
Abstract
Hypothermia reduces excitotoxic neuronal damage after seizures, cerebral ischemia and traumatic brain injury (TBI), while hyperthermia exacerbates damage from these insults. Presynaptic release of ionic zinc (Zn2+), translocation and accumulation of Zn2+ ions in postsynaptic neurons are important mechanisms of excitotoxic neuronal injury. We hypothesized that temperature-dependent modulation of excitotoxicity is mediated in part by temperature-dependent changes in the synaptic release and translocation of Zn2+. In the present studies, we used autometallographic (AMG) and fluorescent imaging of N-(6-methoxy-8-quinolyl)-para-toluenesulfonamide (TSQ) staining to quantify the influence of temperature on translocation of Zn2+ into hippocampal neurons in adult rats after weight drop-induced TBI. The central finding was that TBI-induced Zn2+ translocation is strongly influenced by brain temperature. Vesicular Zn2+ release was detected by AMG staining 1 h after TBI. At 30°C, hippocampus showed almost no evidence of vesicular Zn2+ release from presynaptic terminals; at 36.5°C, the hippocampus showed around 20% to 30% presynaptic vesicular Zn2+ release; and at 39°C vesicular Zn2+ release was significantly greater (40% to 60%) than at 36.5°C. At 6 h after TBI, intracellular Zn2+ accumulation was detected by the TSQ staining method, which showed that Zn2+ translocation also paralleled the vesicular Zn2+ release. Neuronal injury, assessed by counting eosinophilic neurons, also paralleled the translocation of Zn2+, being minimal at 30°C and maximal at 39°C. We conclude that pathological Zn2+ translocation in brain after TBI is temperature-dependent and that hypothermic neuronal protection might be mediated in part by reduced Zn2+ translocation.
Introduction
Systemic hypothermia, which inhibits neuronal release of glutamate, prevents or reduces neuronal death after cerebral ischemia (Busto et al, 1987, 1989; Churn et al, 1990; Dietrich et al, 1993; Hu et al, 1995; Kil et al, 1996; Minamisawa et al, 1990a; Rosomoff, 1959; Tanimoto and Okada, 1987), prolonged seizures (Liu et al, 1993; Lundgren et al, 1994) and traumatic brain injury (TBI) (Clifton et al, 1991), especially if initiated before an insult. Conversely, hyperthermia aggravates the neuronal death produced by those conditions (Dietrich, 1992; Dietrich et al, 1996; Lundgren et al, 1994; Minamisawa et al, 1990b). Hypothermia also has been associated with improved outcome after clinical TBI (Clifton et al, 1993; Marion et al, 1997), although recent data from a clinical trial suggest that therapeutic hypothermia might be ineffective (Clifton et al, 2001).
Many processes related to neuronal injury are temperature-dependent, including the release of glutamate from synaptic vesicles (Arai et al, 1993; Zornow, 1995), intracellular accumulation of Ca2+ (Mitani et al, 1991), modulation of calcium/calmodulin-dependent protein kinase (Churn et al, 1990) and production of free radicals (Kil et al, 1996). Any or all of those temperature dependencies could contribute to the influence of temperature on neuronal injury after TBI.
Evidence implicating Zn2+ as an important contributing cause of excitotoxic neuronal injury has accumulated over the last 15 years (Frederickson et al, 1989; Koh et al, 1996; Suh et al, 2000a, 2004; Tonder et al, 1990). These data raise the possibility that the release of, and postsynaptic accumulation of, toxic amounts of Zn2+ might also be strongly temperature-dependent, thus contributing to the overall temperature sensitivity of the excitotoxic process (Suh et al, 2000b; Tsuchiya et al, 2002). We hypothesized therefore that Zn2+ translocation from presynaptic boutons to postsynaptic neurons would be temperature-dependent after TBI.
We choose the autometallographic (AMG) and the N-(6-methoxy-8-quinolyl)-para-toluenesulfonamide (TSQ) fluorescence method, as it shows both the terminal and the translocated zinc ions (Danscher, 1996; Frederickson and Danscher, 1990; Frederickson et al, 1987, 2000; Suh et al, 1999).
Materials and methods
Weight Drop Traumatic Brain Injury
Male Wistar rats (250 to 350 g) were anesthetized with sodium pentobarbital (50 mg/kg, intraperitoneally), permitted to breathe spontaneously through a natural airway and placed in a stereotaxic frame (David Kopf Instruments, Tujunga, CA, USA). The scalp and temporalis muscles were reflected and a 3.0 mm diameter burr hole was drilled through the skull 3.0 mm lateral to the midline and 4.0 mm caudal to bregma. Traumatic brain injury was performed using a weight drop model (Clark et al, 1994; Suh et al, 2000a). With this method, brief displacement and deformation of brain is induced by dropping a weight through a guide tube such that the weight hits a blunt steel impactor resting on the dura. The instrument was placed in the stereotaxic instrument and a weight of 10 g was dropped from a height of 10 cm. The impactor, shaped so that the distal end would penetrate 1.5 mm below the inner table before descent was arrested by contact of the outer shoulder of the impactor with the dorsal skull surface, was positioned on the intact dura. At 1 or 6 h after TBI, rats were killed and brains were quickly removed and frozen with CO2 gas. All animal surgeries were performed in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 80-23) revised 1996.
Brain Temperature Control
Fifteen rats were randomly assigned to four groups: untraumatized normothermic control (n = 7), TBI + hypothermia (30°C; n = 8), TBI + normothermia (36.5°C; n = 8) and TBI + hyperthermia (39°C; n = 8). Previous experiments had indicated that differences in the number of Zn2+-positive neurons produced by experimental interventions achieve statistical significance with a handful of animals per group. Cranial temperature was measured using a temperature probe (Physitemp Instruments, Inc., Clifton, NJ, USA) inserted into the temporalis muscle. Hypothermia was induced by whole-body immersion in cold water (4.0°C) for > 20 min, then maintained by directing gentle air flow across the head using a rotary fan (Clifton et al, 1991). Normal and elevated brain temperatures were induced by placing the animals on a thermostatically controlled heating pad under a heating lamp. At 30 mins after the designated head temperature was attained, TBI was induced. The targeted brain temperatures were maintained for 1 h after TBI. To show the temperature-dependent vesicular Zn2+ release from presynaptic terminals, the extracellular zinc chelator calcium ethylene-diamine tetraacetic acid (CaEDTA) (100 mmol/L per 5 μL) was injected into the ipsilateral lateral ventricle 10 mins before TBI to prevent re-uptake into presynaptic terminals (Frederickson et al, 2002). In total, 16 rats were killed 1 h after TBI. To see the temperature-dependent Zn2+ accumulation in the hippocampal neurons and subsequent neurodegeneration, in total 12 rats that received TBI without CaEDTA pretreatment were killed 6 h after TBI. The untraumatized (sham operated but maintained at normothermia) rats were killed after an equivalent time interval.
Autometallographic Zinc Staining
Autometallographic stains the same pool of Zn2+ as stained by TSQ, but was also used with a permanent, silver stain. The AMG staining was performed by the postmortem H2S method (gas-AMG method). Briefly, the frozen, unfixed brains were sectioned coronally at 30 μm in a −14°C cryostat, then thawed on to cleaned glass slides and dried by gentle air flow. After exposing the sections to H2S gas for 60 min to precipitate the free Zn2+ as ZnS, the slides were developed for 60 mins. All sections were counterstained with toluidine blue. After rinsing and dehydration, the sections were mounted in DePex mounting medium. Like TSQ, this procedure has also been shown to be a virtually exclusive stain for vesicular Zn2+.
N-(6-Methoxy-8-Quinolyl)-Para-Toluenesulfonamide Fluorescence for Zinc Staining
Temperature-dependent Zn2+ translocation was assessed in 10 μm sections that were stained with TSQ. Staining was performed as previously described (Frederickson et al, 1987; Suh et al, 2000a). Briefly, the frozen, unfixed brains were sectioned coronally in a −14°C cryostat, thawed on gelatin-coated slides and dried by gentle air flow. Sections were stained for free Zn2+ by immersion in a solution of TSQ (4.5 mmol/L) (Molecular Probes, Eugene, OR, USA) in 140 mmol/L sodium barbital and 140 mmol/L sodium acetate buffer (pH 10.5) for 60 secs. After brief rinsing in 0.9% saline, sections were examined under a conventional compound fluorescence microscope (Zeiss Universal: exciter, 355 to 375 nm, dichroic beam splitter, 380 nm, barrier, 420 nm long-pass).
Optical Density Measurements of Zinc Staining
After gas-AMG staining, brain sections were mounted in the light microscope, transilluminated with a fixed intensity of white light (tungsten), and individual images were captured by CCD camera and digitized. Using Photoshop (6.0) the optical intensity was measured by the second observer. Every section that was obtained from an individual brain slice (typically 3 to 5) was used for quantification. Two images were captured from each section, one including the hilus of the dentate gyrus and the other including cornu ammonis (CA)3. The zones were digitized as follows: reference a square of stratum lacunosum-moleculare just overlying the middle of the blade of the dentate gyrus; hilus, the same square, moved top the CA3c-hilar confluence; CA3, the same square centered over the stratum lucidum in CA3a–b (SL). Optical density was calculated conventionally (OD = (log10(incident light/transmitted light))), with ‘incident light’ taken as the intensity of light transmitted through the zinc-free reference zone (in lacunosum – molecular – away from the lateral perforant path innervation), and ‘transmitted light’ taken as the raw intensity reading for individual samples.
Count of N-(6-Methoxy-8-Quinolyl)-Para-Toluenesulfonamide-Positive Neurons
N-(6-Methoxy-8-Quinolyl)-Para-Toluenesulfonamide-positive neurons were counted in photographed fluorescence micrographs of sections that were harvested from 4.0 mm posterior to the bregma. These microscope fields (× 20) included the hippocampal cell layer CA3, the hilus, and the dentate gyrus. These sections were chosen because of the high percentage of TSQ-positive neurons identified in those areas in a previous study of Zn2+ translocation in TBI (Suh et al, 2000a). In total, 10 sections were photographed from each rat. These were then coded (by SWS) and given to a second experimenter (by NFP), who selected a single ‘counting frame’ from each brain region and counted blindly.
Count of Eosinophilic Neurons
Temperature-dependent cell injury was assessed in 10-μm sections that were stained with hematoxylin and eosin (H&E). Collection of sections was started 4 mm posterior to bregma, and every third section was saved until 10 sections were collected. These were then coded (by SWS) and given to a second experimenter (by NFP), who selected a single ‘counting frame’ from each brain region under study. The ‘frame’ was the complete field surrounded by the photoreticule at × 100 total magnification (10 × 10). The hilar frame was set by placing both blades of the dentate gyrus within the space defined by the upper and lower edges of the photoframe and putting the crest of the dentate just touching the appropriate side wall of the photoframe. The CA3 frame was positioned by putting the center of the CA3 pyramidal stratum in the center of the counting frame. A total of 10 frames (one from each section) were counted from each rat. The mean number of eosinophilic neurons per counting frame was then computed for each brain region in each experimental group.
Statistical Analysis
The mean optical intensity of vesicular zinc, the mean number of TSQ-positive neurons and the mean number of eosinophilic neurons at the three temperatures were compared using analysis of variance (ANOVA); post hoc testing was accomplished using Scheffe's test.
Results
Loss of Vesicular Zn2+ Staining from Mossy Fiber (MF) after Traumatic Brain Injury is Temperature-Dependent
Head trauma-induced loss of vesicular zinc ions from MF was shown by the AMG staining methods. In the normothermic (36°C) rats, the Zn2+ depletion from the MF was apparent at 1 h after TBI (Figures 1E and 1F). In the hyperthermic rats, the vesicular Zn2+ depletion from the MF of hilus and CA3 was even greater than normothermic rats. Around 40% to 60% of vesicular Zn2+ was depleted by hyperthermia (39°C) plus TBI. However, hypothermia (30°C) significantly prevented vesicular Zn2+ release after TBI (Figures 1C and 1D). The optical intensity of vesicular Zn2+ in the hilus and CA3 is almost identical to that in normothermic sham-operated rats (without TBI, but pretreated with CaEDTA) (Figures 1 and 2). These findings suggested that vesicular Zn2+ release after TBI is brain temperature dependent.
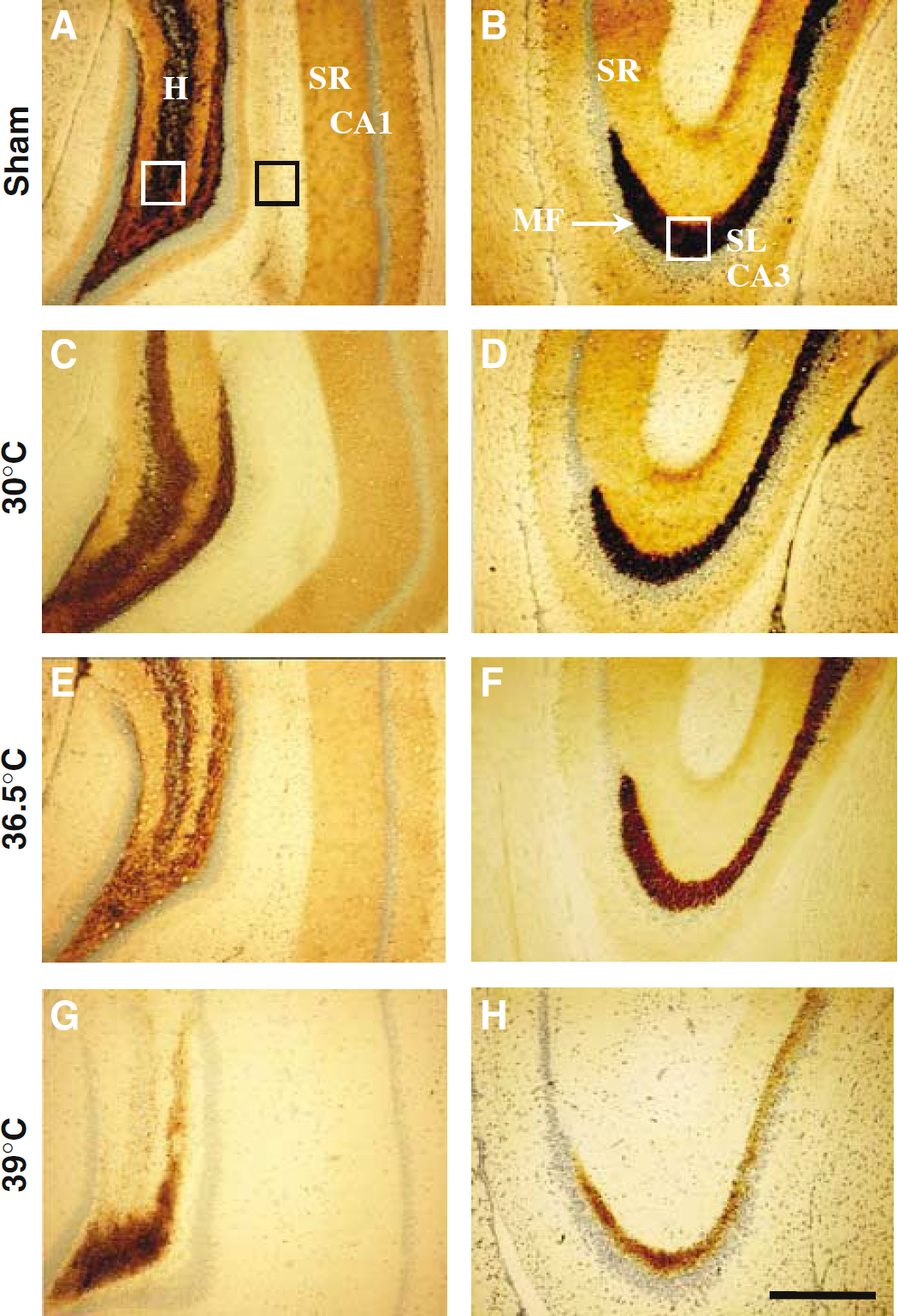
Light microscopic images of temperature-dependent Zn2+ depletion in the hippocampus 1 h after TBI. Gas-AMG-stained hilus and CA3 areas of the rat hippocampus. To prevent Zn2+ re-uptake, all the rats were pretreated with zinc chelator 10 mins before TBI or sham operation. (
The Appearance of Zn2+ in the Hippocampal Neurons after Traumatic Brain Injury is Temperature-Dependent
As has been previously described (Frederickson et al, 1987, 1989; Koh et al, 1996; Tonder et al, 1990), rats without any brain insult showed only few TSQ-labeled neuronal somata (Figures 3A and 3B). At 6 h after TBI, TSQ fluorescent somata were observed in the hilus, dentate gyrus and CA3 pyramidal regions of the hippocampal formation as we have previously shown (Suh et al, 2000a). In the hypothermic group, a significant reduction of TSQ fluorescent neurons was observed in the hilus, dentate gyrus and CA3 pyramidal areas as compared with the normothermic and hyperthermic animals (Figures 3 and 4). N-(6-methoxy-8-quinolyl)-para-toluenesulfonamide-labeled neurons were abundant in the traumatized brains from normothermic and hyperthermic animals (Figures 3E to 3H). Quantitation of TSQ-labeled neurons shows that hypothermia significantly prevented, while hyperthermia potentiated, intracellular Zn2+ accumulation after TBI (Figure 4).
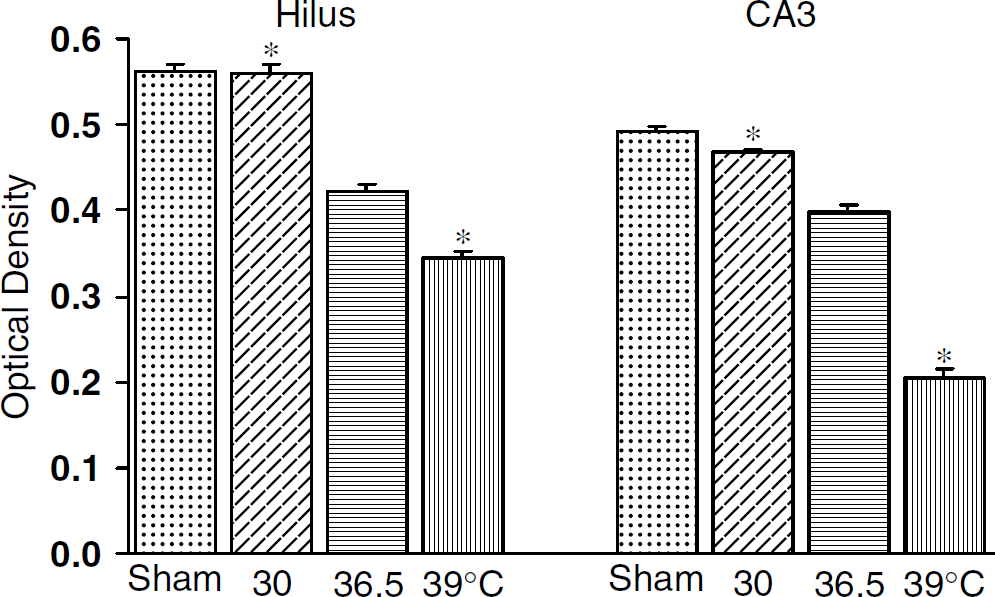
Quantitation of temperature-dependent Zn2+ depletion in the hippocampus 1 h after TBI. The intensity of vesicular Zn2+ (optical density) is measured from Gas-AMG-stained sections with or without TBI. Bar graph represents that vesicular Zn2+ was significantly depleted in normothermic (36.5°C) rat's hippocampus at 1 h after TBI. Hypothermia (30°C) almost completely prevented vesicular Zn2+ release, but hyperthermia (39°C) potentiated it. Average optical density of vesicular Zn2+ represented from normothermic control, n = 4, hypothermia + TBI, n = 4, normothermia + TBI, n = 4 and hyperthermia + TBI, n = 4. Hilus, hippocampal hilus MF area; SR of CA1, stratum radiatum of cornu ammonis area 1; SL of CA3, stratum lucidum of cornu ammonis area 3. ‘*’ represents the difference from normothermia group at P < 0.05. Bars show mean + s.d.
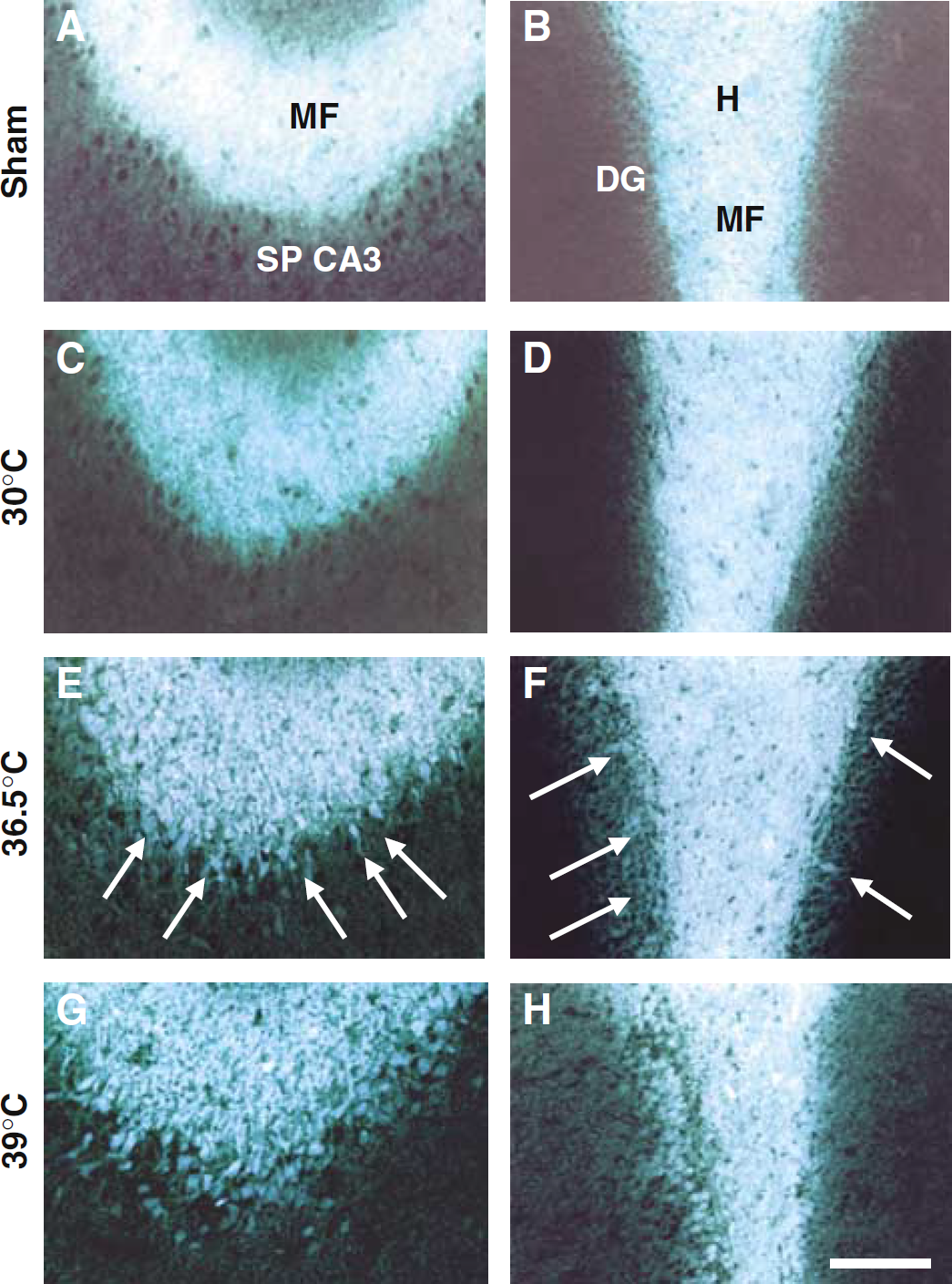
Fluorescence of TSQ-stained hippocampal neurons, caused by weight drop TBI, was completely blocked by hypothermia (30°C), but potentiated by hyperthermia (39°C) in the dentate granule, hilar and CA3 of hippocampus. Fluorescence microscopy images of TSQ-stained CA3 and hilar regions of the rat hippocampus. (
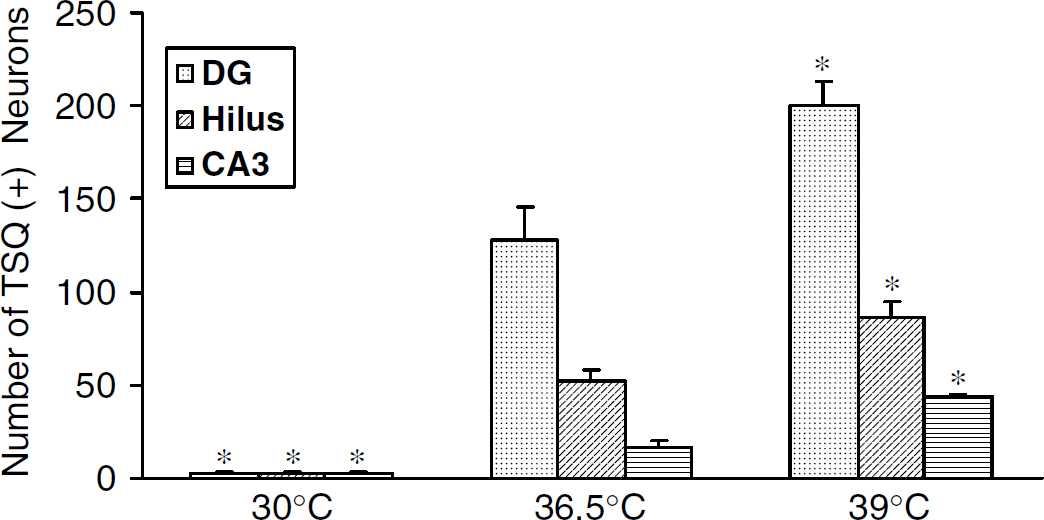
Temperature-dependent Zn2+ translocation into postsynaptic neurons of hippocampus after TBI. Bars show the number of neurons stained with the fluorescent dye TSQ in dentate granule, hilar and hippocampal CA3 neurons (mean) in 6 h after rats were subjected to TBI at low (30°C, n = 4), normal (36.5°C, n = 4) and high (39°C, n = 4) cranial temperatures. Average numbers of TSQ-positive neurons found in single, selected fluorescent microscopic views of four individual rats from each of the three treatment groups. ‘*’ represents the difference from normothermia group at P < 0.05. Bars show mean + s.d.
Degeneration of the Hippocampal Neurons after Traumatic Brain Injury is Temperature-Dependent
Our previous study showed that apparently degenerating (i.e., eosinophilic) neurons were observed in all of the brain regions after TBI that showed fluorescent TSQ staining for Zn2+ in neuronal somata (Suh et al, 2000a). In contrast to the healthy neurons, these eosinophilic neurons were densely stained throughout the perikaryon with bright pink eosin, with dark blue nuclei. In a few individual sections that were examined first under TSQ fluorescence and under H&E, the results indicated that the two methods were staining the same individual neurons (Suh et al, 2000a). The eosino-philia was detectable as early as 6 h after TBI. Eosinophilic neurons (Ito et al, 1975; Suh et al, 2000a) were evident in the brains of rats that were traumatized while at 36.5°C or 39°C, but were virtually absent in hypothermic rats (Figures 5 and 6). In the hyperthermic rats, significantly more eosinophilic neurons were detected than in normothermic rats (Figures 5 and 6).
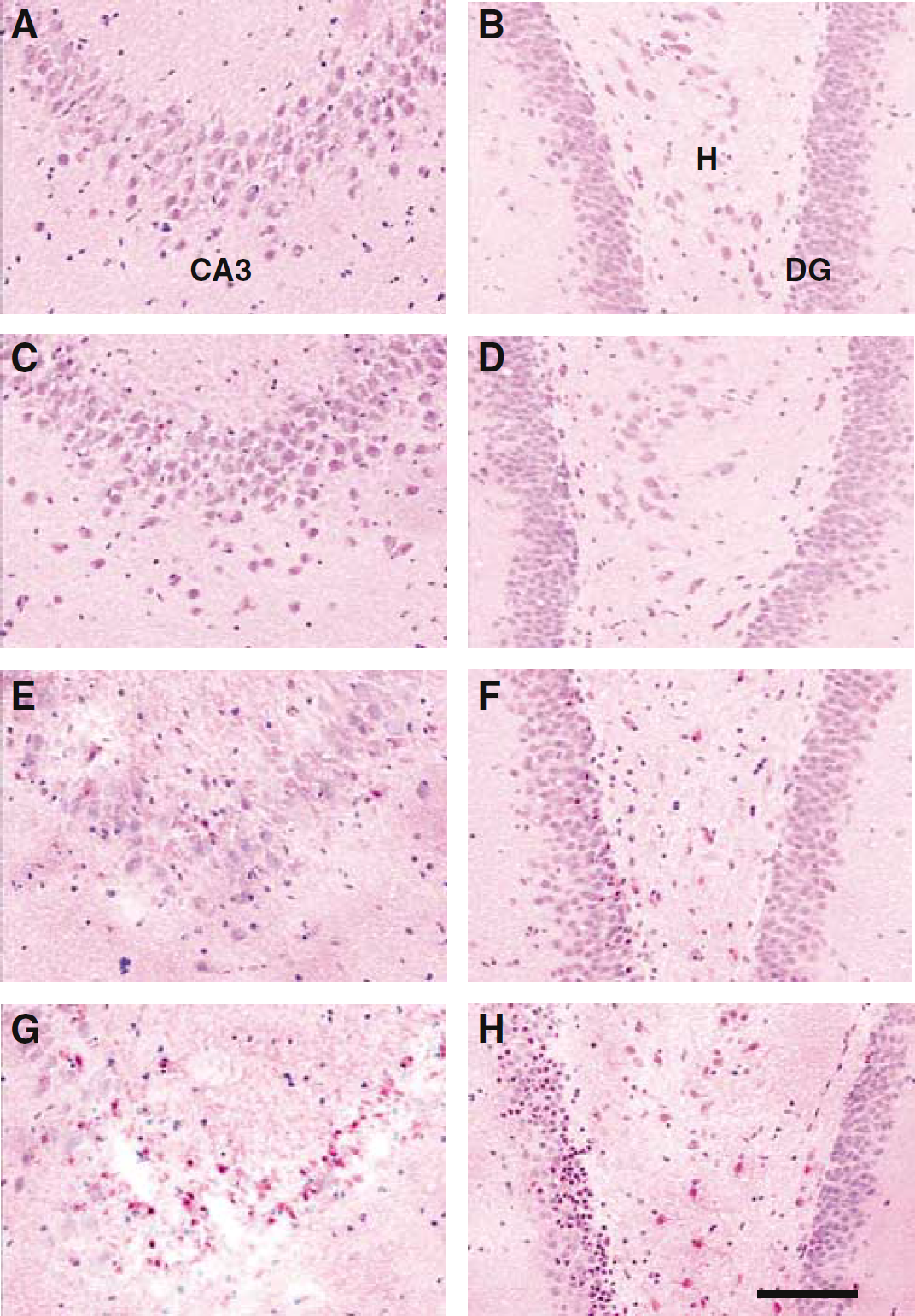
Neurodegeneration was completely blocked by low cranial temperature (30°C) and potentiated by high cranial temperature (39°C) in the hilus and CA3 of hippocampus after TBI. (
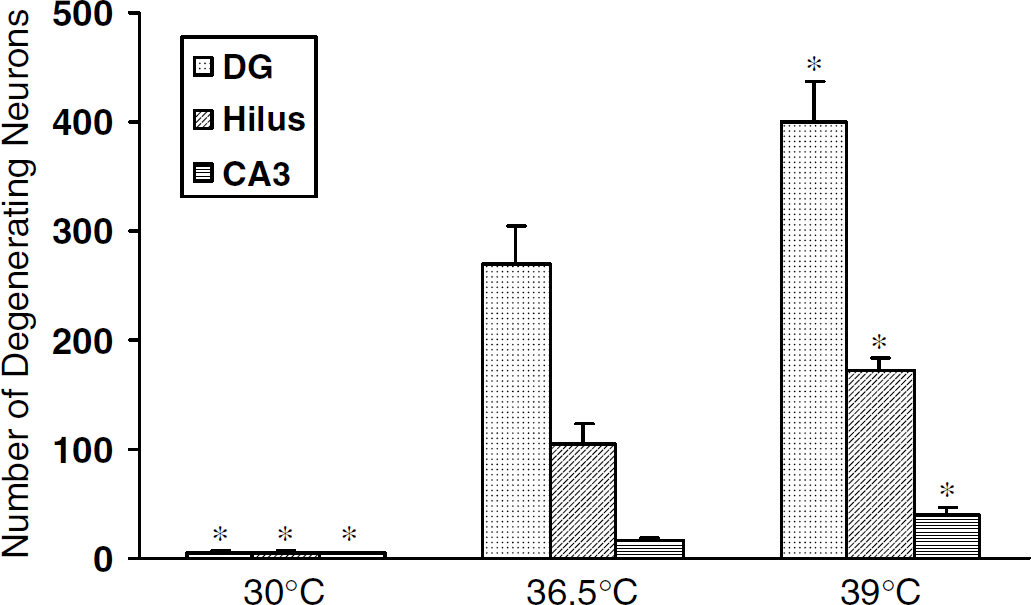
Temperature-dependent neurodegeneration after TBI. Bars show the number of eosinophilic neurons 6 h after TBI in dentate granule, hilar and hippocampal CA3 neurons in low (30°C, n = 4), normal (36.5°C, n = 4) and high (39°C, n = 4) cranial temperatures. ‘*’ represents the difference from normal at P < 0.05. Bars show mean + s.d.
Discussion
The present results are the first to reveal a strong correlation between brain temperature at the time of TBI and the intensity of posttraumatic Zn2+ translocation. Although temperature-dependent neuronal death has been attributed to release of glutamate from synaptic vesicles (Arai et al, 1993), intracellular accumulation of Ca2+ (Mitani et al, 1991) and production of free radicals (Kil et al, 1996), the present findings suggest that Zn2+ translocation from Zn2+-enriched (ZEN) neuronal terminals into postsynaptic neurons is also temperature-dependent. The higher the pre/post-TBI brain temperature, the more vesicular zinc was released at 1 h after TBI, and the more TSQ-positive and eosinophilic neurons were identified 6 h after TBI. Previous studies have shown that Zn2+ translocation is strongly associated with neuronal injury after TBI (Suh et al, 2000a).
Although the presence of chelatable Zn2+in brain has been recognized for decades (Timm, 1958; (Danscher et al, 1973, 1975; Haug, 1975), the potential role of Zn2+ in excitotoxic injury has only been appreciated for slightly more than a decade (Frederickson et al, 1988). Zinc-containing neurons are most densely present in brain regions that are highly vulnerable to ischemic and traumatic damage, that is, in the neocortex, hippocampus and the amygdaloid nuclei. Although the physiologic role of Zn2+ has not been completely elucidated, abundant evidence shows that depolarization of zinc-containing neurons releases Zn2+ in a Ca2+ -dependent fashion from presynaptic vesicles (Howell et al, 1984). In situations that cause neural cell injury, such as seizures (Frederickson et al, 1988, 1989), transient cerebral ischemia (Koh et al, 1996; Tonder et al, 1990), TBI (Suh et al, 2000a) or hypoglycemia (Suh et al, 2004), Zn2+ accumulates in postsynaptic neurons in a process referred to as translocation, because of the assumption that presynaptic Zn2+ is translocated into postsynaptic neurons. In rats subjected to 20 mins of forebrain ischemia (four-vessel occlusion), TSQ staining was reduced in the MFs and enhanced in the dentate hilus within 2 h of ischemia (Tonder et al, 1990). Acid fuchsine again stained most neurons that were also TSQ-positive. Subsequent studies showed that intraischemic hypothermia (29°C) inhibited both Zn2+ accumulation and cell death (Johansen et al, 1993). To date, only one peer-reviewed manuscript addresses the role of Zn2+ neurotoxicity after TBI (Suh et al, 2000a). In that paper, we used a weight drop model of TBI in rats and showed loss of Zn2+ from presynaptic boutons, an abundance of TSQ-positive postsynaptic neurons, high correspondence of TSQ-positive neurons and eosinophilic neurons, and antagonism of Zn2+ accumulation by intracerebroventricular administration of CaEDTA.
Hypothermia, initiated either before or immediately after TBI, substantially ameliorates the biochemical, histopathologic and behavioral sequelae of experimental TBI. Clifton et al (1991) reported marked reduction of mortality after fluid percussion injury (FPI) in rats by preinjury induction of moderate hypothermia (30°C) and improved behavioral outcome if temperature was reduced to 33°C or 30°C 5 mins after FPI. After controlled cortical impact in rats, mild posttraumatic hypothermia (32°C) reduced mortality (Clark et al, 1996). Posttraumatic hypothermia (30°C) reduced acute sensorimotor, neurobehavioral and histopathologic sequelae of FPI (Bramlett et al, 1995; Dietrich et al, 1994; Lyeth et al, 1993) and chronic histopathologic sequelae (Bramlett et al, 1997). Hypothermia (30°C) also provided protection against secondary posttraumatic injury associated with hypotension and hypoxia (Yamamoto et al, 1999). Posttraumatic hypothermia (30°C) reduced and hyperthermia (39°C) increased the inflammatory response to FPI in rats (Chatzipanteli et al, 2000). Moderate hypothermia (30°C) after TBI reduced posttraumatic dysfunction of the blood–brain barrier (Jiang et al, 1992). Hypothermia may exert protective effects through modification of the release of neurotransmitters. Biochemical data, obtained by microdialysis, suggest that the excitotoxic amino-acid glutamate is released in large quantities in response to TBI and to secondary ischemic episodes after TBI in experimental animals and humans (Globus et al, 1995; Katayama et al, 1990; Matsushita et al, 2000; Palmer et al, 1993; Zauner and Bullock, 1995). Although some investigators have reported that hypothermia (30°C to 32°C) dramatically reduced glutamate release after experimental TBI (Globus et al, 1995; Koizumi et al, 1997), others have found evidence of considerable cytoprotection associated with mild hypothermia (32°C to 33°C) despite statistically insignificant changes in glutamate release (Palmer et al, 1993).
The mechanisms underlying the temperature dependence of postsynaptic Zn2+ accumulation after TBI are not fully understood. Temperature-dependent release of Zn2+ from hippocampal MFs has previously been showed after depolarization induced by high potassium (Suh et al, 2000b). Decreased zinc translocation during hypothermia may simply be a result of a general decrease of synaptic Zn2+ release and decrease of permeability of postsynaptic neurons. The stimulation-dependent release of zinc ions from hippocampal MF has been shown after electrical stimulation (Crowford et al, 1973; Danscher et al, 1975; Sloviter, 1985; Howell et al, 1984; Assaf and Chung, 1984) and after mechanical trauma (Suh et al, 2000a). High frequency of electrical stimulation and high extracellular K+ after mechanical trauma cause a release of zinc ions into the synaptic clefts, most likely followed by a ZnT3-dependant reuptake into the presynaptic terminals (Palmiter et al, 1996). The zinc reuptake is fast after release (Howell et al, 1984). We therefore injected CaEDTA, which does not penetrate cellular membranes, before inducing TBI to block Zn2+ re-uptake. After pretreatment with CaEDTA, the zinc depletion became obvious in the normothermic rats 1 h after TBI and hyperthermia significantly potentiated that vesicular zinc depletion. In the hypothermic rat, however, the depletion of the extracellular zinc ions even using CaEDTA had no observable effect after TBI compared with sham-operated rats. The traumatic release of zinc ions out of the ZEN terminals was seemingly stopped by hypothermia. These results strongly support our hypothesis that the temperature-dependent hippocampal neuronal death is closely related to vesicular zinc ion release after TBI.
This study has several limitations. First, the number of animals in each group is small. However, despite the handful number of animals per group, the differences are highly significant because of the small variance between animals managed at similar temperatures. Second, the results of this study could be restricted only to the weight drop model of trauma and to the intensity of trauma in this study. Arguing against this possibility are data suggesting that moderate FPI also is associated with considerable zinc translocation. Third, this study showed that pretraumatic hypothermia reduced Zn2+ translocation; posttraumatic hypothermia should also be studied to determine whether posttreatment also reduces Zn2+ translocation. Fourth, the recent demonstration that mice lacking a zinc transporter ZnT3 and without zinc staining in their axonal boutons show intracellular Zn2+ staining after seizures suggests that caution be used before inferring translocation, from the fact that an ion evidently disappears from one process and simultaneously appears in another (Lee et al, 2000). Therefore, temperature-dependent intracellular Zn2+ release from the subcellular source after TBI should also be tested. Finally, slightly higher temperatures than 30°C should be studied because the range of hypothermia that has been clinically applied during intensive care of head-injured patients is approximately 33°C to 34°C.
Further investigation of the role of Zn2+ translocation in after TBI is indicated. As the brain regions that are most vulnerable to neuronal death after ischemia, seizures and TBI also have the highest content of Zn2+-containing terminals (Frederickson et al, 2000), we speculate that zinc translocation is one of the fundamental mechanisms of brain injury after head trauma. The present results suggest that therapeutic strategies for minimizing neuron loss after TBI could include manipulation of presynaptic release of Zn2+, postsynaptic entry of Zn2+ into cells and intracellular accumulation of Zn2+.
Footnotes
Acknowledgements
The authors gratefully acknowledge the skillful technical assistance of D Jensen, A Meier, K Wiedemann, L Munkø, Neus F Pons, Cathy Jean Frederickson and Aaron Hamby.