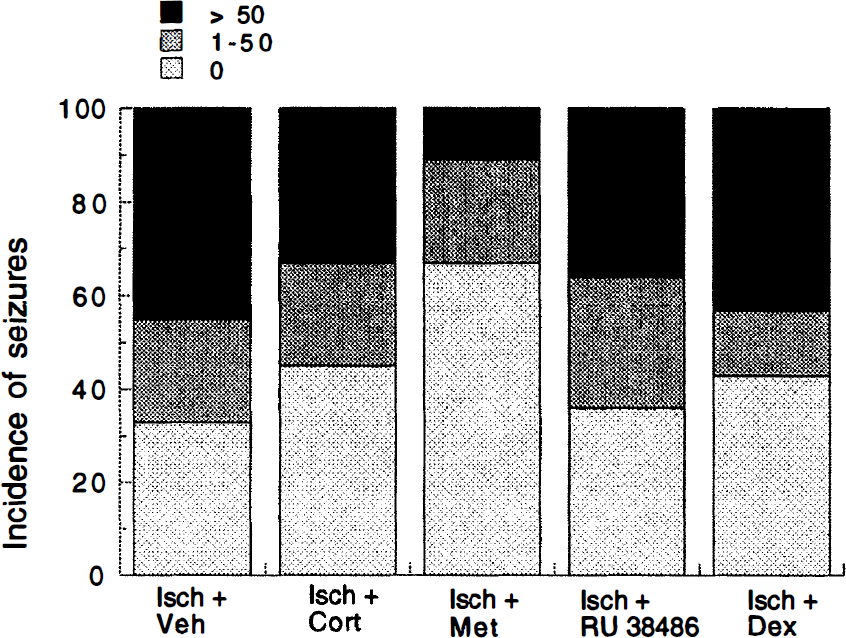
The number of seizures the animals displayed during the first 4 hours after ischemia are categorized into three groups: (1) o convulsions, (2) 1 to 50 convulsions, and (3) more than 50 convulsions. In the metyrapone-treated group, the largest percentage of animals displays no seizures, whereas only one animal is included in the group expressing the largest number of seizures.
Input—output curves of population spike amplitude and slope of the field EPSP
In the 4- to 24-hour period after hypoxia—ischemia, one animal died in the metyrapone- and dexamethasone-treated group. In addition, two animals died in the vehicle-treated group, and three and four animals died in the RU 38486- and corticosterone-treated groups, respectively. In the remaining animals, we studied neuronal functioning in the hippocampal CA1 area after hypoxia—ischemia by establishing synaptic responsiveness at both the ipsilateral and contralateral side to the occlusion. The animals reported on below exhibited a maximal PS amplitude of more than 1.25 m V and a maximal fEPSP slope more negative than −0.40 m V/ms in slices from the contralateral side to the occlusion.
Synaptic responses in slices from the side ipsilateral to the occlusion clearly were affected by ischemia; the maximal slope of the fEPSP was significantly reduced in vehicle-treated animals compared with normoxia animals (Fig. 5D). The steroid treatment did not influence this effect of ischemia: all steroid-treated ischemic animals showed a significant reduction in maximal fEPSP compared with normoxic animals. Recordings in the cell layer also yielded an ischemia-induced reduction of the maximal PS amplitude at the ipsilateral side (Fig. 5B). However, this reduction was absent after treatment with metyrapone. These results support the view that the number (the dendrites) of principal cells that respond to Schaffer collateral/commissural stimulation after hypoxia—ischemia is reduced in animals exposed to ischemia compared with animals exposed to normoxia. For all of the experimental groups, no significant differences were observed with respect to the PS amplitude and fEPSP slope in slices prepared from the contralateral side (Figs. 5A and 5C; [F(5,42) = 0.61; P = 0.69], [F(5,42) = 2.22; P = 0.07], respectively).
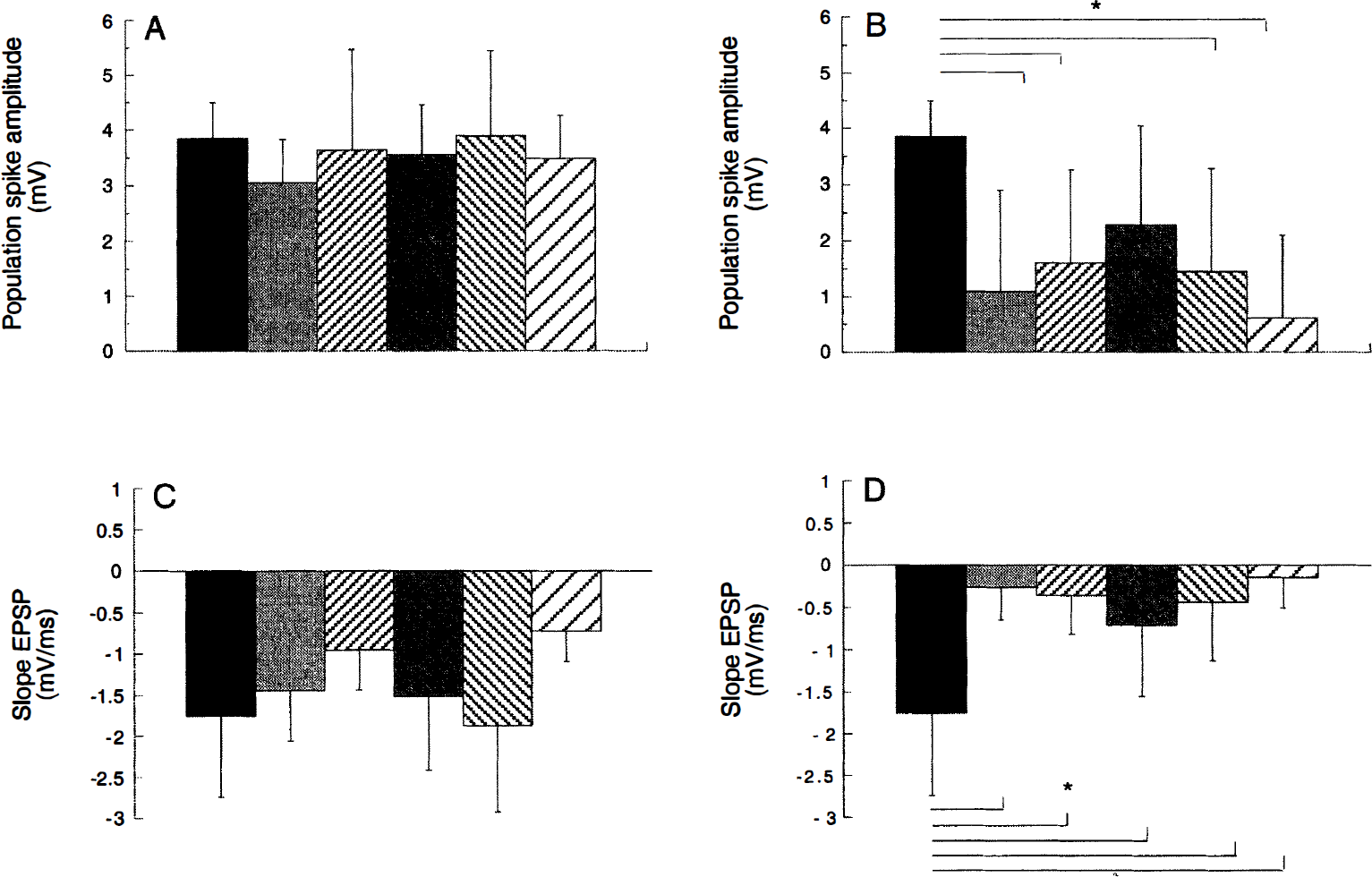
Maximal population spike (PS) amplitude
The data obtained with the IO curve protocol were fitted with a Boltzman equation R(i) = Rmax/(1 + exp((i − ih)/(S)), yielding values for the Rmax, half-maximal stimulation intensity (ih), and a constant S, which is proportional to the slope of the fitted curve. Reliable fits—and thus values for ih and S—could be obtained only from animals where the PS amplitude was more than 0.40 mV and the fEPSP slope was more negative than −0.15 mV/ms. Table 2 shows that in all experimental groups, except the normoxia group, part of the animals were excluded from the fitting analysis at the ipsilateral side based on this criterion. Only groups comprising at least four animals were included for statistical analysis. The dexamethasone group, where only one animal showed a PS amplitude above 0.40 mV and fEPSP slope exceeding −0.15 mV/ms, was excluded from further testing based on this criterion. From the animals where IO curves were successfully fitted, the maximal values of the PS amplitude and fEPSP slope were, on average, reduced at the side ipsilateral to occlusion compared with the normoxia group, but these differences did not attain statistical significance. In addition, no differences in slope factor S were present at the ipsilateral or contralateral side between the experimental groups (Table 2). At the contralateral side, the half-maximal stimulation intensity for the PS amplitude was significantly larger in vehicle-treated ischemic animals compared with the other experimental groups. These results indicate that IO curves of the animals from which bilateral recordings could be made are not different among the groups at the ipsilateral (ischemic) side.
Characteristics of the IO-curves of the population spike (PS) and slope of the fEPSP at the sides contra- and ipsilateral to occlusion
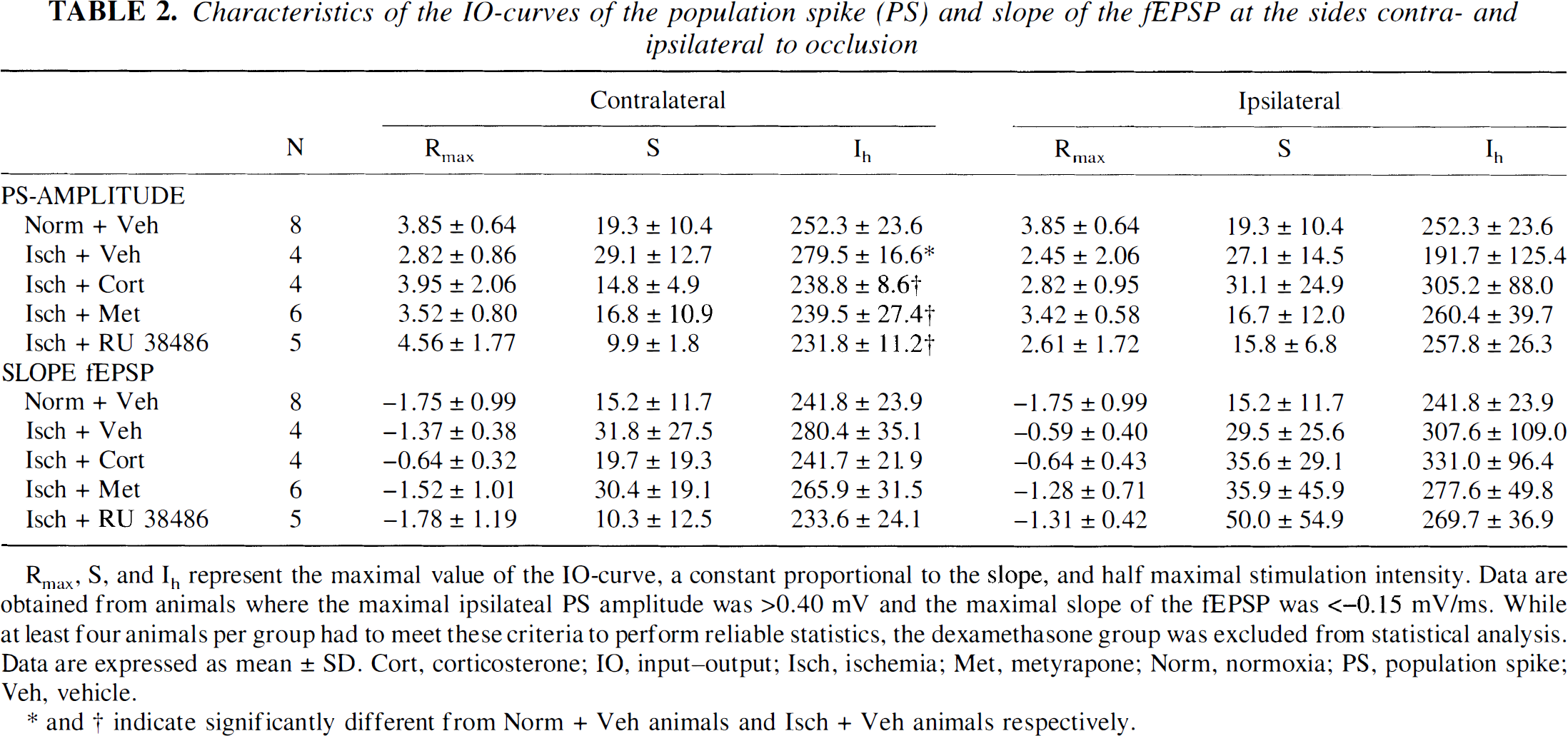
Rmax, S, and Ih represent the maximal value of the IO-curve, a constant proportional to the slope, and half maximal stimulation intensity. Data are obtained from animals where the maximal ipsilateal PS amplitude was >0.40 mV and the maximal slope of the tEPSP was <−0.15 mV/ms. While at least four animals per group had to meet these criteria to perform reliable statistics, the dexamethasone group was excluded from statistical analysis. Data are expressed as mean ± SD. Cort, corticosterone; IO, input—output; Isch, ischemia; Met, metyrapone; Norm, normoxia; PS, population spike; Veh, vehicle.
* and † indicate significantly different from Norm + Veh animals and Isch + Veh animals respectively.
Responses to double-pulse stimulation
The PP ratios were calculated only from the animals from which reliable signals could be obtained at the ipsilateral side (PS amplitude and fEPSP slope more than 0040 mV and more negative than −0.15 m V/ms, respectively). Only groups comprising at least four animals were included for statistical analysis. In general, analysis of these responses in the CA1 pyramidal cell layer, as well as in the stratum radiatum, showed facilitation of the second PS and slope of the fEPSP after stimulation at half-maximal intensity (PPh, Table 3). Stimulation at maximal stimulation intensity (PPm) did not evoke PP potentiation in either the cellular or dendritic layer. At the (ischemic) ipsilateral side, no changes in facilitation were present between the experimental groups at half-maximal or maximal stimulation intensity in either the dendritic or cellular field.
Paired-pulse ratios obtained in the CA1 pyramidal cell layer and stratum radiatium
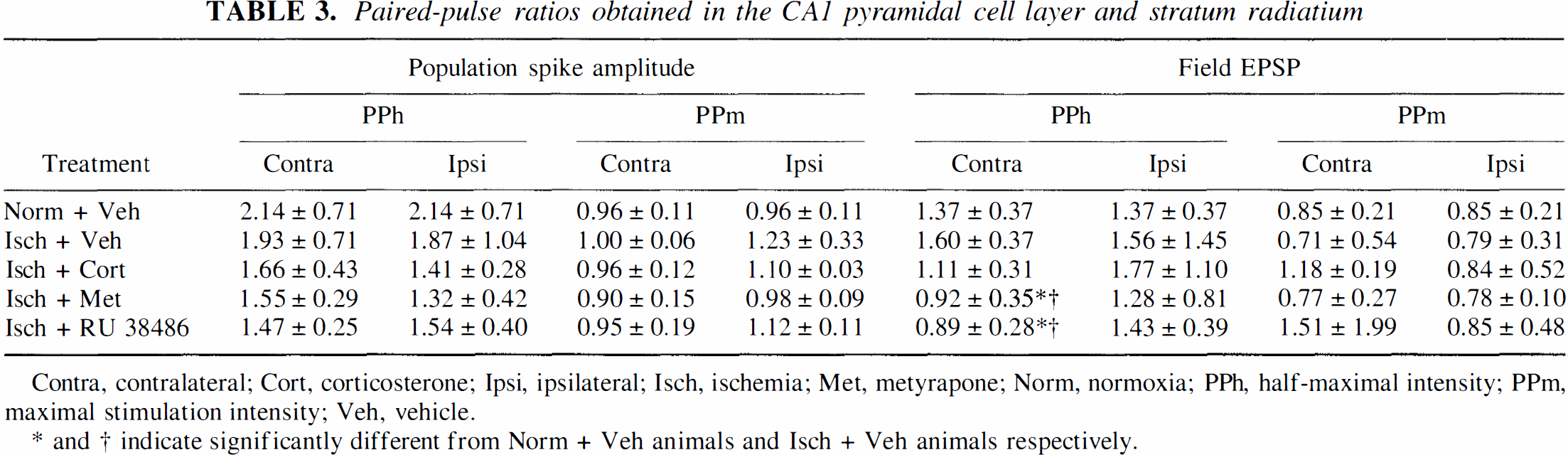
Contra, contralateral; Cort, corticosterone; Ipsi, ipsilateral; Isch, ischemia; Met, metyrapone; Norm, normoxia; PPh, half-maximal intensity; PPm, maximal stimulation intensity; Veh, vehicle.
* and † indicate significantly different from Norm + Veh animals and Isch + Veh animals respectively.
Histology
The Nissl staining revealed histologic changes in the hippocampus at 24 hours after hypoxia—ischemia compared with animals exposed to normoxia. At this time point, a loss of granular cells in the DO was present at the ipsilateral (occluded) side in several animals (compare Figs. 6A with 6B; Table 4). The cell loss was randomly present in restricted parts in both the suprapyramidal and infrapyramidal blade of the granular cell layer. Histo-logic alterations in the hippocampal CA1 area, indicated by reduced density of the cresylviolet staining and irregularly shaped appearance of neurons, were present in the animals that showed severe damage in the DG (compare Figs, 6C with 6D).
The number of animals displaying histological alterations (left) and the amount of degeneration at 24 hours after hypoxia-ischemia (right)
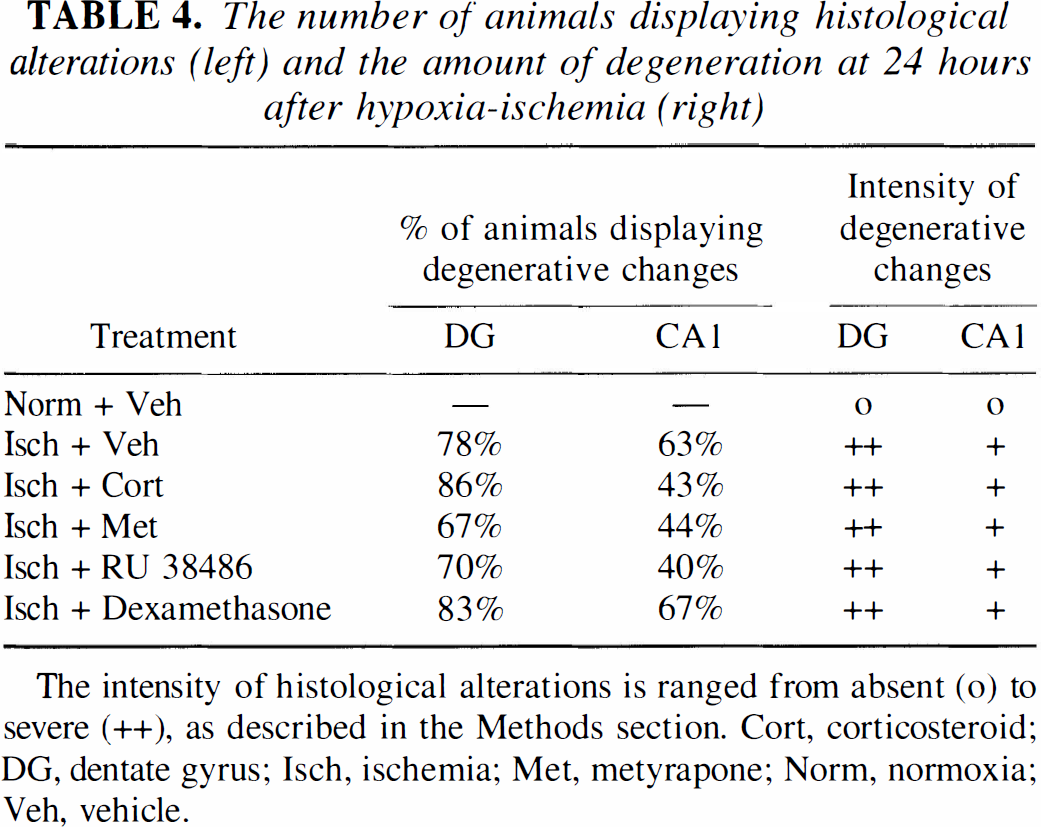
The intensity of histological alterations is ranged from absent (o) to severe (++), as described in the Methods section. Cort, corticosteroid; DG, dentate gyrus; Isch, ischemia; Met, metyrapone; Norm, normoxia; Veh, vehicle.
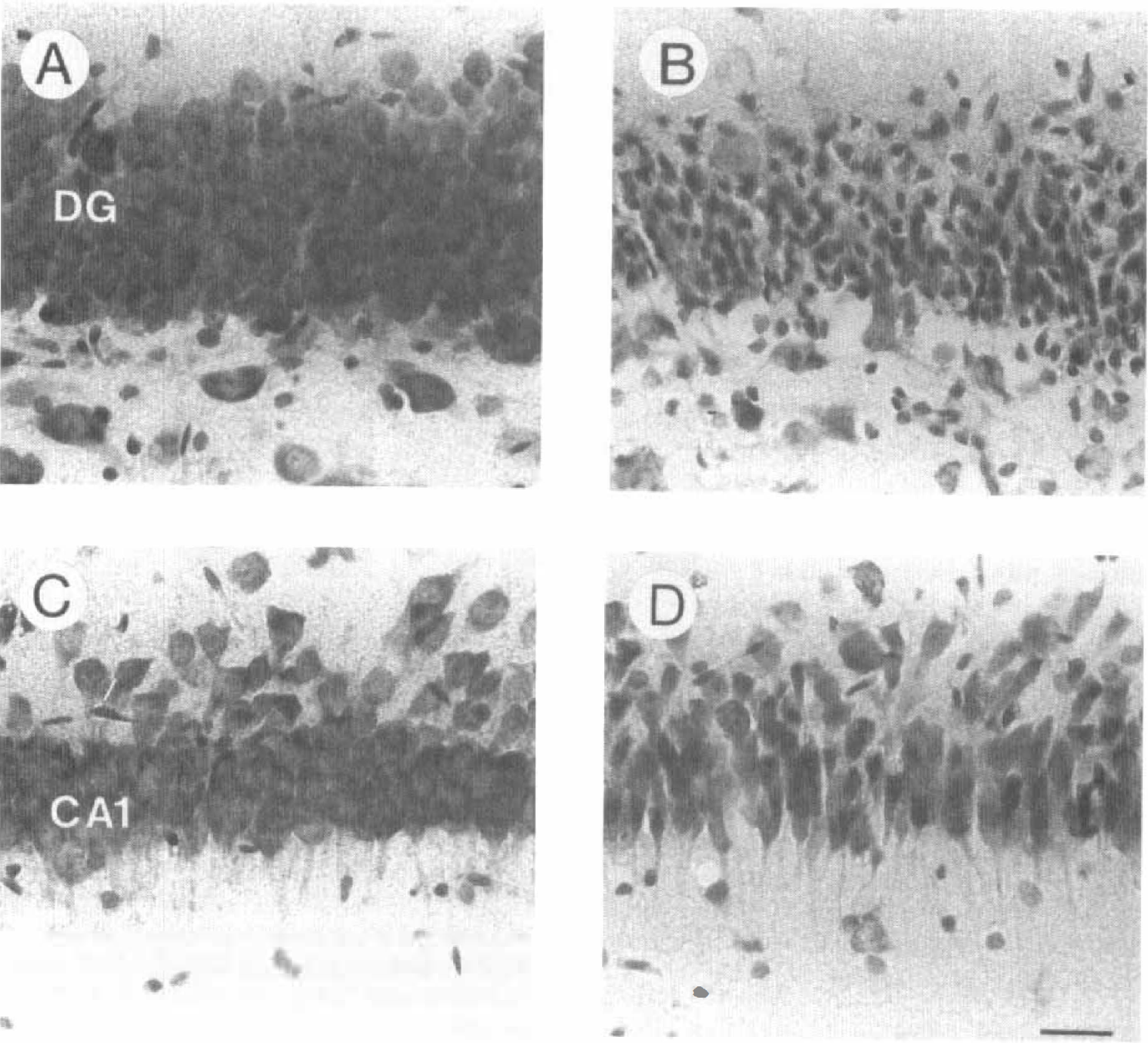
Photographs (×350) showing Nissl stainings of the dentate gyrus
Steroid treatment did not affect the loss of neuronal integrity in the DG 24 hours after ischemia: both the number of animals displaying degenerative changes in the DG and the degree of cell loss in the DG were comparable between the different experimental groups (Table 4). Also in the CA1 area, the prominent histologic alterations were comparable between the experimental groups.
In parallel with the Nissl staining, we observed changes in the cytoskeletal protein MAP2 after hypoxia—ischemia. Normally, MAP2 is localized to dendrites throughout the hippocampus (Figs. 7A and 7C). Exposure to hypoxia—ischemia resulted in a marked loss of MAP2 immunoreactivity that was particularly obvious in the DG molecular layer and to a lesser extent in the hippocampal CA1 area at the side ipsilateral to occlusion (Figs. 7B and 7D). These changes in MAP2 immunoreactivity thereby confirm the cell loss in the DG and changes in the CA1 area as detected with the Nissl staining. This relation is substantiated by individual analysis of the animals, which demonstrates that the changes detected with Nissl staining show a similar pattern as changes in MAP2 immunoreactivity per animal. In agreement with the Nissl staining, both the number of animals that displayed a reduction in MAP2 immunore-activity and the amount of reduction were comparable between the experimental groups exposed to hypoxia—ischemia.
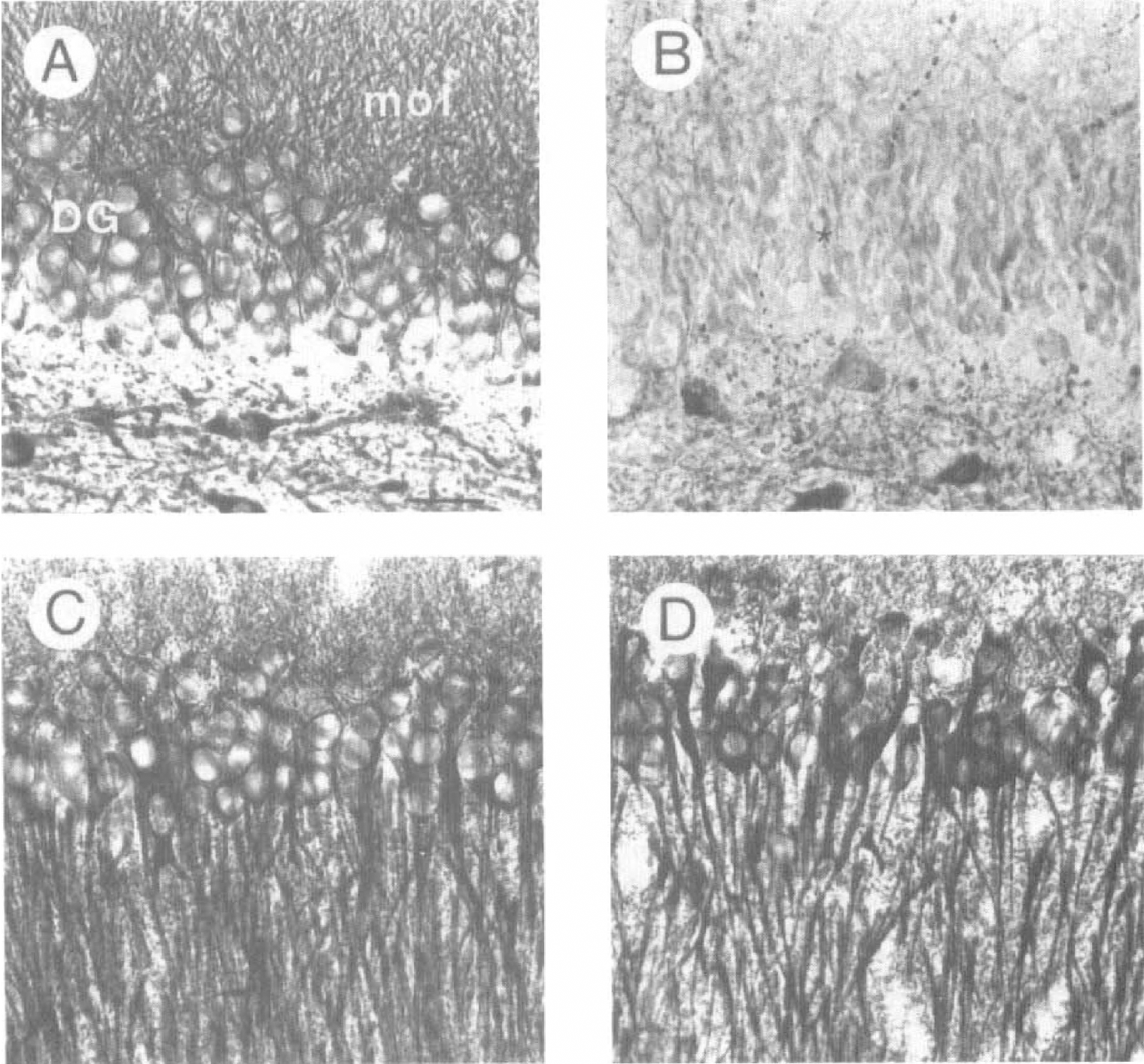
Loss of microtubule-associated protein 2 (MAP2) immunoreactivity in the CA1 pyramidal cell layer and dentate gyrus (magnification ×350). Photographs
DISCUSSION
Previous studies in animals demonstrate that high corticosteroid hormone levels, presumably using GR, potentiate brain damage in response to ischemia (Sapolsky and Pulsinelli, 1985;(Morse and Davis, 1990; Sapolsky, 1996). Preventing the rise in corticosteroid levels during ischemia has been reported to reduce cell loss and seizures after ischemia (Krugers et al., 1995; Smith-Swintosky et al., 1996; Adachi et al., 1998; Krugers et al., 1998). The glucocorticoid endangerment of neurons is thought to involve an energy-dependent (Sapolsky, 1986; Virgin et al., 1991) elevation of glutamate concentrations and the subsequent mobilization of toxic intracellular Ca2+ levels (Elliott et al., 1993b; Stein-Behrens et al., 1992, 1994; Adachi et al., 1998). From the therapeutic point of view, it is interesting that removal of the adrenal gland after the ischemic insult also prevents neuronal loss (Sapolsky and PulsineIli, 1985; Morse and Davis, 1990). The current study was initiated to establish (1) whether therapeutically more relevant steroid modulations after hypoxia—ischemia also are effective in preserving neuronal integrity, and (2) whether synaptic function in the hippocampus is preserved along with neuronal integrity. To test this, we treated animals with a steroid synthesis inhibitor or GR (ant)agonists and examined both synaptic transmission and neuronal integrity in the hippocampal CA1 area 24 hours after hypoxia—ischemia as a function of steroid treatment after ischemia.
We found that the steroid synthesis inhibitor metyrapone exerted a (moderately) beneficial effect. First, metyrapone reduced the incidence and severity of seizure activity immediately after ischemia. Second, the metyrapone-treated group exhibited the highest survival rate (90%) over the 24-hour survival period and consisted of the largest percentage of animals from which bilateral recordings could be made (67%). Finally, metyrapone prevented the ischemia-induced reduction of the PS amplitude (but not of the fEPSP slope) in the CA1 hippocampal field. Taken together, these results indicate that metyrapone administered after ischemia at least initially exerts a beneficial effect. Based on the evidence that metyrapone prevents the ongoing production of corticosterone (Smith-Swintosky et al., 1996; Krugers et al., 1995), we speculate that this effect of metyrapone is related to low plasma corticosteroid levels after ischemia. Measuring plasma corticosterone levels at several time points after ischemia will be necessary to substantiate this. We did not perform such detailed measurements, since the procedures to obtain blood samples in combination with ischemia severely compromises the viability of animals (unpublished observations). Notice, though, that the protection by metyrapone was only partial, since 24 hours after ischemia, metyrapone treatment resulted in the highest incidence of seizures and did not prevent the severe loss of neuronal structure.
Since metyrapone, presumably by maintaining low plasma corticosteroid levels after ischemia, reduces deleterious effects of ischemia, we predicted that the synthetic OR-agonist dexamethasone administered after ischemia should have similar effects. This hypothesis is based on recent findings that dexamethasone is more efficient to activate OR in the pituitary than in the brain based on its poor ability to cross the blood—brain barrier (Meijer et al., 1998). If so, dexamethasone would promote negative feedback action on the hypothalamopituitary—adrenal axis activity after stress at the level of the pituitary, thereby reducing the production of endogenous corticosteroids, while at the same time the low dose of the synthetic glucocorticoid is not able to substantially activate brain OR. Our current results do not reveal beneficial effects of dexamethasone: animals treated with the synthetic OR-agonist showed severe seizure activity, substantial loss of hippocampal integrity, and poor synaptic transmission 24 hours after the insult. These data extend earlier findings that peripheral dexamethasone treatment increases ischemia-associated structural damage (Koide et al., 1986; Packan and Sapolsky, 1990; Adachi et al., 1998). One explanation for these observations could be that injection with the low dosage of dexamethasone used here (10 μg/mL) only suppresses plasma corticosterone levels with a substantial delay after administration (Meijer et al., 1998). This means that high levels of endogenous corticosterone immediately after ischemia can occupy OR and subsequently endanger hippocampal neurons. This also could explain why dexamethasone-treated animals in many aspects resembled the animals that received a high dose of corticosterone after ischemia.
Because high plasma corticosterone levels, associated with extensive activation of OR (McEwen et al., 1986; De Kloet et al., 1991), are thought to be involved in the endangering effect of steroids (Packan and Sapolsky, 1990), blockade of OR with RU 38486 after ischemia should potentially preserve neuronal structure along with synaptic responsiveness. This prediction is supported by observations that RU 38486 protects neurons in vitro against oxidative stress (Behl et al., 1997). However, we found that RU 38486 in vivo, administered after ischemia, preserved neither neuronal integrity nor synaptic functioning. The poor efficacy of RU 38486 may be explained in several ways. First, the rise in steroids that accompanies the ischemic insult (Krugers et al., 1998) presumably activates OR. Therefore, administration of the OR-antagonist RU 38486, which requires time to block OR, at a time point when OR in the brain are already activated may be inefficient. Second, peripherally injected RU 38486 in control animals is much more efficient in blocking OR at the level of the pituitary than in the brain (Van Haarst et al., 1997). In addition, by interfering with the negative feedback action of corticosterone at the level at the pituitary, RU 38486 treatment may cause a prolonged elevation of plasma corticosteroid levels (Ratka et al., 1989). Finally, we should consider the possibility that OR do not play an important role in the ischemia-induced impairment of synaptic transmission and loss of neuronal integrity. If so, the beneficial effects of metyrapone may result from properties other than inhibiting the corticosteroid synthesis. This notion is supported by the inability of both dexamethasone and RU 38486 to alter the outcome of ischemic pathology compared with vehicle-treated controls. Moreover, preliminary evidence suggests that metyrapone treatment in control animals induces anesthesia-like effects (Krugers et al., 1998), possibly from a metyrapone-induced accumulation of neuroactive steroids such as 11-deoxycorticosterone and its metabolite 3α-21-dihydroxy-5α pregnan-20-one. Since these metabolites are known to potentiate GABAA receptor-mediated responses (Lambert et al., 1995) and block voltage-dependent Ca2+ channels (Ffrench-Mullen and Spence, 1991), they may influence the outcome of ischemia.
Whereas preliminary studies show that adrenalectomy after ischemia can exert beneficial effects on cell structure (Sapolsky and Pulsinelli, 1985; Morse and Davis, 1990), no data were available about the functional properties in brain areas that are known to be affected by ischemia. In the current study, we clearly demonstrate that not only neuronal structure, but also synaptic transmission, is significantly impaired after ischemia. In general, the impaired synaptic functioning appears to be linked to histologic changes. The notion that the impaired synaptic function might be related to histologic changes rather than changes in neuronal excitability is supported by the absence of group differences in PP responses. Although the methods used allow only qualitative analysis of gross morphologic changes, both the Nissl and the MAP2 staining indicate that animals exposed to ischemia reveal a high degree of structural damage, confirming earlier studies (Krugers et al., 1995, 1998; Smith-Swintosky et al., 1996; Adachi et al., 1998). The findings that structural damage after ischemia was not only confined to the cell layers, but also present in the dendritic field of the hippocampal CA1 area, may explain the reduction in fEPSP slope in the CA1 dendritic field. The extent of neuronal loss after ischemia was not influenced by the steroid treatment, suggesting that synaptic functioning after ischemia generally is impaired along with loss of neuronal structure, despite steroid treatment. Only metyrapone preserved synaptic functioning in the hippocampal cell layer after ischemia despite loss of neuronal integrity. Hence, this indicates that factors other than neuronal integrity are involved in the preservation of synaptic functioning after treatment with metyrapone.
Whereas the overall data support a correlation between neuronal damage and loss in synaptic function after hypoxia—ischemia, detailed evaluation of the data indicates that histologic damage is not a reliable predictor for loss in synaptic function (and vice versa): approximately 20% of the animals showing clear damage in the CA1 area still displayed PS amplitudes exceeding 0.40 mV at the ipsilateral side, whereas 20% of the animals showing no damage were found to have a loss of synaptic function ipsilateral to the occlusion (Table 5). Conversely, loss of synaptic function was in approximately 15% of the animals not associated with histologic damage. The dissociation between electrophysiologic and histologic observations stresses the need for histologic and electrophysiologic investigations performed in parallel when interpreting histologic changes in terms of functional alterations.
Relationship between histological alterations in the hippocampal CA1 area and electrophysiological recordings

In a considerable proportion of animals histological damage was not linked to loss in function and vice versa.
In summary, our current study shows that treatment with the steroid synthesis inhibitor metyrapone has beneficial effects on ischemia-related pathologic changes and impairment of synaptic transmission, even when the compound is administered after the ischemic insult. From the therapeutic point of view, our results suggest that treatment with steroid synthesis inhibitors like metyrapone after ischemia can be useful because they may prolong the window for therapeutic intervention.
Footnotes
Acknowledgments
The authors thank Lisette van Essen for her work on the corticosterone assays and Dr. Ron de Kloet for critically reading the manuscript.